

Abstract
The predominant neuronal glutamate transporter, EAAC1 (for excitatory amino acid carrier-1), is localized to the dendrites and somata of many neurons. Rare presynaptic localization is restricted to GABA terminals. Because glutamate is a precursor for GABA synthesis, we hypothesized that EAAC1 may play a role in regulating GABA synthesis and, thus, could cause epilepsy in rats when inactivated. Reduced expression of EAAC1 by antisense treatment led to behavioral abnormalities, including staring–freezing episodes and electrographic (EEG) seizures. Extracellular hippocampal and thalamocortical slice recordings showed excessive excitability in antisense-treated rats. Patch-clamp recordings of miniature IPSCs (mIPSCs) conducted in CA1 pyramidal neurons in slices from EAAC1 antisense-treated animals demonstrated a significant decrease in mIPSC amplitude, indicating decreased tonic inhibition. There was a 50% loss of hippocampal GABA levels associated with knockdown of EAAC1, and newly synthesized GABA from extracellular glutamate was significantly impaired by reduction of EAAC1 expression. EAAC1 may participate in normal GABA neurosynthesis and limbic hyperexcitability, whereas epilepsy can result from a disruption of the interaction between EAAC1 and GABA metabolism.
Keywords: EAAC1, transport, antisense, GABA, metabolism, epilepsy
Glutamate transport is the major mechanism controlling extracellular glutamate levels, preventing excitotoxicity, and averting neural damage associated with epilepsy. (McBean and Roberts, 1985; Rothstein et al., 1992, 1993, 1994, 1996;Robinson et al., 1993a,b; Tanaka et al., 1997). Glutamate transporters are localized to the membranes of synaptic terminals and astroglial processes that ensheath synaptic complexes (Hertz, 1979; Kanner and Schuldiner, 1987; Danbolt et al., 1992; Kanai et al., 1993; Rothstein et al., 1994, 1996; Conti et al., 1998; He et al., 2000). GLAST (for glutamate–aspartate transporter [EAAT-1 (for excitatory amino acid transporter-1)] (Storck et al., 1992; Arriza et al., 1994) and GLT-1 (for glutamate transporter-1) (EAAT-2) (Pines et al., 1992;Arriza et al., 1994) are astroglial glutamate transporters, and EAAC1 (for excitatory amino acid carrier-1) (EAAT-3) (Kanai and Hediger, 1992; Arriza et al., 1994; Shashidharan et al., 1994; Kanai et al., 1995; Bjoras et al., 1996; Nakayama et al., 1996; Velaz-Faircloth et al., 1996; Eskandari et al., 2000), EAAT-4 (Fairman et al., 1995), and EAAT-5 (Arriza et al., 1997) are neuronal proteins. Astroglial glutamate transporters are responsible for at least 80% of the high-affinity glutamate transport and the majority of synaptic inactivation.
EAAC1 is highly concentrated in the somata and dendrites of many neurons, especially those in the hippocampus, striatum, cerebellum, and olfactory bulb. There is little evidence for glutamate transporter proteins in the presynaptic terminal, with one exception. EAAC1 has been localized to inhibitory GABAergic neurons, including cerebellar Purkinje cells (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000), which are highly concentrated in presynaptic GABAergic terminals. Because of its unusual localization to GABA terminals, we hypothesized that the loss of EAAC1 could alter presynaptic GABA metabolism, perhaps by altering a precursor supply of glutamate. In this study, we now show that EAAC1 contributes to new synthesis of inhibitory neurotransmitter GABA and that loss of EAAC1 produces “staring” epilepsy.
MATERIALS AND METHODS
Intraventricular antisense administration. All animal experiments were performed with approval by the Johns Hopkins Animal Care and Use Committee. Male Sprague Dawley rats (250–350 gm) were anesthetized with 4% chloral hydrate (1 ml/100 gm). Antisense oligonucleotides (oligodeoxynucleotides) were infused intraventricularly by mini-osmotic pumps as described previously, including oligonucleotides to EAAC1, GLAST, and GLT-1 (Rothstein et al., 1996). Lyophilized oligonucleotides were reconstituted in artificial CSF (ACSF) (Wagner, 1994; Wahlestedt, 1994; Rothstein et al., 1996) at a concentration of 2.5 mg/ml, dialyzed (Rothstein et al., 1996), and then filtered (0.22 μm) before use. Four EEG screw electrodes were placed on the skull during the same procedure (one frontal and one parietal on each side). Antisense and sense oligodeoxynucleotides were delivered at a rate of 1–10 μg/hr over a 9–12 d period.
Video–EEG recording. Four EEG electrodes (Plastics One, Roanoke,VA) were mounted on the skull with cranioplastic cement during the stereotaxic placement of the cannula (one frontal and one parietal on each side, 2 and 3 mm from the midline skull suture, respectively). Rats underwent daily EEG recording in a chamber that allowed unrestrained movement while recording. Signals were recorded using Grass EEG machine model 8–16 with amplifiers with a bandpass filter set between 1 and 70 Hz. Sensitivity was set to 7 μV/mm, and paper speed was set to 30 cm/sec. Two channels were recorded. Bipolar and referential recordings were performed while the rat was awake and mobile in the chamber. EEG was sampled for 20 min or until seizure activity occurred. If seizure activity occurred, observation was continued until 5 min of non-epileptiform EEG activity had been recorded. Clinical behavior was monitored by video camera during the EEG recording. EEG recording and interpretation was done with the recorder–interpreter blinded to the treatment.
Extracellular thalamocortical and hippocampal entorhinal cortical field potential slice recording. Four antisense EAAC1 experimental and four sense EAAC1 control rats were decapitated 10 d after infusion. Thirteen antisense and 10 sense thalamocortical slices were recorded. Rat thalamocortical slices were prepared using the slice angle developed by Agmon and Connors (1991) (Coulter and Lee, 1993). Connections were verified by microscopic examination of slices. Once cut and visualized, slices were transferred to an incubator, in which they were kept submerged in warmed (35°C) oxygenated medium until use. The slice medium was composed of (in mm): 130 NaCl, 3 KCl 3, 1.25 NaH2PO4, 0 MgCl2, 26 NaHCO3, and 10 dextrose. A separate incubator allowed preexposure of individual slices to low Mg2+ medium before recording. Slices were incubated for at least 1 hr after dissection and for 1 hr or more in low Mg2+ medium before recording. Before recording, slices were transferred to an interface-type recording chamber, in which they were maintained at 35°C. Differential AC-coupled extracellular recordings were conducted using 2 MΩ insulated tungsten electrodes (bandpass filtered at 10–3000 Hz). Recording was conducted from cortex in one channel and thalamus in the second channel.
Five antisense EAAC1 experimental and three sense EAAC1 control rats were decapitated 10 d after infusion for hippocampal entorhinal cortex (HEC) slice experiments, and eight antisense and five sense slices were recorded. In brief, brain slices were prepared using previously reported methods (Rafiq et al., 1993). Rats were anesthetized with halothane and decapitated, and the brain was quickly removed and chilled for 1–2 min in a modified sucrose-based ACSF (SACSF) composed of (in mm): 200 sucrose, 3 KCl, 1.25 NA2PO4, 0.9 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose (equilibrated with 95% O2–5% CO2). After a 1 min wash in cold SACSF, the two hemispheres were dissected by midsagittal dissection. Hemispheres were immediately returned to oxygenated, cold SACSF until needed for slicing. Each hemisphere was individually blocked and sectioned in an inclined 12° transverse plane, modified from Jones and Heinemann (1988), with the use of a vibratome (Lancer 1000; Vibratome, St. Louis, MO). Brain slices were subsequently transferred to a holding chamber in which they were kept submerged in ACSF containing (in mm): 130 NaCl, 3 KCl, 1.25 NA2PO4, 0.9 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose (warmed to 32°C and vigorously bubbled with 95% O2–5% CO2 for a period of 1–2 hr). Before recording, slices were transferred to an interface-type recording chamber, in which they were perfused with 35°C ACSF at 1–1.5 ml/min for extracellular recording experiments. Extracellular field potential recordings were made with the use of insulated tungsten electrodes, placed in the pyramidal cell body layers of area CA1, CA3, and the dentate gyrus of each slice (Jones and Heinemann, 1988; Coulter and Lee, 1993; Rafiq et al., 1993). All slice recordings and interpretation was done with the recorder–interpreter blinded to the treatment.
Quantification of excitability. EEG and hippocampal slice recordings were analyzed by manually counting spikes (Daly and Pedley, 1990) during the whole record for each rat and slice on the 10th treatment day. Spike count in the EEG recordings of all EAAC1 antisense-treated rats were compared with spike count in the EEG recordings of three controls: EAAC1 sense-, GLT-1 antisense-, and GLAST antisense-treated rats. The average number of spikes per minute in each group was compared [ANOVA, followed by Fisher's protected least significant difference (PLSD) for spikes per minute]. All of the spontaneous interictal spikes (SISs) in the hippocampal slice recordings were counted and compared between the EAAC1 antisense- and EAAC1 sense-treated rats (Student's t test). The thalamocortical slice recordings were analyzed by manual measurement of the duration of discharges in seconds and calculating the average duration of discharges (seconds discharge per minute recording) in each slice and then comparing the averages of EAAC1 antisense- and EAAC1 sense-treated rats (Student's t test).
Patch recording in brain slices. Male Sprague Dawley rats were used in all experiments. Recordings were obtained from visually identified pyramidal neurons in stratum pyramidale of area CA1 of the rat hippocampus. For the purpose of this study, animals were divided into four groups: EAAC1 antisense-treated rats, controls (sense-treated rats and naive rat controls), and GLAST antisense-treated rats. Brain slices were prepared using previously reported methods (Rafiq et al., 1993). In brief, rats were anesthetized with halothane and decapitated, and the brain was quickly removed and chilled for 1–2 min in a modified SACSF) composed of (in mm): 201 sucrose, 3.2 KCl, 1.25 NaHPO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose 10 (equilibrated with 95% O2–5% CO2 at 32.5°C). The brain was glued, frontal side down, to a glass platform with cyanoacrylate cement, and coronal whole brain slices (225 μm) were sectioned using a vibratome (Lancer 1000). Brain slices were subsequently hemisected, transferred to a holding chamber, and incubated in warm (35°C) normal ACSF containing 126 mm NaCl substituted for sucrose and allowed to equilibrate for at least 2 hr before being transferred to the recording chamber.
Whole-cell voltage-clamp recordings were conducted at room temperature from visually identified CA1 pyramidal neurons using infrared differential interference contrast or Hoffman modulation contrast video microscopy (Stuart et al., 1993; Cohen et al., 2000). Cells were voltage clamped at −60 mV, and signals were recorded and amplified with an Axopatch 1D (Axon Instruments, Foster City, CA), filtered at 2 kHz, digitized, sampled at 44 kHz with a pulse code modulator digitizer (Neuro-Corder DR-890; Neurodata Instruments, New York, NY), and stored on videotape for off-line analysis. Electrodes were fabricated from thick-walled borosilicate glass (World Precision Instruments, Sarasota, FL) and pulled to a resistance between 2 and 6 MΩ when filled with an internal solution composed of (in mm): 135 CsCl, 10 HEPES, 2 MgCl2, and 4 MgATP, pH 7.25 (CsOH) on a two-stage puller (model PP-83, Narishige, East Meadow, NY). To isolate GABAA-mediated events, tetrodotoxin (TTX) (400 nm) and the excitatory amino acid antagonistsd-2-amino-5-phosphonopentanoic acid (d-AP-5) (50 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (6 μm) were added to the superfusing ACSF.
Recorded miniature IPSCs (mIPSCs) were reacquired using Dempster software (Strathclyde, Glasgow, UK), which collects events using a manually controlled threshold detector and is capable of detecting events as small as 2–3× the baseline noise. To attempt to minimize cases of inadequate space clamp, neurons were used for analysis only when series resistance (Rs) was <20 MΩ, and at least 80% series resistance compensation was achieved.Rs was checked frequently throughout experiments, and neurons in which Rsincreased >20% were discarded. The kinetics of mIPSCs, i.e., amplitude, rise, and decay times, were analyzed using cumulative probability histograms. mIPSC frequency was determined using Mini-Analysis software (Synaptosoft, Leonia, NJ). Reagents were purchased from the following vendors: all salts and diazepam were from Sigma (St. Louis, MO); d-AP-5 and CNQX were from Research Biochemicals (Natick, MA); and TTX was from Calbiochem (La Jolla, CA). All drugs were made as stock solutions and then diluted to their final concentration in the bathing medium. Statistical significance between cumulative probability distributions in control and drug conditions in individual neurons was assessed at thep < 0.05 confidence level using the Kolmogorov–Smirnov nonparametric statistical test. Two-tailed unpaired Student's t tests were performed to determine statistical significance at the p < 0.05 confidence level when comparing different treatment groups. All mIPSCs recordings and interpretation was done with the recorder–interpreter blinded to the treatment.
GABA levels. Dissected brain regions were homogenized in 0.1N perchloric acid. After thiol derivatization, levels of GABA were determined using HPLC (0.18 m sodium acetate and 41% acetonitrile buffer, pH 5.0; mobile phase at a flow rate of 2.5 ml/min) using a 5 nm reverse-phase column (ODS Biocal; Bio-Rad, Hercules, CA) coupled with electrochemical detection (BioAnalytical Systems, West Lafayette, IN). Retention time for GABA was ∼6.5 min as confirmed with [14C]GABA standard. Tissue protein levels were determined by a Coomassie blue assay. Levels of total GABA are expressed as nanomoles per milligram of protein.
GABA metabolism. Supernatants (30 μl) from [14C]glutamate uptake experiments were mixed with 0.1N perchloric acid (165 μl), 5 μl of 5-aminovaleric acid (45 μm solution in 0.1N perchloric acid), and 800 μl of “working reagent” [opthaldialdehyde (OPA) solution], and the solution was incubated at room temperature for 6 min. The working reagent was prepared by dissolving 67.1 mg of OPA in 50 ml of methanol, 56 μl of t-butylthiol, and 15 ml of 1m carbonate buffer, pH 9.6, and the mixture was brought to a final volume of 100 ml with ∼30 ml of water. Exactly 6 min after the addition of the OPA solution to the sample, 50 μl of the derivatized sample was analyzed by HPLC. Authentic GABA standards allowed quantification of total GABA levels in the samples. Fractions of each sample were collected (30 sec/fraction), and 300 μl aliquots were mixed with 3 ml of scintillation cocktail and [14C] activity was analyzed by liquid scintillation spectroscopy (Wallac, Turku, Finland). Specific activity of [14C]GABA was calculated for each sample. Tissue protein levels were determined by a Coomassie blue assay with bovine serum albumin as standard. [14C]GABA-specific activities were compared in antisense- and sense-treated samples, in experiments of naive rats with and withoutdl-threo-β-hydroxy-aspartic acid (THA) and in experiments of naive rats treated with 6-diazo-5-oxo-l-norleucine (DON) and dihydrokainate (DHK) at times t = 0 min andt = 30 min.
Tissue preparation and [C14]glutamate uptake. Antisense- and sense-treated rats were decapitated 10 d after antisense infusion, and brains were rapidly removed and placed into chilled, oxygenated (95% O2–5% CO2) Krebs'–Ringer's bicarbonate (KRB) buffer (in mm: 119 NaCl, 4.8 KCl, 1.7 CaCl2, 1.2 MgCl2, 1.2 KH2PO4, 23.8 NaHCO3, and 5.5 glucose, pH 7.4). After 30 sec, the brain was placed on a chilled aluminum block (4°C), and hippocampus and thalamus were dissected from 1 mm coronal sections, followed by slicing into 0.25 mm prisms (McIlwain tissue chopper; Brinkman Instruments). Prisms were suspended in chilled fresh KRB buffer in microvials (Eppendorf) on ice. DON, an inhibitor of phosphate-activated glutaminase (Sigma), was added to the microvial to a final concentration of 10 mm. The microvials containing antisense- and sense (control)-treated hippocampi and thalami were incubated with shaking at 37°C for 15 min. The vials were then centrifuged (500 rpm, 5 min), and DON was washed out using the chilled KRB buffer. In some experiments, after washing out DON, THA was added to hippocampal prisms in the microvials to a final concentration of 1 mm. The vials were incubated for 15 min in 37°C, and the reaction was stopped on ice. While on ice in chilled fresh KRB oxygenated buffer, glutamate (20 μm final concentration), [14C]glutamate (∼1 μCi), anddl-gabaculline (20 μmfinal concentration) were added and incubated at 37°C for 30 min (Behar and Boehm, 1994; Sibson et al., 1998). The reaction was terminated by chilling on ice and adding 0.4N perchloric acid. Some tissue was reserved for the HPLC assay of GABA. The tissue was homogenized, frozen, and thawed at 37°C twice to break the cell membranes, and the homogenate was centrifuged at 14,000 rpm for 20 min at 4°C. The remaining pellet was dissolved in 200 μl of 1N NaOH for protein assay. The supernatant fraction was filtered through 0.45 μm pore syringe filter and stored in −70°C until GABA analysis.
Immunoblots. Antisense- and sense-treated rats were decapitated after 10 d of intraventricular treatment, and the brains were rapidly removed and placed on a chilled aluminum block (4°C). Coronal sections of brain were sliced at 1–2 mm intervals from the occipital pole to the olfactory bulbs. Immunoblots of the tissue homogenates were prepared with affinity-purified polyclonal oligopeptide antibodies to EAAC1 as described previously (Rothstein et al., 1994, 1996)
RESULTS
Antisense knockdown of EAAC1
Previous studies have demonstrated reliable and specific knockdown of EAAC1 expression after chronic intraventricular administration of antisense oligonucleotides (Rothstein et al., 1996). By 8–10 d after administration of EAAC1 antisense oligonucleotides, expression of EAAC1 was reduced by >60% (Fig.1a). Sense oligonucleotides (Fig. 1a) had no effect on EAAC1 expression and neither did random oligonucleotides or antisense to other glutamate transporters: GLT-1 or GLAST (Rothstein et al., 1996). Loss of EAAC1 leads to a small loss in total tissue glutamate transporter activity (Rothstein et al., 1996).
Fig. 1.

Antisense EAAC1 treatment leads to loss of hippocampal EAAC1 expression and epilepsy. a, Immunoblots from hippocampal tissue of three antisense-treated rats (4, 5, 6) have reduced expression of EAAC1 by 60–70% compared with the hippocampus of three sense controls (1, 2,3). b, e, EEG of an awake ambulating adult rat 1 or 2 d after infusion of antisense (b) or sense (e). A normal mixture of frequencies without spikes was seen in both animals.c, f, Recordings of the same rats (as inb and e, respectively) on day 4 or 5. Runs of high-voltage polyspike discharges for ∼2.5 sec, with return to baseline background EEG, can be seen in the antisense-treated rat (c) but not in the sense-treated rat (f). d, g, On days 9 or 10, the maximal antisense effect was seen as prolonged continuous high-voltage spikes, spike and wave complexes, background slowing, and decreased mixture of frequencies in the antisense-treated rat (d) but not in the sense-treated rat (g). h, The mean number of spikes per minute on days 9 or 10 in antisense EAAC1 compared with three controls (GLT-1 antisense-, GLAST antisense-, and EAAC1 sense-treated animals) is significantly increased. *p < 0.005 between the study group and each of the control groups but not between the different controls; ANOVA with Fisher's PLSD for mean spikes per minute.
Subacute loss of EAAC1 leads to epilepsy: video–EEG recording
To evaluate the epileptogenicity of EAAC1 antisense treatment, EEG and video were recorded in awake and ambulating rats (n= 32). Up to 4 d after oligonucleotide treatment, no differences in behavior or the EEG patterns were noted between the study group and the control treatments (EAAC1 sense-, GLAST antisense-, and GLT-1 antisense-treated). The EEG consisted of a normal mixture of frequencies without spikes (Fig. 1b,e) (GLT and GLAST antisense controls were not different from untreated or sense treated animals; data not shown). Beginning on the fourth day after EAAC1 antisense infusion, brief staring episodes and “freezing”-like postures were observed that were not seen in EAAC1 sense controls or in GLAST antisense-treated and GLT-1 antisense-treated animals. The GLT-1 and GLAST antisense-treated animals showed some motor slowing but no staring–freezing episodes. The behavioral episodes of the EAAC1 antisense-treated rats correlated with EEG changes, marked by brief runs of rhythmic spikes, again not seen in the controls (Fig. 1c,f) (GLT-1 and GLAST antisense controls were not different; data not shown). These behavioral manifestations deteriorated daily, coupled with worsening EEG. On the 10th day of infusion (a time point corresponding to a maximal 70–90% loss of EAAC1 protein) (Rothstein et al., 1996), the behavioral changes consisted of prolonged freezing and staring episodes with occasional tonic posturing of the forepaws. The EAAC1 sense control rats did not show behavioral changes, and the GLT-1 and GLAST antisense controls manifested severe motor weakness without posturing, freezing, or staring episodes. Corresponding to the long staring and freezing episodes (as recorded by simultaneous video–EEG recording), the EEG recording showed prolonged runs of 4–6 sec spikes and spike and wave complexes, along with slowing and reduced fast activity of the background. No epileptiform EEG changes were recorded in the controls. In two of seven GLT-1 antisense controls, there were a few spikes, but no runs of spikes, rhythmic spikes, or spike and wave complexes were seen (Fig. 1d,g) (GLT-1 and GLAST antisense controls were not different; data not shown). The mean number of spikes per minute in the EAAC1 antisense-treated group (12.03;n = 6) was significantly higher then the mean number of spikes per minute in the three other control groups: sense EAAC1 (0.98;n = 5), antisense GLT-1 (2.99; n = 6), and antisense GLAST (0.57; n = 4) (Fig. 1h) (ANOVA; p = 0.034). Fisher's PLSD for spikes per minute showed a significant difference between the study group antisense EAAC1 and each of the control groups: sense EAAC1 (p = 0.0016), antisense GLT-1 (p = 0.0051), and antisense GLAST (p = 0.0020), without a difference among the different control groups. This finding implies significant and specific epileptiform activity associated with EAAC1 antisense treatment.
Extracellular thalamocortical field potential slice recording
To explore the physiological basis of EAAC1 antisense oligonucleotide epileptogenesis, extracellular slice recording and intracellular recording were used to test the hypothesis that hyperexcitation associated with the EAAC1 antisense treatment was attributable to decreased inhibition. Extracellular field potentials from thalamocortical and hippocampal slices of rat brains, relevant to both absence and partial complex seizures, respectively, were recorded in study animals and controls.
Extracellular thalamocortical slice recordings showed excessive excitability from the EAAC1 antisense rats compared with sense-treated controls. Recordings from thalamocortical slices in the study group showed prolonged runs of spontaneous bursts of spike discharges (as displayed from the thalamic channel) (Fig.2a), which, at close scrutiny, showed 6–7 sec spikes and slow-wave complexes (data not shown). Because of the use of magnesium free medium, these runs were occasionally present in sense control slices but were much less frequent and very short in duration. Typical control thalamocortical slices showed occasional brief spontaneous bursting spike discharges (Fig. 2b). Quantitative analysis comparing the mean duration of these spike discharges (seconds discharges per minute recording) in the thalamocortical slices of antisense-treated rats (8.1;n = 13) to slices of sense controls (2.8;n = 10) showed significant hyperexcitability of the study group (p = 0.013) (Fig.2c).
Fig. 2.
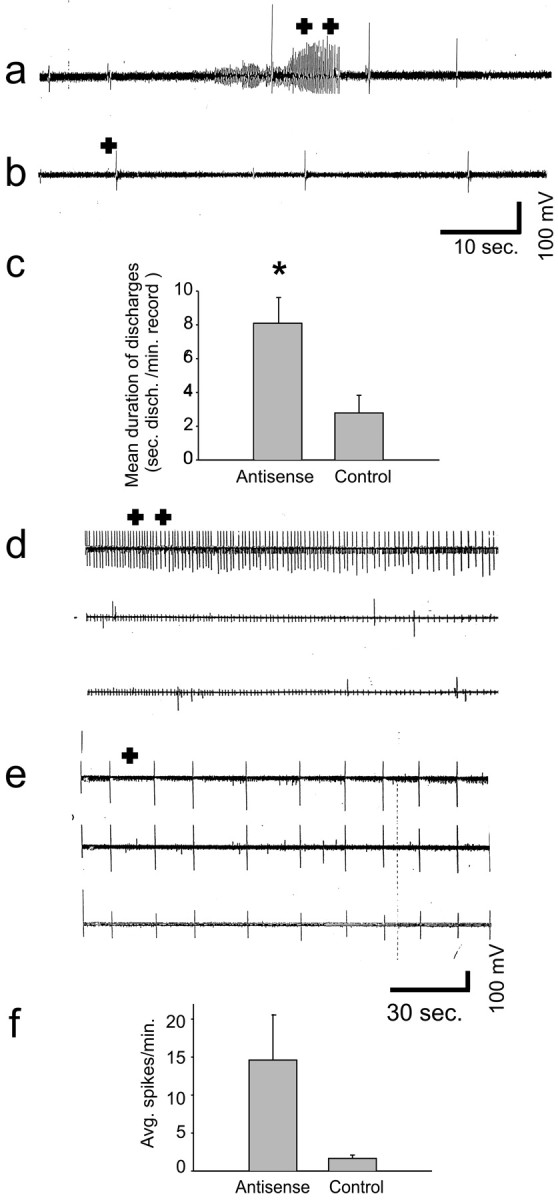
Thalamocortical and HEC slices of EAAC1 antisense-treated rats are hyperexcitable. a,b, Thalamocortical slice field potential recordings showing a 10 sec run of spontaneous bursting rhythmic spike discharges (a; + +) in antisense-treated tissue but not in the controls (b) in which occasional spontaneous spike discharges were seen (b; +) (a andb are recorded at 5 mm/sec paper speed).c, The mean duration of spike discharges (seconds per minute) in the antisense-treated thalamocortical slices was significantly increased compared with control (*p < 0.05).d, e, HEC slice field potential recordings showing the occurrence of very frequent SISs in the antisense-treated rat slice (d; + +) and much less frequent SISs (e; +) in the sense-treated rat slice (d and e are recorded at 1 mm/sec).f, Mean number of spikes per minute in the HEC slices of antisense-treated rats was increased.
Extracellular HEC slice recording
Extracellular hippocampal slice recordings showed increased excitability from EAAC1 antisense rats compared with sense controls. The typical recording of HEC slices in the EAAC1 antisense group showed very frequent SISs (Fig. 2d), whereas in recordings of EAAC1 sense-treated rats, such spikes were found much less frequently (Fig.2e). The mean number of spikes per minute in all of the HEC slices recorded in antisense-treated rats (14.6; n = 5) was increased compared with sense-treated controls (1.66;n = 3), but the difference was not statistically significant (p = 0.16) (Fig.2f). There was a strong correlation between the mean number of spikes per minute in hippocampal slices and the mean number of spikes per minute on EEG (r = 0.97;p < 0.0001; data not shown).
Whole-cell visualized slice patch recording
The limbic hyperexcitability observed in extracellular recording in HEC slices from EAAC1 antisense-treated animals (see above) suggests that the reduction of functional levels of EAAC1 transporter protein may promote seizure generation by a decrease of releasable GABA. To test this hypothesis directly, mIPSCs, which are mediated by the spontaneous release of single GABA quanta, in hippocampal area CA1 in EAAC1 antisense-treated, EAAC1 sense-treated, GLAST antisense-treated, and naive animals, were recorded. GLT-1 antisense oligonucleotide-treated animals were not used in this analysis because the hippocampus was severely damaged by this treatment (Rothstein et al., 1996). The median mIPSC amplitude recorded in CA1 pyramidal neurons from EAAC1 antisense oligonucleotide-treated animals (n = 10; number of events was 2486) was significantly smaller (−29.8 ± 3 pA; p < 0.05; unpairedt test) than that recorded in EAAC1 sense oligonucleotide-treated (−39.4 ± 2 pA; n = 5; number of events was 1023), GLAST antisense oligonucleotide-treated (−38.5 ± 2 pA; n = 6; number of events was 1140), and naive (−37.1 ± 1; n = 3; number of events was 879) controls (Fig.3a1–a3,b1,c1). The 90% decay time (t90) was significantly faster in the EAAC1 antisense-treated group compared with both GLAST antisense and EAAC1 sense groups (Fig. 3b2,c2). However, caution should be used in interpretation of this result because the 50% decay times (t50) were not significant between the three groups (t50 of 8.97 ± 2, 10.13 ± 2, 10.5 ± 1 for EAAC1 antisense-, EAAC1 sense-, and GLAST antisense-treated, respectively). The current measured corresponding to the t90 decay time is very close to our limit of detection. Because of our lack of confidence in the t90 decay time measurement, we calculated the weighted decay τ values for EAAC1 antisense-, sense-, and GLAST antisense-treated groups. The weighted decay τ values were calculated by dividing the area of the events by their peak amplitudes, and the median values were compared. In similar manner to the t90 decay time data, we found that the EAAC1 antisense-treated group decayed significantly faster (p < 0.05) than both EAAC1 sense- and GLAST antisense-treated groups. The median weighted τ values were 12.75 ± 1.9 (n = 10), 15.25 ± 1.0 (n = 5), and 16.83 ± 3.1 (n = 6) for EAAC1 antisense-, EAAC1 sense-, and GLAST antisense-treated groups, respectively. mIPSC mean frequency was similar in all three groups recorded (3.2 ± 2.1 Hz for GLAST antisense, 3.44 ± 1.7 Hz for EAAC1 sense, and 3.8 ± 1.1 Hz for EAAC1 antisense).
Fig. 3.

The hyperexcitability of hippocampal slices of EAAC1 antisense-treated rats may be secondary to decreased inhibition in CA1 pyramidal neurons. a, Representative mIPSCs (average of 50) from EAAC1 sense (a1), EAAC1 antisense (a2), and GLAST antisense (a3) CA1 pyramidal neurons. b, Cumulative frequency amplitude (b1) and t90 decay time (b2) histograms for neurons in a.c, Histogram of mean mIPSC amplitudes (c1) and t90 decay times (c2) for the three populations. *p< 0.05 denotes significant differences between EAAC1 antisense-treated and both EAAC1 sense- and GLAST antisense-treated groups.AS, Antisense; S, sense.
Regional brain GABA
Because of the decreased inhibition demonstrated by the voltage-clamp recording studies described above, total GABA was measured in the hippocampus and thalamus of antisense-treated animals compared with sense controls and also in other brain regions close to and far from the intraventricular oligonucleotide infusion (Rothstein et al., 1994). HPLC analysis of selected brain regions indicated that GABA levels were decreased 50% (p < 0.05) in the hippocampus and mildly decreased (nonsignificantly) in the thalamus of EAAC1 antisense but not in controls (sense, n = 4; antisense, n = 7) (Fig.4a).
Fig. 4.

Total GABA is decreased significantly in the hippocampus (Hipp) and nonsignificantly in the thalamus (Thal) of EAAC1 antisense-treated rats, suggesting a role for EAAC1 in GABA synthesis in normal rat hippocampi.a, Histograms of mean total GABA levels in different brain regions (Crblm) comparing antisense-treated rats (gray) to sense controls (black). Antisense EAAC1 treatment leads to a loss of hippocampal GABA significantly (*p < 0.05) and thalamic GABA nonsignificantly. b, THA significantly decreases the [14C]GABA-specific activity measured in hippocampal tissue of normal rats (*p < 0.05).c, The addition of the glutaminase inhibitor DON with glutamate to hippocampal tissue produces a nonsignificant increase in [14C]GABA-specific activity. d, Addition of DON and DHK with glutamate to hippocampal tissue produces a significant increase in [14C]GABA-specific activity (*p < 0.05).
New [14C]GABA metabolism in normal rats
To determine whether changes in GABA reflect a coupling of EAAC1-mediated glutamate transport and GABA metabolism, new synthesis of GABA in EAAC1 antisense-treated rats was measured using [U-14C]glutamate in the presence of selective enzyme inhibitors to block glutamate repletion from astrocytic glutamine and GABA catabolism. New glutamate synthesis from astrocytic glutamine was blocked with the glutaminase inhibitor DON, and GABA catabolism was prevented by gabaculine, a potent and irreversible inhibitor of GABA transaminase. Application of the GABA transaminase inhibitor permitted the experiments to be conducted under conditions of net GABA synthesis. To test whether any potential GABA labeling from [14C]glutamate was transporter dependent in the control hippocampal tissue and because no specific pharmacological inhibitor of EAAC1 currently exists, it was necessary to use the nonspecific glutamate transport inhibitor THA.
Newly synthesized [14C]GABA from [U-14C]glutamate in hippocampal tissue from normal rats incubated in the presence of DON and gabaculine was significantly lower [specific activity, 2.225 (n = 3) vs 6.072 (n = 6); p = 0.003] in the presence of THA (Fig. 4b), suggesting that GABA synthesis is dependent on glutamate transport. To test the effect of DON alone on GABA synthesis, DON was added to hippocampal tissue acquired from normal rats at time 0 (without adding glutamate), and this was compared with DON, which was added to other hippocampal tissue from control rats at time 0 and was followed by addition and incubation with cold and [14C]glutamate and gabaculine for 30 min. Newly synthesized [14C]GABA is reflected by the difference in [14C]GABA-specific activity between the tissue with added glutamate compared with the tissue with no added glutamate, and there was a nonsignificant difference [increment from specific activity, 0.2733 (n = 6) vs 0.3967 (n = 6); p = 0.19] (Fig.4c). This difference could be explained by new GABA synthesis from glutamate taken up into synaptic terminals by EAAC1. Alternatively, the enzymatic inhibition of glutaminase, by DON, may have been incomplete, and the difference could be explained by astroglial glutamine supply. That is, [14C]glutamate may have been transported into astrocytes by the astroglial transporter GLT-1, converted to [14C]glutamine, and then shuttled to the GABA neuron. However, when DHK, a specific and potent blocker of GLT-1, in addition to DON was added to hippocampal tissue from control rats at time 0 (without adding glutamate) and this was compared with DHK and DON, which were added to other hippocampal tissue from normal rats at time 0 and was followed by addition and incubation with cold and [14C]glutamate and gabaculine for 30 min, there was a significant difference [increment from specific activity, 0.172 (n = 11) vs 0.338 (n = 11); p = 0.01]. Newly synthesized [14C]GABA is reflected by the difference in [14C]GABA-specific activity between the tissue with added glutamate compared with the tissue with no added glutamate, and there was a significant difference, i.e., increased GABA labeling this time (Fig. 4d).
The opposite result, i.e., decreased GABA labeling, would be expected if GABA was labeled from glutamine synthesized from [14C]glutamate uptake by astroglia attributable to DHK blockade of this uptake. Thus, increased GABA labeling after application of DHK suggests that inhibition of the astroglial transporter led to an increase in extracellular glutamate, with more now available for transport through EAAC1 and GABA synthesis. This suggests that EAAC1 is important for GABA synthesis in normal rats.
New [14C]GABA metabolism in antisense-treated rats and sense controls
To more fully evaluate the role of EAAC1-mediated transport in GABA metabolism, new GABA synthesis was measured in hippocampal slices prepared from EAAC1 sense- and antisense-treated rats in the presence of DON and gabaculine. [14C]GABA-specific activity in hippocampal tissue of antisense-treated rats was significantly lower (1.65; n = 7) compared with sense controls (4.03;n = 6; p = 0.017) (Fig.5a). The newly formed [14C]GABA over the 30 min incubation period was increased nonsignificantly in the antisense-treated tissue [from 1.85 to 2.66 nmol/mg protein (× 10−5); n = 6 (Fig.5c)] compared with a significant increase [from 1.51 to 5.4 nmol/mg protein (× 10−5);n = 6; p = 0.02 (Fig. 5b)] in the sense-treated tissue. Furthermore, the rate of [14C]GABA synthesis was approximately five times slower [0.027 nmol/mg protein (× 10−5)/min; n = 6]in the antisense-treated compared with the rate of synthesis in the EAAC1 sense-treated [0.13 nmol/mg protein (× 10−5)/min; n = 6 (Fig.5d)] tissue.
Fig. 5.

[14C]GABA-specific activity and synthesis rate are reduced in EAAC1 antisense-treated rats.a, [14C]GABA-specific activity after incubation of hippocampal tissue with [14C]glutamate is significantly decreased in EAAC1 antisense-treated rats (*p < 0.05). b, [14C]GABA is significantly increased with addition of [14C]glutamate in EAAC1 sense-treated animals (*p < 0.05). c, [14C]GABA is not increased significantly with added [14C]-labeled glutamate in EAAC1 antisense-treated animals. d, The rate of [14C]GABA synthesis with added [14C]glutamate is five times faster in tissue of EAAC1 sense-treated rats compared with tissue of EAAC1 antisense-treated rats.
DISCUSSION
Together, these in vivo and in vitro studies demonstrate that EAAC1 antisense-treated rats develop epilepsy and limbic hyperexcitability and that this hyperexcitability may be attributable, in part, to a reduction in new GABA synthesis in the hippocampus. These studies also suggest that glutamate transporters in general, and EAAC1 specifically, have a role in synthesis and release of new neurotransmitter GABA in the hippocampus of normal naive rats.
The EEG and behavioral monitoring have shown that EAAC1 antisense-treated rats develop behavioral changes manifested as staring–freezing episodes, which occur simultaneously with EEG epileptiform changes. These changes are specific and maximal at the time of maximal EAAC1 knockdown. The phenotype of the rats in this study differs from the EAAC1-deficient mice reported by Peghini et al. (1997). EAAC1 null mice have a deficit of the transporter protein during ontological development allowing for compensatory responses, whereas antisense knockdown results in the loss of this transporter during adulthood. Noteworthy in EAAC1 null mice was the decrease in locomotor activity with episodes of locomotor arrest (Peghini et al., 1997); however, simultaneous EEG recording to rule out seizures were not reported in their study. In the present study, extracellular field potential recordings from both thalamocortical and hippocampal slices of antisense-treated rats showed hyperexcitability. The hippocampal hyperexcitability was correlated with the EEG hyperexcitability. Based on this finding and because of the large decrease in GABA synthesis, whole-cell patch-clamp recordings of CA1 pyramidal neurons were performed. The data suggest that EAAC1 antisense-induced hyperexcitability may be attributed to a decrease in mIPSC amplitude, but not mIPSC frequency, in CA1 pyramidal neurons.
A balance of excitation and inhibition is essential for the maintenance of normal function in the brain. Golan et al. (1996) showed that GABA concentration determines the efficacy of inhibition. In the present study, a decrease in total GABA was observed in the hippocampus of antisense-treated rats but not in sense controls. This decrease was not significant in other regions, although it was present in the thalamus also. Inhibiting GABA synthesis causes seizures, and some of the effects of anticonvulsants occur through interference with enzymes associated with GABA metabolism (Petroff et al., 1996a,b).
High-affinity glutamate transporter subtypes have been found to be specifically localized to both neuronal and astroglial membranes. Under normal conditions, these proteins maintain low extracellular levels of glutamate. A series of studies suggest that the astroglial transporters, GLT-1 in particular, are primarily responsible for the synaptic inactivation of glutamate and for preventing excitotoxic injury (Bergles and Jahr, 1997; Otis and Jahr, 1998; Otis and Kavanaugh, 2000). Nevertheless, EAAC1 is the predominant neuronal transporter present widely throughout the CNS dendrites and somata of large and small pyramidal neurons. However, its function in normal synaptic biology has eluded investigators. Molecular anatomic studies suggested that this protein was unexpectedly localized to presynaptic GABA terminals (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000). Subsequently, preliminary antisense knockdown studies suggested a relationship between this protein and tissue GABA levels (Rothstein et al., 1996).
GABA is synthesized primarily from the α-decarboxylation of glutamate by glutamate decarboxylase (Martin and Rimvall, 1993). An alternate pathway for GABA synthesis via putrescine has been described previously (Seiler and Al-Therib, 1974). Although the contribution of this pathway to GABA synthesis appears to be small in the mature rat brain [∼1% of total GABA synthesis (Noto et al., 1986)], putrescine has been shown to be a GABA precursor in the developmentally immature retina (Yamasaki et al., 1999). GABA carbon, which is lost from GABAergic neurons, must be replenished from other cells because mature neurons do not possess the necessary enzymes for de novo synthesis. Glutamine produced in astrocytes is a major precursor of GABA, although few quantitative studies of the precursors of GABA in vivo have been reported. Neostriatal microinjections of methionine sulfoximine, an inhibitor of glutamine synthetase, resulted in only a ∼50% reduction of GABA synthesis (Paulsen et al., 1988), suggesting that a pathway(s) other than glutamine may also supply glutamate precursors for GABA synthesis in this brain region. The large decrease in GABA levels and GABA synthesis from extracellular [14C]glutamate in the hippocampus after knockdown of EAAC1 indicate that direct transport of glutamate into GABAergic neurons can provide precursors for GABA synthesis. Furthermore, the electrophysiologic studies suggest that the loss of EAAC1 leads to decreased mIPSCs, consistent with decreased presynaptic release of GABA.
Together, these metabolic and electrophysiologic studies clearly document a relationship between the presynaptic glutamate transporter and the inhibitory transmitter GABA. In turn, these metabolic studies strongly suggest that the mechanism of epilepsy in the EAAC1 antisense knockdown rats is mediated by decreased GABA synthesis and, therefore, decreased CNS inhibition. The data suggest that, in normal rat brain, EAAC1 may have an important role in regulating GABA synthesis and release synthesis. Recently, GTRAP3–18 (for glutamate transporter-associated protein 3-18), an EAAC1 inhibitory modulator, was described (Lin et al., 2001), suggesting that, under normal conditions, EAAC1 may be modulated, perhaps to regulate presynaptic GABA synthesis. Preliminary studies also show that increased GTRAP3–18, through inhibition of EAAC1 (like antisense), produces epilepsy (Sepkuty et al., 2001). Overall, these studies suggest a novel interaction between excitatory amino acid transporters and an inhibitory amino acid neurotransmitter system. Furthermore, they raise new possibilities of manipulating GABA metabolism through direct or indirect modulation of EAAC1 (e.g., GTRAP3–18) and may provide novel therapeutic modalities for the treatment of epilepsy.
Footnotes
This work was supported by National Institutes of Health Grants NS 40151, NS36465, and NS33958 (J.D.R.) and NS 32403 and NS 38572 (D.A.C.).
Correspondence should be addressed to Jeffrey D. Rothstein, Johns Hopkins University, Department of Neurology, Meyer 6-109, 600 North Wolfe Street, Baltimore, MD 21287-7247. E-mail: jrothste@jhmi.edu.
REFERENCES
- 1.Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- 2.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behar KL, Boehm D. Measurement of GABA following GABA-transaminase inhibition by gabaculine: a 1H and 31P NMR spectroscopic study of rat brain in vivo. Magn Reson Med. 1994;31:660–667. doi: 10.1002/mrm.1910310612. [DOI] [PubMed] [Google Scholar]
- 5.Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 6.Bjoras M, Gjesdal O, Erickson JD, Torp R, Levy LM, Ottersen OP, Degree M, Storm-Mathisen J, Seeberg E, Danbolt NC. Cloning and expression of a neuronal rat brain glutamate transporter. Mol Brain Res. 1996;36:163–168. doi: 10.1016/0169-328x(95)00279-2. [DOI] [PubMed] [Google Scholar]
- 7.Cohen AS, Lin DD, Coulter DA. Protracted postnatal development of inhibitory synaptic transmission in rat hippocampal area CA1 neurons. J Neurophysiol. 2000;84:2465–2476. doi: 10.1152/jn.2000.84.5.2465. [DOI] [PubMed] [Google Scholar]
- 8.Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M. EAAC1, a high-affinity glutamate transporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- 9.Coulter DA, Lee CJ. Thalamocortical rhythm generation in vitro: extra- and intracellular recordings in mouse thalamocortical slices perfused with low Mg2+ medium. Brain Res. 1993;631:137–142. doi: 10.1016/0006-8993(93)91199-3. [DOI] [PubMed] [Google Scholar]
- 10.Daly DD, Pedley TA. Current practice of clinical electroencephalography, Ed 2, pp 275–276. Raven; New York: 1990. [Google Scholar]
- 11.Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]-coupled l-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- 12.Eskandari S, Kreman M, Kavanaugh MP, Wright EM, Zampighi GA. Pentameric assembly of a neuronal glutamate transporter. Proc Natl Acad Sci USA. 2000;97:8641–8646. doi: 10.1073/pnas.97.15.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 14.Golan H, Talpalar AE, Schleifstein-Attias D, Grossman Y. GABA metabolism controls inhibition efficacy in the mammalian CNS. Neurosci Lett. 1996;217:25–28. doi: 10.1016/0304-3940(96)13061-5. [DOI] [PubMed] [Google Scholar]
- 15.He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- 16.Hertz L. Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol. 1979;13:277–323. doi: 10.1016/0301-0082(79)90018-2. [DOI] [PubMed] [Google Scholar]
- 17.Jones RS, Heinemann U. Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium-free medium in vitro. J Neurophysiol. 1988;59:1476–1496. doi: 10.1152/jn.1988.59.5.1476. [DOI] [PubMed] [Google Scholar]
- 18.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 19.Kanai Y, Smith CP, Hediger MA. A new family of neurotransmitter transporters: the high-affinity glutamate transporters. FASEB J. 1993;7:1450–1459. doi: 10.1096/fasebj.7.15.7903261. [DOI] [PubMed] [Google Scholar]
- 20.Kanai Y, Bhide PG, DiFiglia M, Hediger MA. Neuronal high-affinity glutamate transport in the rat central nervous system. NeuroReport. 1995;6:2357–2362. doi: 10.1097/00001756-199511270-00020. [DOI] [PubMed] [Google Scholar]
- 21.Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- 22.Lin CG, Orlov I, Ruggiero AM, Dykes-Hoberg M, Lee A, Jackson M, Rothstein JD. Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3–18. Nature. 2001;410:84–88. doi: 10.1038/35065084. [DOI] [PubMed] [Google Scholar]
- 23.Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- 24.McBean GJ, Roberts PJ. Neurotoxicity of glutamate and dl-threo-hydroxyaspartate in the rat striatum. J Neurochem. 1985;44:247–254. doi: 10.1111/j.1471-4159.1985.tb07137.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama T, Kawakami H, Tanaka K, Nakamura S. Expression of three glutamate transporter subtype mRNAs in human brain regions and peripheral tissues. Brain Res Mol Brain Res. 1996;36:189–192. doi: 10.1016/0169-328x(95)00297-6. [DOI] [PubMed] [Google Scholar]
- 26.Noto T, Hashimoto H, Nakao J, Kamimura H, Nakajima T. Spontaneous release of gamma-aminobutyric acid formed from putrescine and its enhanced Ca2+-dependent release by high K+ stimulation in the brains of freely moving rats. J Neurochem. 1986;46:1877–1880. doi: 10.1111/j.1471-4159.1986.tb08507.x. [DOI] [PubMed] [Google Scholar]
- 27.Otis TS, Jahr CE. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otis TS, Kavanaugh MP. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulsen RE, Odden E, Fonnum F. Importance of glutamine for gamma-aminobutyric acid synthesis in rat neostriatum in vivo. J Neurochem. 1988;51:1294–1299. doi: 10.1111/j.1471-4159.1988.tb03099.x. [DOI] [PubMed] [Google Scholar]
- 30.Peghini P, Janzen J, Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997;16:3822–3832. doi: 10.1093/emboj/16.13.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petroff OA, Rothman DL, Behar KL, Mattson RH. Low brain GABA level is associated with poor seizure control. Ann Neurol. 1996a;40:908–911. doi: 10.1002/ana.410400613. [DOI] [PubMed] [Google Scholar]
- 32.Petroff OA, Behar KL, Mattson RH, Rothman DL. Human brain gamma-aminobutyric acid levels and seizure control following initiation of vigabatrin therapy. J Neurochem. 1996b;67:2399–2404. doi: 10.1046/j.1471-4159.1996.67062399.x. [DOI] [PubMed] [Google Scholar]
- 33. Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain l-glutamate transporter. Nature 360 1992. 464 467 [Erratum (1992) 360:768] [DOI] [PubMed] [Google Scholar]
- 34.Rafiq A, DeLorenzo RJ, Coulter DA. Generation and propagation of epileptiform discharges in a combined entorhinal cortex/hippocampal slice. J Neurophysiol. 1993;70:1962–1974. doi: 10.1152/jn.1993.70.5.1962. [DOI] [PubMed] [Google Scholar]
- 35.Robinson MB, Sinor JD, Dowd LA, Kerwin JF., Jr Subtypes of sodium-dependent high-affinity l-[3H]glutamate transport activity: pharmacologic specificity and regulation by sodium and potassium. J Neurochem. 1993a;60:167–179. doi: 10.1111/j.1471-4159.1993.tb05835.x. [DOI] [PubMed] [Google Scholar]
- 36.Robinson MB, Djali S, Buchhalter JR. Inhibition of glutamate uptake with l-trans-pyrrolidine-2, 4-dicarboxylate potentiates glutamate toxicity in primary hippocampal cultures. J Neurochem. 1993b;61:2099–2103. doi: 10.1111/j.1471-4159.1993.tb07447.x. [DOI] [PubMed] [Google Scholar]
- 37.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 38.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 40.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke J, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 41.Seiler N, Al-Therib MJ. Putrescine catabolism in mammalian brain. Biochem J. 1974;144:29–35. doi: 10.1042/bj1440029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sepkuty JS, Orlov I, Ruggerio A, Rothstein JD. Increased expression of endogenous GTRAP3–18 causes epilepsy, possibly through its inhibitory effect on EAAC1 mediated glutamate transport. Soc Neursoci Abstr. 2001;27:710.8. [Google Scholar]
- 43.Shashidharan P, Huntley GW, Meyer T, Morrison JH, Plaitakis A. Neuron-specific human glutamate transporter: molecular cloning, characterization and expression in human brain. Brain Res. 1994;662:245–250. doi: 10.1016/0006-8993(94)90819-2. [DOI] [PubMed] [Google Scholar]
- 44.Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA. 1998;95:316–321. doi: 10.1073/pnas.95.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 48.Velaz-Faircloth M, McGraw TS, Alandro MS, Fremeau RT, Jr, Kilberg MS, Anderson KJ. Characterization and distribution of the neuronal glutamate transporter EAAC1 in rat brain. Am J Physiol. 1996;270:C67–C75. doi: 10.1152/ajpcell.1996.270.1.C67. [DOI] [PubMed] [Google Scholar]
- 49.Wagner RW. Gene inhibition using antisense oligodeoxynucleotides. Nature. 1994;372:333–335. doi: 10.1038/372333a0. [DOI] [PubMed] [Google Scholar]
- 50.Wahlestedt C. Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- 51.Yamasaki EN, Barbosa VD, De Mello FG, Hokoc JN. GABAergic system in the developing mammalian retina: dual sources of GABA at early stages of postnatal development. Int J Dev Neurosci. 1999;17:201–213. doi: 10.1016/s0736-5748(99)00002-7. [DOI] [PubMed] [Google Scholar]