

Abstract
Gene promoter hypermethylation in sputum is a promising biomarker for predicting lung cancer. Identifying factors that predispose smokers to methylation of multiple gene promoters in the lung could impact strategies for early detection and chemoprevention. This study evaluated the hypothesis that double-strand break repair capacity and sequence variation in genes in this pathway are associated with a high methylation index in a cohort of current and former cancer-free smokers. A 50% reduction in the mean level of double-strand break repair capacity was seen in lymphocytes from smokers with a high methylation index, defined as ≥ 3 of 8 genes methylated in sputum, compared to smokers with no genes methylated. The classification accuracy for predicting risk for methylation was 88%. Single nucleotide polymorphisms within the MRE11A, CHEK2, XRCC3, DNA-Pkc, and NBN DNA repair genes were highly associated with the methylation index. A 14.5-fold increased odds for high methylation was seen for persons with ≥ 7 risk alleles of these genes. Promoter activity of the MRE11A gene that plays a critical role in recognition of DNA damage and activation of ATM was reduced in persons with the risk allele. Collectively, ours is the first population-based study to identify double-strand break DNA repair capacity and specific genes within this pathway as critical determinants for gene methylation in sputum, that is, in turn, associated with elevated risk for lung cancer.
Keywords: promoter methylation, DNA double strand break, single nucleotide polymorphism, DNA repair capacity, association study
Introduction
Lung cancer, the leading cause of cancer mortality in both men and women in the United States, now accounts for approximately 30% of all deaths from cancer (1). The lack of a validated screening approach for early detection and the resistance of advanced-stage tumors to therapy are largely responsible for the 5-year survival rate of 14% for lung cancer patients (2). The discovery of field cancerization in the respiratory tract of smokers prompted studies leading to the discovery that inactivation of genes such as p16 by promoter hypermethylation occurs in precursor lesions to non-small cell lung cancer (3). This finding suggested that methylation, when detected in exfoliated cells within sputum, could serve as a biomarker for the early stages of lung carcinogenesis (4). To test this hypothesis, our group examined a large panel of genes for their ability to predict lung cancer in a nested case-control study. A combination of six genes was identified whose methylation in sputum predicted lung cancer prior to clinical diagnosis with both a sensitivity and specificity of 65% (5). A better understanding of the susceptibility factors that predispose smokers to the acquisition of multiple epigenetic alterations in key cellular regulatory genes within the respiratory epithelium could improve prediction of lung cancer risk and impact strategies for early detection and chemoprevention.
The precise mechanisms by which carcinogens disrupt the cells' capacity to maintain the normal epigenetic code during DNA replication and repair are largely unknown. Smoking accounts for > 90% of lung cancer. Carcinogens within tobacco induce single- and double-strand breaks (DSBs) in DNA, and reduced capacity for repair of DNA damage has been associated with lung cancer (6). Accumulating evidence suggests that extensive DNA damage, manifested through DSBs, could in part be responsible for the acquisition of aberrant gene promoter methylation during lung carcinogenesis. For example, the prevalence of promoter methylation of the p16 gene was significantly greater in adenocarcinomas from workers occupationally exposed to plutonium, an exposure that predominantly produces DSBs, than in cancer from unexposed smokers (7). The prevalence of p16 methylation increased with increasing plutonium exposure. In a second study, the prevalence of methylation of the estrogen receptor α gene promoter was greater in plutonium-induced adenocarcinomas in rodent lung tumors compared to tumors induced by NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone], diesel exhaust, or carbon black exposures which mainly induce single-strand breaks of DNA (8). These studies support the hypothesis that DSBs may play a key role in the development of aberrant gene promoter hypermethylation. The purpose of this study was to test the hypothesis that a high methylation index (defined as the methylation of three or more gene-specific promoters detected in sputum) is associated with a reduced capacity to repair DSBs. We also hypothesize that sequence variation in genes from the DSB repair pathway will predict for high methylation index.
Materials and Methods
Study population and sample collection
The Lovelace Smokers Cohort (n = 1860) was established in 2001 to conduct longitudinal studies on molecular markers of respiratory carcinogenesis in biological fluids such as sputum from people at risk for lung cancer. The institutional review board of the Lovelace Respiratory Research Institute approved a protocol outlining the procedures for subject enrollment, biological specimen collection (sputum and blood), processing, and data management. Cohort subjects were recruited through advertisement in local newspaper, radio and television programs, and were mainly residents in the Albuquerque metropolitan area. Enrolled subjects were between 40 and 75 years of age with at least a 20-pack year smoking history. All participants signed the consent form. At enrollment, individual information about medical, family, and smoking, exposure history, and quality of life was collected through a computer-based system. Induced sputum and blood were collected and pulmonary function testing was performed. Blood was processed within 2 h after blood draw to isolate lymphocytes and plasma. Cyropreservation of lymphocytes began in 2005.
Cytologically adequate sputum samples from 824 cohort subjects were evaluated for gene promoter methylation of eight genes as described below. High methylation index was defined as the methylation of three or more gene-specific promoters in sputum. We selected persons from our cohort that exhibited a high (cases) or low (controls [0 of 8 genes]) methylation index. To increase the stringency for case selection, GATA4, which was most commonly methylated in sputum, was excluded as one of the three methylated genes needed for case classification and 131 of 824 cohort subjects met this criteria. Cases were frequency matched by gender to controls. Cases (n = 131) and controls (n = 130) were selected for the genetic association study. Among the 131 cases, 77 had adequate number of cryopreserved lymphocytes for the mutagen sensitivity assay. Seventy-eight controls were selected from the 130 controls, with frequency matching by gender maintained, for the mutagen sensitivity assay.
Sputum cytology and nested methylation-specific PCR
Sputum samples were stored in Saccomanno's fixative. Three slides were made for each sputum sample to check for adequacy defined as the presence of deep lung macrophages or Curschmann's spiral (9). The methylation specific PCR assay was only performed on cytologically adequate sputum samples. Eight genes (p16, MGMT, DAPK, RASSF1A, PAX5 α, PAX5 β, GATA4 and GATA5) were selected for analysis of methylation in sputum based on our previous studies establishing their association with risk for lung cancer (5, 10-12). Nested MSP was used to detect methylated alleles in DNA recovered from the sputum samples as described (5, 10-12).
Evaluation of double-strand break repair capacity (DSBRC) in peripheral lymphocytes
PHA-stimulated lymphocytes were treated with bleomycin to evaluate the generation of chromosome aberrations as an index of DSBRC (13). Briefly, cryopreserved lymphocytes were thawed and cultured in RPMI1640 medium supplemented with FBS (20%) and PHA (1.5%) at a cell density of < 0.5 × 106 /ml. Sixty-seven hours after PHA stimulation, the culture was split into two T25 flasks and treated with bleomycin or vehicle for 5 h. The final concentration for bleomycin in culture medium was 3 U/l, a concentration defined through dose-response studies using isolated lymphocytes from cohort subjects and two lymphoblastoid cells lines: GM02782 (mutant ATM) and GM00131 (wild-type ATM) (data not shown). The dose selected was within the linear dose-response range and caused obvious genotoxicity, but minimal cytotoxicity. One hour before harvest, colcemid was added to the cultures at a final concentration of 0.06 mg/ml. Slides were prepared according to conventional procedures and 100 well-spread metaphases were examined for chromatid breaks. Samples were assayed as a batch, and slides were scored by a person blinded to case-control status. The criteria of Hsu et al. (13) were used to record the aberrations: a chromatid break was scored as one break and each isochromatid break set was scored as two breaks. Chromosome/chromatid gaps, chromosome-type aberrations (dicentrics, ring, and acentric fragments) or chromatid exchanges were recorded, but not added to the frequencies of chromatid breaks. On rare occasions, a metaphase with > 12 breaks was observed on a slide with bleomycin treatment. When this occurred, the number breaks was recorded as 12. The DSBRC was expressed as the mean number of chromatid breaks per cell.
The means of spontaneous chromatid breaks per cell derived from 100 metaphases of untreated cells were 0.013 in cases and 0.021 in controls, which were similar to the spontaneous frequency reported in the literature (13) and < 1/15 the mean number of breaks seen in bleomycin-treated cells (0.32). Therefore, for statistical comparisons, the spontaneous breaks were not subtracted from the breaks observed following treatment with bleomycin.
SNP selection and genotyping by illumina platform for 16 genes in the double-strand break repair pathway and related cell cycle control genes
A total of 294 SNPs were selected for 16 candidate genes from DSBR and cell cycle control pathways. Tag SNPs (n = 245) were derived from Latino and White data from University of Southern California plus phase 1 HapMap for whites for 15 genes. Tag SNPS were selected using r2 ≥ 0.8 with nonsynonymous SNPs retained as the tag SNPs (14). One additional SNP for bins with at least six or more SNPs was selected as a redundant SNP in case of genotyping failure. For the remaining gene, NBN, 49 SNPs were selected using dbSNPs based on a SNP density of 1−3 SNPs/kb depending on the haplotype block structure, validation status, Illumina design score, and functional potential of the SNPs. The number of SNPs selected for each of these 16 genes is shown in Supplemental Table 1, and a SNP list is available on request. These SNPs were genotyped by the Illumina Golden Gate Assay for 261 DNA samples isolated from lymphocytes of cases and controls.
Selection of subjects and construction of MRE11A promoter constructs
Five common haplotypes (6−34%) were constructed based on the 14 tag SNPs assayed for MRE11A in the population (15). A Bayesian statistical method implemented in the program PHASE (Version 2.1) was used to reconstruct the haplotypes from the SNPs in the MRE11A gene for the 261 subjects. Two subjects homozygous for the haplotype that contained the RS7117042 SNP associated with high methylation index were selected. The other four people selected were each homozygous for one of the other four haplotypes. The MRE11 promoter fragment (−2541 to −5 with +1 being the translational start site) was amplified from lymphocyte DNA from these six subjects. The promoter fragment was directionally subcloned into the pGL2-basic Luciferase Reporter Vector (Promega, Madison, WI) upstream of the luciferase coding sequence. Five clones from each person were commercially sequenced to identify variants within the promoter region (Sequetech, Mountain View, CA).
Transient transfection and reporter gene assays
The Calu 6 lung tumor-derived cell line was used for transient transfections. Cells (1.5 × 105) were plated into 6-well dishes and transfected the following day. Plasmid DNA (1 μg) and the pSV-β-Galactosidase control vector (0.5 μg, Promega) were co-transfected into cells with FuGENE 6 transfection reagent (ROCHE Diagnostics, Indianapolis, IN) at a FuGENE:DNA ratio of 3:1. A promoter-less pGL2-basic vector and the pGL2-control vector that contains the SV40 promoter were used as negative and positive controls, respectively. Forty-eight hours after transfection, cells were harvested and lysed. Immediately after lysing, cell extracts were assayed in a luminometer for luciferase activity using the Lumionskan Ascent luminometer (Thermo Electron, Milford, MA) for luciferase activity using the Luciferase Assay System (Promega). β-galactosidase activity in cell lysates was measured using the Galacto-Star Reporter Gene Assay System (Tropix, Bedford, MA). Promoter activity was calculated as the ratio of activities of luciferase and β-galactosidase. Transfections were done in duplicate in four independent experiments.
Statistical analysis
The two-sample t-test, Wilcoxon rank sum test, and χ2 test were employed to compare the mean or distribution of several demographic variables and DSBRC results between cases and controls as appropriate. Because the DSBRC data and the number of spontaneous breaks were not normally distributed, analysis was also performed on log-transformed data. The results based on log-transformed data were similar to those based on untransformed data so only results based on untransformed data are shown. Analysis of covariance and logistic regression were used to assess the association between selected variables such as SNPs and case-control status, and the outcome variable, DSBRC with adjustment of covariates selected a priori (age at sputum collection, sex, race, current smoking status, and pack years). DSBRC was dichotomized for logistic regression models using the upper quartile of DSBRC in control participants. The selection of the upper quartile of DSBRC in controls as the cutt-off value was based on the distribution of DSBRC in cases and controls. Analysis of covariance and logistic regression models, stratified by status were also examined for different associations between SNPs and DSBRC by case-control status. A ROC curve was also drawn to compare the sensitivity and specificity of DSBRC induced by bleomycin for classifying cases (16). Multivariate unconditional logistic regression assessed the association between SNPs and the outcome of case-control status, with the same covariates outlined above. Model results are presented as ORs with 95% CIs for having ≥ 3 methylated genes. Logistic regression modeling was extended to generalized logit models to more precisely examine the high methylation index. ORs and 95% CIs for the risk of having 3, 4 or ≥ 5 methylated genes with 0 methylated genes as the reference group was obtained with adjustment for the same covariates.
The call rate for each SNP was assessed prior to data analysis. For the 294 SNPs assayed, 42 were deemed unsuitable because they were monomorphic, had MAF < 0.05, low yield (< 80%), or showed a highly significant distortion from Hardy-Weinberg equilibrium (p < 0.0001). These SNPs were removed from analysis. Four models were tested: co-dominant, dominant, additive, and recessive. Because of power limitations, only results for the additive model are presented for each SNP, and common homozygote, heterozygote, and rare homozygote were coded as 0, 1, and 2, respectively. A logistic regression model was used to calculate the ORs and 95% CIs for each individual SNP with adjustment for age, sex, ethnicity, and smoking selected a priori. A ROC curve was drawn to evaluate the classification accuracy of this panel of variables for promoter methylation. An analysis excluding the 23% of study subjects that were not of non-Hispanic white origin had no effect on the identified associations. Therefore, all 261 subjects were included in the data analysis.
Monte Carlo estimates of exact p-values were calculated by permuting the case-control status for all subjects 10,000 times to adjust for multi-comparisons. False positive report probability (FPRP) was also calculated to address the robustness of our findings for individual SNPs (17). In assigning a prior probability for these genes, we considered the strong association between DSBRC and risk for promoter methylation and the stringent r2 value (0.8) for selecting tag SNPs. On the basis of the evidence for associations between SNPs in CHEK2, XRCC3, DNA-PKc, NBN, LIG4, and XRCC2 and several cancers (18-25), we assigned a relatively high prior probability range (0.1−0.25) for SNPs of these six genes. In contrast, for MRE11A, Ku80, RAD50, and CHEK1, a relatively low prior probability range (0.01−0.1) was assigned because there are no studies that have addressed the association of variants within these genes to cancer. All data analyses were performed with SAS/STAT and SAS/GENETICS 9.1.3.
Results
Gene Methylation in Sputum
Gene promoter methylation was assessed in sputum from 824 members of the Lovelace Smokers Cohort, a cohort of current and former cancer-free smokers (Table 1). Methylation of an eight-gene panel that included p16, O6-methylguanine-DNA methyltransferase (MGMT), death associated protein kinase (DAPK), ras effector homolog 1 (RASSF1A), GATA4, GATA5, PAX5 α, and PAX5 β was evaluated. Methylation of these genes has been associated with increased risk for lung cancer (5,10-12). The prevalence of methylation ranged from 1.2% for RASSF1A to 31% for GATA4 and was not associated with family history for lung cancer (Supplemental Table 2). Nineteen percent of cohort members were methylated for three or more genes (Supplemental Table 2). Our previous nested case-control study within the Colorado Cohort revealed that methylation of ≥ 3 genes from a 6-gene panel (excluding GATA4 and PAX5 α) was associated with a 6.5-fold increased risk for lung cancer (5).
Table 1.
Characteristics of study participants: mutagen sensitivity assay and genetic association study.
| Variables | Cohort members with methylation results | Mutagen sensitivity assay |
Genetic association study |
||||
|---|---|---|---|---|---|---|---|
| Cases | Controls | P value | Cases | Controls | P value | ||
| Total | 824 | 77 | 78 | 131 | 130 | ||
| Age at enrollment, mean ± SD | 56.7 ± 9.7 | 59.6 ± 9.2 | 55.1 ± 9.4 | 0.003* | 57.2 ± 9.9 | 55.0 ± 9.7 | 0.067* |
| < 51 (%) | 33 | 22 | 38 | 0.005† | 31 | 38 | 0.104† |
| 51−63 | 38 | 34 | 41 | 37 | 41 | ||
| ≥ 63 | 29 | 44 | 21 | 32 | 21 | ||
| Gender (%) | |||||||
| Female | 79 | 62 | 62 | 0.918† | 72 | 72 | 0.921† |
| Male | 21 | 38 | 38 | 28 | 28 | ||
| Race (%) | |||||||
| Non-Hispanic White | 76 | 74 | 73 | 0.510† | 76 | 77 | 0.733† |
| Hispanic | 17 | 17 | 22 | 18 | 18 | ||
| Others | 6 | 9 | 5 | 7 | 5 | ||
| Smoking history | |||||||
| Current (%) | 55 | 47 | 63 | 0.045† | 49 | 57 | 0.172† |
| Pack years, mean ± SD | 40.5 ± 21.5 | 42.8 ± 25.0 | 39.5 ± 22.6 | 0.393* | 42.2 ± 24.3 | 40.4 ± 21.4 | 0.524* |
| Duration, mean ± SD | 33.7 ± 9.7 | 33.4 ± 9.7 | 32.6 ± 8.9 | 0.569* | 33.3 ± 9.9 | 32.9 ± 9.4 | 0.804* |
| Chronic airway obstruction (%)‡ | 26 | 36 | 26 | 0.181† | 36 | 32 | 0.458† |
| Spontaneous chromatid breaks/cell, mean ± SD | — | 0.013 ± 0.015 | 0.021 ± 0.025 | 0.085§ | |||
Two-sided two-sample t test between cases (methylated group) and controls (unmethylated group).
χ2 test for differences between cases and controls.
Chronic airway obstruction is defined as post-bronchodilator FEV1/FVC % < 70%.
Two-sided Wilcoxon rank sum test between cases and controls.
Repair capacity associates with methylation index
The mutagen sensitivity assay was used to assess double-strand break repair capacity (DSBRC) (13). The number of chromatid breaks induced in lymphocytes following exposure to bleomycin, a radiomimetic agent, was used to measure DSBRC. We selected persons from our cohort who exhibited a high (cases [≥ 3 methylated genes]) or low (controls [zero of eight genes methylated]) methylation index because of the increased risk for lung cancer seen in our Colorado nested, case-control study when 3 or more genes were methylated in sputum. Cryopreserved lymphocytes were available for assessment of DSBRC for 77 cases and 78 controls. Demographics and smoking history for cases and controls are detailed in Table 1. A highly statistically significant difference was seen in DSBRC (p < 0.001) between cases and controls with a mean number of chromosome breaks per cell of 0.47 ± 0.11 and 0.32 ± 0.10, respectively (Fig. 1A). The mean number of bleomycin-induced chromatid breaks per cell was significantly higher in cases than in controls when subjects were stratified by age, sex, race, chronic airway obstruction, pack years, and smoking status indicating that none of these covariates were major confounders for the strong association seen between DRC and methylation index (Supplemental Table 3). We further classified the cases into three groups based on the number of methylated genes (3, 4 and ≥5 methylated genes) and found that the number of chromatid breaks per cell induced by bleomycin increased with the increasing number of methylated genes in sputum (p < 0.001; Fig. 1B). Age did not differ in cases with 3, 4 and ≥ 5 methylated genes. Finally, after adjusting for sex, race, current smoking status, cigarette pack years, seeding number of lymphocytes, cryopreservation time, and log-transformed spontaneous chromatid breaks per cell, age was the only factor significantly associated with chromatid breaks induced by bleomycin in both cases and controls (Supplemental Table 3). The reduction of DNA repair capacity with age is well established and supports the accuracy of the mutagen sensitivity assay in this study (13).
Fig. 1.

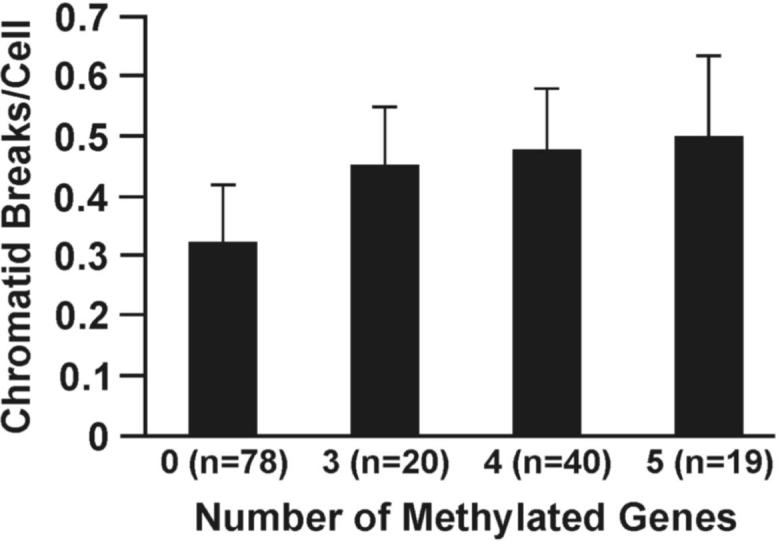


DNA repair capacity is associated with gene promoter methylation in sputum. (A) Bleomycin treatment causes an increased number of chromatid breaks/cell in lymphocytes from cases (methylated group) compared to controls (unmethylated group; p < 0.0001). (B) Positive association between number of methylated genes and chromatid breaks/cell. Sample size for each group is indicated in parentheses. (C) ROC curve comparing sensitivity and specificity of DNA repair capacity for classifying cases and controls. The covariates included in the ROC curve were age at sputum collection, sex, race, current smoking status, and pack years. (D) The distribution of chromatid breaks/cell by case-control status is depicted.
A receiver operator characteristic (ROC) curve was generated to determine how well DSBRC distinguished cases from controls. The ROC curve demonstrates that DSBRC significantly (p < 0.0001) increased the classification accuracy from 66% to 88% for predicting risk for promoter methylation (Fig. 1C). With the sensitivity set at 80%, the false positive rate was < 20%.
The odds ratio (OR) and 95% confidence interval were calculated to further characterize the association between methylation index and DSBRC. The OR associated with an increment of 0.1 chromatid breaks per cell for having ≥ 3 methylated genes was 6.6 (95% CI = 3.7 to 13.2) for bleomycin treatment after adjustment for selected covariates. When the number of methylated genes was used as a multinomial response variable, the OR associated with an increment of 0.1 chromatid breaks/cell for methylation of 3, 4 and ≥ 5 genes was 5.3 (95% CI, 2.7 − 11.4), 7.1 (95% CI, 3.8 − 14.8), and 8.5 (95% CI, 4.2 − 19.3), respectively. A histogram detailing the distribution of chromatid breaks/cell by case-control status revealed that 75% of controls compared to 18% of cases accumulated < 0.38 breaks/cell (Fig. 1D). We chose to dichotomize the number of DSBs per cell at the 75th percentile for controls and this gave an adequate overlap in the distribution of breaks/cell in cases and controls. The results did not differ significantly with other cut points. When methylation index was compared to chromatid breaks/cell, the ORs for detecting methylation increased monotonically from 10 to 15 to 26 (Table 2). Overall, chromatid breaks ≥ 0.38 per cell were associated with a 14.5-fold increased risk of having ≥ 3 methylated genes in sputum (Table 2).
Table 2.
Analysis of the relationship between chromatid breaks per cell categorized by upper quartile of control subjects and risk of methylation.
| Variable | Chromatid breaks/cell |
Crude OR (95% CI) | Adjusted OR (95% CI)† | |
|---|---|---|---|---|
| < 0.38* | ≥ 0.38 | |||
| Methylation index‡ | (No. of subjects) | |||
| 0 | 58 | 20 | 1.0 (reference) | 1.0 (reference) |
| 3 | 5 | 14 | 8.1 (2.6−25.4) | 9.8 (2.9−33.6) |
| 4 | 7 | 32 | 13.3 (5.1−34.7) | 15.1 (5.4−42.6) |
| ≥ 5 | 2 | 17 | 24.7 (5.2−116.2) | 25.7 (5.1−129.5) |
| Case-control§ | ||||
| Controls | 58 | 20 | 1.0 (reference) | 1.0 (reference) |
| Cases | 14 | 63 | 13.1 (6.0−23.2) | 14.5 (6.3−33.8) |
Upper quartile of breaks/cell induced by bleomycin in control subjects.
Obtained from models with adjustment for age at sputum collection, sex, race, smoking status and packyears.
Using generalized logit model.
Using logistic regression model.
SNPs within DNA repair genes and risk for methylation
The finding that DNA repair capacity strongly predicts for high methylation index, combined with its high heritability, suggests that variants in genes involved in repair should also be predictive (26). We selected 16 candidate genes from the DSBR and cell cycle control pathways for tag SNP-based genotyping (Supplemental Table 1). A total of 294 SNPs were evaluated for 131 cases and 130 controls that included the subset evaluated in the mutagen sensitivity assay.
Forty-four SNPs were associated with risk for promoter methylation (p < 0.15) with adjustment for covariates (Supplemental Table 4). Because of the relatively high correlation between SNPs in these genes, we tested which SNP, or set of SNPs, was most significantly associated with risk for promoter methylation by using a step-wise logistic regression model. The underlined SNPs in Supplemental Table 4 were selected from each gene (p < 0.15) to represent the allelic status for those genes. These 16 SNPs were then included with the covariates in one model and step-wise selection was used to identify the SNPs with the lowest p-value. Ten SNPs from different genes were identified with 4 SNPs associated with increased risk for promoter methylation (ORs, 1.6−4.0) and 6 SNPs with reduced risk for promoter methylation (ORs, 0.4−0.7) (Table 3). Monte Carlo estimates of exact p-values were calculated by permuting the case-control status for all subjects 10,000 times to adjust for multi-comparisons. The exact p-value for five SNPs (rs7117042, rs5762763, rs2295146, rs7830743 and rs6998169) was < 0.05 (Table 3). This result indicates that if a similar study were repeated under a null distribution (i.e., no SNPs associated with risk for promoter methylation), an association similar to that observed with any of these five SNPs would occur by chance < 5% of the time. The calculated FPRP was below 0.2 for 4 SNPs (rs7117042, rs5762763, rs2295146, and rs7830743) under the assigned prior probability range (Table 3). Findings with a FPRP ≤ 0.2 are considered to be noteworthy.
Table 3.
Summary of associations between genes and promoter methylation using step-wise logistic regression.
| Gene/SNPs* | OR∥ | 95%CI | P-value | Permuted P-value† | Statistical power‡ | Prior probability of FPRP§ |
||
|---|---|---|---|---|---|---|---|---|
| 0.25 | 0.1 | 0.01 | ||||||
| MRE11A / rs7117042 | 3.97 | 1.77 − 8.89 | 0.0008 | 0.0008 | 0.37 | 0.006 | 0.019 | 0.176 |
| CHEK2 / rs5762763 | 1.89 | 1.20 − 2.97 | 0.0064 | 0.0058 | 0.88 | 0.021 | 0.061 | 0.419 |
| XRCC3 / rs2295146 | 0.54 | 0.35 − 0.83 | 0.0051 | 0.0073 | 0.83 | 0.018 | 0.052 | 0.378 |
| DNA-PKc / rs7830743 | 0.38 | 0.18 − 0.80 | 0.0117 | 0.0142 | 0.70 | 0.048 | 0.131 | 0.623 |
| NBN / rs6998169 | 0.47 | 0.23 − 0.93 | 0.0452 | 0.0308 | 0.89 | 0.132 | 0.314 | 0.834 |
| LIG4 / rs1151402 | 0.68 | 0.44 − 1.06 | 0.0859 | 0.1078 | 0.98 | 0.208 | 0.441 | 0.897 |
| XRCC2 / rs3218400 | 0.55 | 0.28 − 1.06 | 0.0751 | 0.0823 | 0.92 | 0.197 | 0.424 | 0.890 |
| Ku80 / rs828911 | 1.55 | 1.02 − 2.37 | 0.0416 | 0.059 | 0.95 | 0.116 | 0.283 | 0.813 |
| RAD50 / rs2244012 | 1.64 | 0.94 − 2.76 | 0.0864 | 0.1132 | 0.98 | 0.209 | 0.442 | 0.897 |
| CHEK1 / rs537046 | 0.64 | 0.37 − 1.12 | 0.1176 | 0.1091 | 0.97 | 0.267 | 0.522 | 0.923 |
Age, sex, ethnicity, smoking status and pack years were selected a priori and forced in the model. Step-wise selection was only used to select genetic susceptibility factors. The p-values for both entry and inclusion of a variable in each round of variable selection were set at 0.1.
Case and control status was permuted 10,000 times to adjust for multi-comparison.
Statistical power is the power to detect an odds ratio of 2.0 for individual tag SNPs under an additive model.
FPRPs are calculated based on Wacholder et al. (9). Prior probabilities were set at 0.1−0.25 for rs5762763, rs2295146, rs7830743, rs6998169, rs1151402 and 218400, and at 0.01−0.1 for rs828911, rs7117042, rs7906967, rs2244012 and rs537046. Findings with FPRP ≤ 0.2 were considered noteworthy.
ORs were calculated using an additive model where common homozygote, heterozygote, and rare homozygote are coded as 0, 1 and 2, respectively.
ROC curves were generated to evaluate the classification accuracy of this panel of SNPs to distinguish cases from controls. The area under the curve increased from 57% (covariates only) to 72% (covariates with the 5 most significant SNPs) and to 75% (covariates with all 10 SNPs, Fig. 2A). The difference between the area under the curve with only covariates and the two models that included both covariates and multiple SNPs is highly significant (p < 0.001). Restricting this analysis to include only cases and controls in which DSBRC was determined resulted in an area of 82% that increased to 93% when repair capacity was included in the model. In order to test the hypothesis that the identified SNPs in different genes would work additively to influence risk for promoter methylation, the joint effect of each SNP, inclusive of both putative susceptibility alleles, was evaluated. When the 5 SNPs with the strongest association with risk for promoter methylation were included, persons with 5, 6, or ≥ 7 alleles were found to have a 2.5-, 2.8-, and 14.4-fold increased risk, respectively for ≥ 3 methylated genes in sputum compared to those with ≤ 4 alleles (Table 4). The inclusion of 10 SNPs did not greatly increase the ability to classify persons with a high methylation index. This outcome was not surprising, because the added SNPs were only weakly associated with risk for promoter methylation.
Fig. 2.


SNPs in repair genes are associated with gene promoter methylation in sputum and promoter activity of the MRE11A gene. (A) ROC curve comparing sensitivity and specificity of SNPs within DNA repair genes for classifying cases and controls. (B) Difference in MRE11A promoter activity by haplotype. Values are mean ± SD from transfection of two constructs containing each haplotype four times. * p < 0.05 compared to ACGACTG.
Table 4.
Association between number of risk alleles and promoter methylation in the Lovelace Smokers Cohort.
| No. of high-risk alleles | Cases (%) N=128 | Controls (%) N=130 | ORs (95%CI)* |
|---|---|---|---|
| Top 5 SNPs† | |||
| ≤ 4 | 9 (7.0) | 29 (22.3) | 1.00 (reference) |
| 5 | 36 (28.1) | 45 (34.6) | 2.54 (1.06−6.53) |
| 6 | 37 (28.9) | 44 (33.9) | 2.84 (1.18−7.33) |
| ≥ 7 | 46 (35.9) | 12 (9.2) | 14.39 (5.37−42.45) |
| All 10 SNPs‡ | |||
| ≤ 10 | 13 (10.3) | 50 (39.1) | 1.00 (reference) |
| 11 | 26 (20.6) | 26 (20.3) | 4.09 (1.78−9.79) |
| 12 | 31 (24.6) | 34 (26.6) | 3.68 (1.69−8.40) |
| ≥ 13 | 56 (44.4) | 18 (14.1) | 13.73 (6.08−33.21) |
Unconditional logistic regression with adjustment for age, sex, ethnicity, smoking status, and pack years.
Top 5 SNPs include rs7117042, rs5762763, rs2295146, rs7830743 and rs6998169.
All 10 SNPs include rs7117042, rs5762763, rs2295146, rs7830743, rs6998169, rs1151402, rs3218400, rs828911, rs2244012 and rs537046.
Reduced activity of the MRE11A promoter
The genes included in the prediction model have biological plausibility. Two of the five genes whose sequence variation is associated with methylation, NBN and XRCC3, have shown association with lung cancer (18,27). SNPs within the DNA-PKc and CHEK2 genes have been associated with breast and other cancers, while no studies have been conducted with MRE11A (19,28). Assessment of the functional potential of the SNPs identified from our study for these genes revealed that DNA-PKc/rs7830743 is a nonsynonymous SNP changing amino acid residue 3434 from Ile to Thr in exon 73. This amino acid substitution is predicted to change the secondary structure and may influence the serine/threonine protein kinase activity of this protein (29). We have shown that reduced DNA-PK activity is associated with risk for lung cancer and sensitivity to cell killing by bleomycin (30), thus supporting an important role for this gene in lung cancer and aberrant gene promoter methylation. The SNPs from the other four genes are neither nonsynonymous or in high linkage disequilibrium with any nonsynonymous SNP with known function. However, MRE11A/rs7117042 and NBN/rs6998169 are predicted to locate in the middle of the sequence, forming DNA triplexes that could inhibit DNA transcription (31). To begin addressing function of these SNPs, we tested whether MRE11A/rs7117042 is associated with a reduction in promoter activity.
Two subjects homozygous for the haplotype containing rs7117042 and 4 subjects, each homozygous for one of the other four common haplotypes were selected for assessment of promoter activity (Supplemental Table 5 and described under Methods). Sequencing of the 2500 bp promoter construct revealed three haplotypes (ACGACTG, GCACTAT, and AGGCTTG), with each haplotype present in two subjects. The most distinct sequence difference was the G to C change at −590 bps. We genotyped 100 subjects selected randomly from our study population for this SNP and found that the G allele was in complete linkage disequilibrium with the T allele of the risk SNP, rs7117042, identified to be most strongly associated with high methylation index. The highest promoter activity was seen in constructs containing the ACGACTG haplotype. With this haplotype as the reference, a 23% and 38% reduction in promoter activity was seen for the GCACTAT and AGGCTTG haplotypes, respectively (Fig. 2B). These results show that the risk tag SNP is associated with a marked reduction in transcription of the MRE11A gene. MRE11A has a critical role in recognition of double-strand break damage. It complexes with Rad50 and Nbs1 to directly sense the double-strand breaks, binds to the DNA, modifies the ends via 3′ to 5′ exonuclease activity, recruits ATM to the damaged DNA template, and dissociates the ATM dimer (32). Therefore, a reduction in level of the MRE11A protein could have a major impact on DSBRC.
Discussion
These results indicate a strong link between reduced DSBRC and risk for methylation in sputum. Our studies transcend from a functional assay for DNA repair to specific genotypes, and finally demonstrate an activity deficit of the MRE11A gene that plays a critical role in recognition of double-strand break DNA damage and activation of the ATM gene (32). The mechanism underlying this association could in part, be mediated by the genes that are recruited to sites of DSBs, and the resultant modification of chromatin to facilitate repair. One of the earliest responses to DSB damage is phosphorylation by ataxia telangiectasia mutated kinase (ATM) of the histone H2AX which then facilitates accumulation of repair/signaling proteins and also SWI/SNF complexes that have been implicated in transcriptional silencing to chromatin regions distal to a DSB (33,34). A recent study demonstrated activation of H2AX by ATM in the A549 lung tumor-derived cell line by tobacco smoke (35).
Another key contributing factor for aberrant de novo methylation during DNA damage is the rapid recruitment of DNMT1 to sites of DNA damage (36). Le Gac et al. (37) found that in cells treated with doxorubicin which induces DSBs, DNMT1 is recruited by activated p53 and binds to functional Sp1 sites within promoters of the survivin, cdc2, and cdc25 genes. Moreover, the transcriptional repressor HDAC1 and the repressive chromatin mark H3K9me2 were also found at these promoters following DNA damage (37,38). Subsequent in vitro studies showed that following DSB DNA damage induced by doxorubicin, DNMT1 complexed with p53 was recruited to the survivin gene promoter followed by de novo methylation and gene silencing (39). Cuozzo et al. (40) provides even stronger support for a mechanistic link between DNA damage and methylation. In that study, a recombinant plasmid containing a 1-SCE1 restriction site within one copy of two inactivated tandem repeated green fluorescent protein (GFP) genes was introduced into Hela, or mouse embryonic stem cells. The restriction endonuclease 1-Sce1 was added to the cell to induce a DSB in the GFP gene at this site. Rapid gene silencing associated with homologous recombination and DNA methylation of the recombinant gene was seen and could be blocked by treatment with the demethylating agent, 5-aza-deoxycytidine. Chromatin immunoprecipitation revealed that DNMT1 was bound specifically to the homologous GFP DNA. Together, these in vitro studies strongly support a direct mechanistic link between DNA damage and induction of de novo methylation by DNMT1. Our population-based studies now provide for the first time, an in vivo association between DRC and gene promoter methylation, both through a functional assay and genetic variants in genes within the double-strand break repair pathway. Thus, in the absence of efficient repair, the recruitment of p53, DNMT1, and transcriptional repressors to many genes such as p16 that also contain Sp1 sites within its promoter could lead to de novo methylation and gene silencing.
The identification of DSBRC and specific genes within this pathway as a critical determinant for gene promoter hypermethylation has important implications for basic and translational science. Our study substantiates that DNA damage that has long been recognized as an initiating event for mutagenesis, is also likely a major factor in initiating aberrant promoter hypermethylation. Other DNA damage response pathways such as apoptosis, nucleotide and base excision repair may also contribute to the induction of aberrant promoter hypermethylation. A major priority for our research is to replicate the provocative findings in this study along with our emerging methylation gene panel in a prospective population-based study. Genetic variants associated with promoter hypermethylation could be used to identify young smokers who would be most susceptible to induction of preneoplasia, and thus, should receive chemoprevention. In addition, the integration of these genetic variants with detection of gene promoter hypermethylation in sputum in long-term heavy smokers could lead to the first diagnostic test for incident lung cancer and impact long-term survival from this fatal disease.
Acknowledgments
These studies were supported by U01 CA097356 and the State of New Mexico as a direct appropriation from the Tobacco Settlement Fund. We thank Trisstin Maroney for her technical assistance.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Notes: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Conflict of interest statement: Steven Belinsky is a consultant to Oncomethylome Sciences. Under a licensing agreement between Lovelace Respiratory Research Institute and Oncomethylome Sciences, nested methylation-specific PCR was licensed to Oncomethylome Sciences and the author is entitled to a share of the royalties received by the Institute from sales of the licensed technology. The Institute, in accordance with its conflict-of-interest policies, is managing the terms of these arrangements.
Supplementary Material
Supplemental Table 1. Number of SNPs in the 16 genes evaluated for association with gene methylation.
Supplemental Table 2. Prevalence of gene promoter methylation in sputum from 824 cohort members.
Supplemental Table 3. Chromatid break per cell induced by bleomycin between cases and controls stratified by covariates.
Supplemental Table 4. Individual SNPs associated with risk for promoter methylation at p values ≤ 0.15 for the 16 genes evaluated in this study.
Supplemental Table 5. Common haplotypes of the MRE11A gene.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA. Cancer J. Clin. 2006;56:106–30. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, Isselbacher KJ, editors. Harrison's Principles of Internal Medicine. 16th ed McGraw-Hill: 2004. pp. 506–16. [Google Scholar]
- 3.Belinsky SA, Nikula KJ, Palmisano WA, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. U. S. A. 1998;95:11891–6. doi: 10.1073/pnas.95.20.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 5.Belinsky SA, Liechty KC, Gentry FD, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–44. doi: 10.1158/0008-5472.CAN-05-3408. [DOI] [PubMed] [Google Scholar]
- 6.Shen H, Spitz MR, Qiao Y, et al. Smoking, DNA repair capacity and risk of nonsmall cell lung cancer. Int. J. Cancer. 2003;107:84–8. doi: 10.1002/ijc.11346. [DOI] [PubMed] [Google Scholar]
- 7.Belinsky SA, Klinge DM, Liechty KC, et al. Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis. 2004;25:1063–7. doi: 10.1093/carcin/bgh096. [DOI] [PubMed] [Google Scholar]
- 8.Belinsky SA. Silencing of genes by promoter hypermethylation: key event in rodent and human lung cancer. Carcinogenesis. 2005;26:1481–7. doi: 10.1093/carcin/bgi020. [DOI] [PubMed] [Google Scholar]
- 9.Saccomanno G. Diagnostic Pulmonary Cytology. 2nd ed Amer. Society of Clinical Pathologists; Chicago: 1986. [Google Scholar]
- 10.Palmisano WA, Divine KK, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000;60:5954–8. [PubMed] [Google Scholar]
- 11.Belinsky SA, Palmisano WA, Gilliland FD, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62:2370–7. [PubMed] [Google Scholar]
- 12.Palmisano WA, Crume KP, Grimes MJ, et al. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer Res. 2003;63:4620–5. [PubMed] [Google Scholar]
- 13.Hsu TC, Johnston DA, Cherry LM, et al. Sensitivity to genotoxic effects of bleomycin in humans: possible relationship to environmental carcinogenesis. Int. J. Cancer. 1989;43:403–9. doi: 10.1002/ijc.2910430310. [DOI] [PubMed] [Google Scholar]
- 14.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 2004;74:106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 16.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 17.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J. Natl. Cancer Inst. 2004;96:434–42. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan Q, Shen M, Berndt SI, et al. Smoky coal exposure, NBS1 polymorphisms, p53 protein accumulation, and lung cancer risk in Xuan Wei, China. Lung Cancer. 2005;49:317–23. doi: 10.1016/j.lungcan.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Wang LE, Bondy ML, Shen H, et al. Polymorphisms of DNA repair genes and risk of glioma. Cancer Res. 2004;64:5560–3. doi: 10.1158/0008-5472.CAN-03-2181. [DOI] [PubMed] [Google Scholar]
- 20.Manuguerra M, Saletta F, Karagas MR, et al. XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am. J. Epidemiol. 2006;164:297–302. doi: 10.1093/aje/kwj189. [DOI] [PubMed] [Google Scholar]
- 21.Einarsdóttir K, Humphreys K, Bonnard C, et al. Effect of ATM, CHEK2 and ERBB2 TAGSNPs and haplotypes on endometrial cancer risk. Hum. Mol. Genet. 2007;16:154–64. doi: 10.1093/hmg/ddl451. [DOI] [PubMed] [Google Scholar]
- 22.Kilpivaara O, Alhopuro P, Vahteristo P, Aaltonen LA, Nevanlinna H. CHEK2 I157T associates with familial and sporadic colorectal cancer. J. Med. Genet. 2006;43:e34. doi: 10.1136/jmg.2005.038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roddam PL, Rollinson S, O'Driscoll M, Jeggo PA, Jack A, Morgan GJ. Genetic variants of NHEJ DNA ligase IV can affect the risk of developing multiple myeloma, a tumour characterised by aberrant class switch recombination. J. Med. Genet. 2002;39:900–5. doi: 10.1136/jmg.39.12.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goode EL, Dunning AM, Kuschel B, et al. Effect of germ-line genetic variation on breast cancer survival in a population-based study. Cancer Res. 2002;62:3052–7. [PubMed] [Google Scholar]
- 25.Tempfer CB, Hefler LA, Schneeberger C, Huber JC. How valid is single nucleotide polymorphism (SNP) diagnosis for the individual risk assessment of breast cancer? Gynecol. Endocrinol. 2006;22:155–9. doi: 10.1080/09513590600629175. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Spitz MR, Amos CI, et al. Mutagen sensitivity has high heritability: evidence from a twin study. Cancer Res. 2006;66:5993–6. doi: 10.1158/0008-5472.CAN-06-1007. [DOI] [PubMed] [Google Scholar]
- 27.Matullo G, Dunning AM, Guarrera S, et al. DNA repair polymorphisms and cancer risk in non-smokers in a cohort study. Carcinogenesis. 2006;27:997–1007. doi: 10.1093/carcin/bgi280. [DOI] [PubMed] [Google Scholar]
- 28.Johnson N, Fletcher O, Palles C, et al. Counting potentially functional variants in BRCA1, BRCA2 and ATM predicts breast cancer susceptibility. Hum. Mol. Genet. 2007;16:1051–7. doi: 10.1093/hmg/ddm050. [DOI] [PubMed] [Google Scholar]
- 29.Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure. 2005;13:243–55. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Auckley DH, Crowell RE, Heaphy ER, et al. Reduced DNA-dependent protein kinase activity is associated with lung cancer. Carcinogenesis. 2001;22:723–7. doi: 10.1093/carcin/22.5.723. [DOI] [PubMed] [Google Scholar]
- 31.Conde L, Vaquerizas JM, Dopazo H, et al. PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res. 2006;34:W621–5. doi: 10.1093/nar/gkl071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 34.Park JH, Park EJ, Lee HS, et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006;25:3986–97. doi: 10.1038/sj.emboj.7601291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka T, Huang X, Jorgensen E, et al. ATM activation accompanies histone H2AX phosphorylation in A549 cells upon exposure to tobacco smoke. BMC Cell Biol. 2007;8:26. doi: 10.1186/1471-2121-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8905–9. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Gac G, Esteve PO, Ferec C, Pradhan S. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J. Biol. Chem. 2006;281:24161–70. doi: 10.1074/jbc.M603724200. [DOI] [PubMed] [Google Scholar]
- 38.Esteve PO, Chin HG, Pradhan S. Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J. Biol Chem. 2007;282:2615–25. doi: 10.1074/jbc.M606203200. [DOI] [PubMed] [Google Scholar]
- 39.Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1000–5. doi: 10.1073/pnas.0407729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuozzo C, Porcellini A, Angrisano T, et al. DNA Damage, Homology-Directed Repair, and DNA Methylation. PLoS Genet. 2007;3:e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Number of SNPs in the 16 genes evaluated for association with gene methylation.
Supplemental Table 2. Prevalence of gene promoter methylation in sputum from 824 cohort members.
Supplemental Table 3. Chromatid break per cell induced by bleomycin between cases and controls stratified by covariates.
Supplemental Table 4. Individual SNPs associated with risk for promoter methylation at p values ≤ 0.15 for the 16 genes evaluated in this study.
Supplemental Table 5. Common haplotypes of the MRE11A gene.