

Abstract
In breast cancer cells, cytoplasmic localization of the estradiol receptor α (ERα) regulates estradiol-dependent S phase entry. We identified a nuclear export sequence (NES) in ERα and show that its export is dependent on both estradiol-mediated phosphatidylinositol-3-kinase (PI3K)/AKT activation and chromosome region maintenance 1 (CRM1). A Tat peptide containing the ERα NES disrupts ERα–CRM1 interaction and prevents nuclear export of ERα- and estradiol-induced DNA synthesis. NES-ERα mutants do not exit the nucleus and inhibit estradiol-induced S phase entry; ERα-dependent transcription is normal. ERα is associated with Forkhead proteins in the nucleus, and estradiol stimulates nuclear exit of both proteins. ERα knockdown or ERα NES mutations prevent ERα and Forkhead nuclear export. A mutant of forkhead in rhabdomyosarcoma (FKHR), which cannot be phosphorylated by estradiol-activated AKT, does not associate with ERα and is trapped in the nucleus, blocking S phase entry. In conclusion, estradiol-induced AKT-dependent phosphorylation of FKHR drives its association with ERα, thereby triggering complex export from the nucleus necessary for initiation of DNA synthesis and S phase entry.
Introduction
Steroid receptors, in addition to regulating the transcription of specific genes (Mangelsdorf et al., 1995), trigger rapid effects in the extra nuclear compartment (for review see Migliaccio et al., 2007a). It is expected that hormone action depends on the integration of these different receptor activities (Vicent et al., 2006).
Localization and action of estradiol receptor α (ERα) are regulated by multiple factors, including interaction with signaling effectors or other proteins, such as the metastatic tumor antigen or the modulator of nongenomic action of steroid receptors (MNAR; Kumar et al., 2002; Vadlamudi et al., 2005; Gururaj et al., 2006). Deregulation of these processes causes ERα mislocalization and may trigger tumor progression. Overexpression of the EGF receptor is a hallmark of aggressive human breast cancers (Slamon et al., 1989), and cross talk between ERα and growth factor signaling has emerged as a critical factor in endocrine resistance through the control of the subcellular localization of ERα signaling components (Saporita et al., 2003; Gururaj et al., 2006). ER association with MNAR may sequester ER in the cytoplasm/membrane (Vadlamudi et al., 2005), thus enhancing the nongenomic effects of ERα and resulting in tumorigenesis, as well as the anti-hormone resistance of breast cancer cells (Vadlamudi et al., 2005; Gururaj et al., 2006). Expression of a shortened form of the metastatic tumor antigens sequesters ERα in the cytoplasm and leads to malignant phenotypes by enhancing ERα cytoplasmic functions in hormone-dependent breast cancer cells (Kumar et al., 2002). These data imply that cytoplasmic localization of ERα provides a mechanism to control signal transduction–dependent functions, such as DNA synthesis and anchorage-dependent growth of target cells. They suggest that the ERα localization has functional implications in breast cancer progression.
Steroid receptors undergo nucleocytoplasmic shuttling (for review see DeFranco, 2001). In the presence of a ligand, glucocorticoid receptors (GR), androgen receptors (AR), thyroid hormone receptors (TR), and progesterone receptors (PgR) rapidly shuttle between the nuclei and cytoplasm (DeFranco, 2001). Hormone withdrawal from cells induces accumulation of GR and AR in the cytoplasm (Tyagi et al., 2000). ERα also shuttles from the nuclei to the cytoplasm, and the antiestrogen, ICI 182, 780, disrupts this process (Dauvois et al., 1993). Ligand binding and protein–protein interactions can significantly influence the nucleocytoplasmic shuttling of the chimeric GFP-ERα (Maruvada et al., 2003). However, although the mechanism of nuclear import of steroid receptors is well documented, the mechanism of export is still unclear (DeFranco, 2001).
The best-characterized nuclear export pathway uses chromosome region maintenance 1 (CRM1) as a receptor for proteins with leucine-rich nuclear export signals (NESs; Fornerod et al., 1997; Fukuda et al., 1997). Leptomycin B (LMB), through covalent binding to CRM1, inhibits CRM1-dependent nuclear export (Kudo et al., 1998). Conflicting data have been reported on inhibition of steroid receptor export by LMB treatment. Although it is generally accepted that steroid receptors lack classical leucine-rich NESs, they do have sequences with limited homology to NESs (Liu and De Franco, 2000). Because the exact spacing of the leucine/hydrophobic residues in the NES of each protein is subject to variation, it can be difficult to define a NES on this basis alone (Nigg, 1997). Under some circumstances, LMB treatment does indeed inhibit the nuclear export of steroid receptors (Savory et al., 1999; Prufer and Barsony, 2002; Maruvada et al., 2003; Saporita et al., 2003).
Here, we find that in MCF-7 cells, ERα is a nucleocytoplasmic shuttling protein whose traffic out of nuclei is regulated by estradiol activation of the phosphatidylinositol-3-kinase (PI3K)–AKT pathway and depends on CRM1. By combining different approaches, we identified a hormone-dependent and CRM1-directed NES in the ERα hormone-binding domain. The potential interest of these findings is highlighted by the observation that the ERα NES shows significant homology with sequences of other steroid receptors. A peptide mimicking the NES-ERα specifically sequesters the receptor in the nuclei and interferes in hormone-triggered S phase entry in these cells. Site-directed mutagenesis of the ERα NES inhibits forkhead in rhabdomyosarcoma (FKHR) nuclear export, the estradiol-induced cytoplasmic relocalization of receptor, and DNA synthesis. Targeting ERα by siRNA retains FKHR in the nuclear compartment of estradiol-treated MCF-7 cells.
This study provides a new link between the rapid extranuclear action of estradiol and ERα nuclear export, as well as a proliferative role of receptor nuclear export, which is closely associated to FKHR export.
Results
Estradiol regulates ERα shuttling in MCF-7 cells
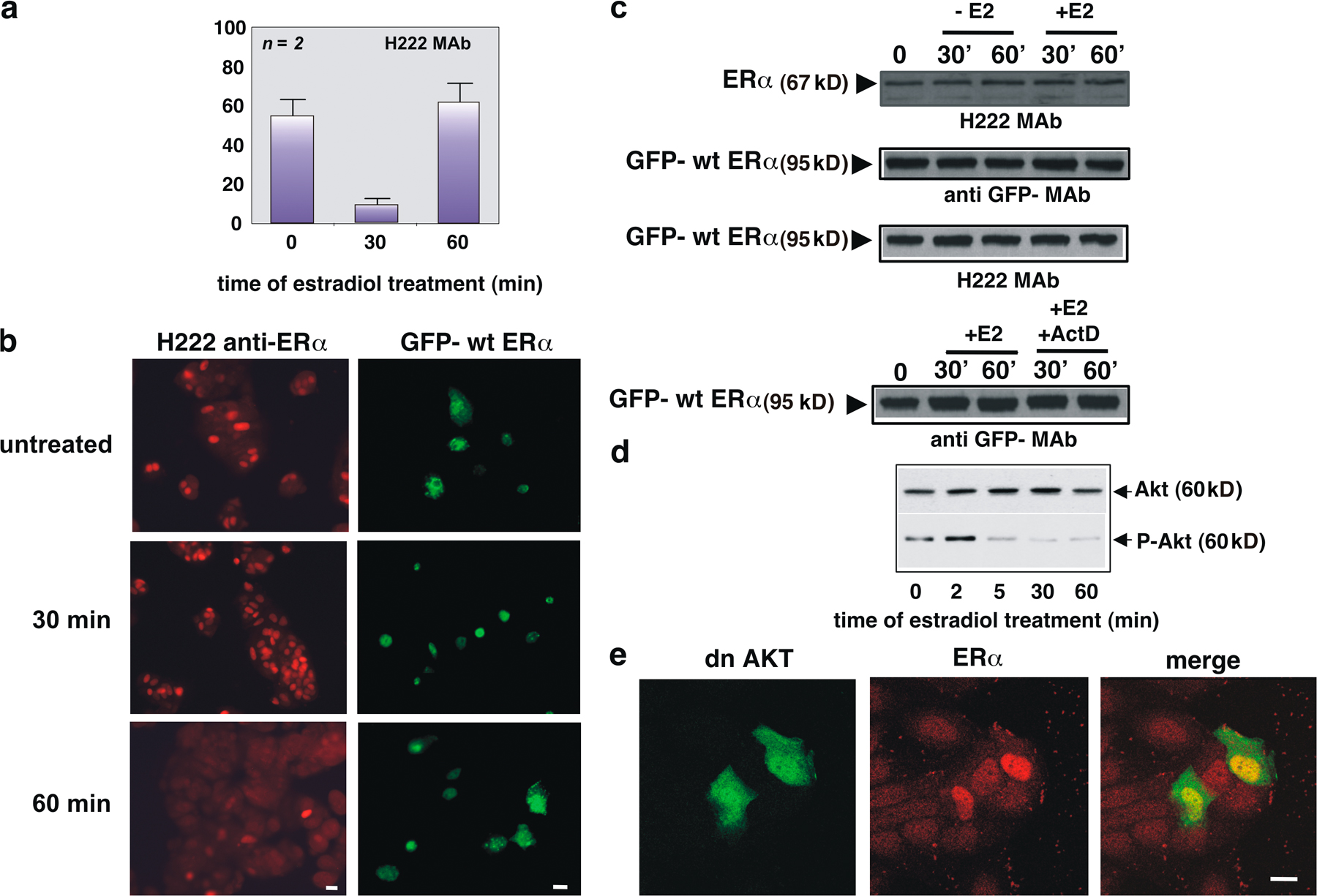
Localization of endogenous ERα in quiescent MCF-7 cells was verified by immunofluorescence using two different antibodies directed against either a C-terminal (H222 mAb; Katzenellenbogen et al., 1987) or N-terminal epitope of ERα (A314 mAb; Abbondanza et al., 1998). Fig. 1 a shows that, regardless of the antibody used, estradiol treatment of MCF-7 cells induces nuclear translocation of ERα after 30 min, followed by a decrease of the nuclear receptor to the basal level by 1 h. Representative images of one of these experiments are presented in the supplemental materials, together with the parallel change in cytoplasmic redistribution of the receptor (Fig. S1 a and Fig. S1 b, left, available at http://www.jcb.org/cgi/content/full/jcb.200712125/DC1). Immunoblot analysis of cell lysates does not reveal any significant change in ERα level (Fig. S1 c), which suggests that the observed decrease in ERα nuclear localization is caused by nuclear export of the receptor.
Figure 1.

Estradiol induces nuclear export of ERα, which is regulated by the PI3K–AKT pathway and depends on CRM1. Quiescent MCF-7 cells were used. (a) Cells were untreated or treated with 10 nM estradiol (E2) for the indicated times (min). ERα localization was analyzed by immunofluorescence using the indicated antibodies. (b and c) Cells were transfected with GFP-wtERα then left untreated or treated with the indicated compounds. OH-tamoxifen (Tx; AstraZeneca) was used at 0.1 μM; actinomycin D (Act D) was added at 5 μg/ml, 1 h before estradiol stimulation. GFP-wtERα localization was determined by fluorescence. (d) Cells were transfected with the indicated plasmids and left untreated or treated with 10 nM estradiol for the indicated times (min). The Myc-tagged pSG5, Δp85α, or Myc-His–tagged dominant-negative AKT ectopically expressed in MCF-7 cells were visualized by immunofluorescence, as described in Materials and methods. ERα localization was analyzed by immunofluorescence using the rat H222 anti-ERα mAb. (e) Cells were transfected with GFP-wtERα and then left untreated or treated with 10 nM estradiol in the absence or presence of LMB (at 5 ng/ml). LMB was added 30 min before the hormone. Cells were also treated with LMB in the absence of hormone. GFP-wtERα localization was determined by fluorescence. (a, b, c, d, and e) Cells that fell into the category of exclusively nuclear fluorescence were scored, and data was expressed as a percentage of total cells (in a) or transfected cells (in b, c, d, and e). Data were derived from at least 1,000 scored cells. The results of several independent experiments were averaged; means and SEM are shown. n represents the number of experiments. (f) Images from one experiment in panel e were captured. Panels show GFP-wtERα localization in MCF-7 cells stimulated for 60 min with estradiol (E2) in the absence or presence of LMB. Bar, 5 μm. (g) 35S-labeled HA-CRM1 was incubated with recombinant ERα in the absence or presence of 10 nM estradiol. The purified recombinant RanQ69L (at 1 μM) was included in the incubation mixture of each sample. Proteins were immunoprecipitated with rabbit polyclonal anti-ERα antibody. Eluted proteins were immunoblotted with anti-ERα antibody (top) or revealed by fluorography (bottom).
Quiescent MCF-7 cells were next transiently transfected with a full-length ERα cDNA subcloned into the GFP plasmid (pEGFP-HEG0; HEG0 represents the wtERα). In Fig. 1 b, the quantitative count of cells with nuclear fluorescence shows that hormone treatment triggers nuclear accumulation of GFP-ERα after 30 min in the absence of any significant change in GFP-wtERα expression (Figs. 1 b and S1 c). By 1 h, the nuclear GFP-wtERα decreases to the basal level, whereas no trafficking of GFP-wtERα was detectable in untreated cells (Fig. 1 b). Representative images of one of these experiments are presented in Fig. S1 b (right). In addition, GFP alone was insensitive to estradiol treatment when overexpressed in MCF-7 cells (unpublished data). This pattern of trafficking is similar to that observed for endogenous ERα. Furthermore, the partial antagonist, 4-OH-tamoxifen, prevents the observed nucleocytoplasmic shuttling of GFP-wtERα in response to hormonal treatment (Fig. 1 b).
To exclude the potential contribution of de novo GFP-ERα synthesis to its reemergence in the cytoplasm 1 h after estradiol treatment, we included actinomycin D in the medium during hormone stimulation. No significant change in the estradiol-induced redistribution of GFP-wtERα (Fig. 1 c) or in protein expression was observed (Fig. S1 c). Therefore, the chimeric protein in cytoplasm is from nuclear export rather than from de novo synthesis. Because actinomycin D inhibits the nuclear import of the NES-containing REV protein (Henderson, 2000), and the GFP-wtERα dynamic redistribution is not modified by actinomycin D (Fig. 1 c), it is likely that cytoplasmic relocalization of the chimera depends on nuclear export rather than inhibition of nuclear import.
Because data in Fig. 1 (a–c) show that ERα export shortly occurs upon estradiol treatment of MCF-7 cells, we hypothesized a role for a rapid action of estradiol in the ERα intracellular traffic. Activation of the PI3K–AKT pathway, for instance, modulates the trafficking of various proteins, including FKHR, nuclear factor κB, glycogen synthase kinase-3 β, Mdm2, p27 (for review see Kau and Silver, 2003), and PTEN (phosphatase and tensin homologue deleted on chromosome; Liu et al., 2007). Because of the rapid activation of this pathway by estradiol treatment of breast cancer cells (Fig. S1 d; Castoria et al., 2001, 2004), we verified its contribution to the ERα nuclear export. Quiescent MCF-7 cells were then transiently transfected with the dominant-negative form of the regulatory subunit of PI3K, p85α (Δp85α; Dhand et al., 1994), or the catalytically inactive version of AKT (K179M; dominant-negative AKT). In Fig. 1 d, the count of transfected cells showing ERα nuclear fluorescence indicates that overexpression of either Δp85α or dominant-negative AKT induces nuclear retention of ERα in 60-min estradiol-treated MCF-7 cells. Transfection of cells with the Myc-tagged pSG5 control plasmid does not interfere in the trafficking of ERα. Images of one experiment in Fig. 1 d are presented in Fig. S1 e.
To determine if CRM1 plays a role in ERα nuclear exit, MCF-7 cells were treated with LMB in addition to estradiol. This antifungal compound blocks the nuclear export of NES-containing proteins by preventing their association with the CRM1 export receptor (Kudo et al., 1998). A quantitative analysis of transfected cells shows that LMB treatment results in maintenance of nuclear GFP-wtERα 1 h after estradiol stimulation (Fig. 1, e and f). LMB treatment alone does not have an effect on the localization of GFP-wtERα (Fig. 1, e and f). Thus, the effect of LMB treatment on ERα localization suggests that this receptor, when bound to the hormone, is exported from nuclei through the CRM1–exportin pathway. Estradiol induces a strong interaction between recombinant ERα and in vitro translated CRM1 (Fig. 1 g) when active Ran (Ran-GTP; Askjaer et al., 1999) was included as a GTPase-deficient RanQ69L mutant (Bischoff et al., 1994).
This set of experiments shows that estradiol activation of the PI3K–AKT pathway modulates nuclear export of ERα and that CRM1 contributes to estradiol-mediated ERα nuclear export in MCF-7 cells.
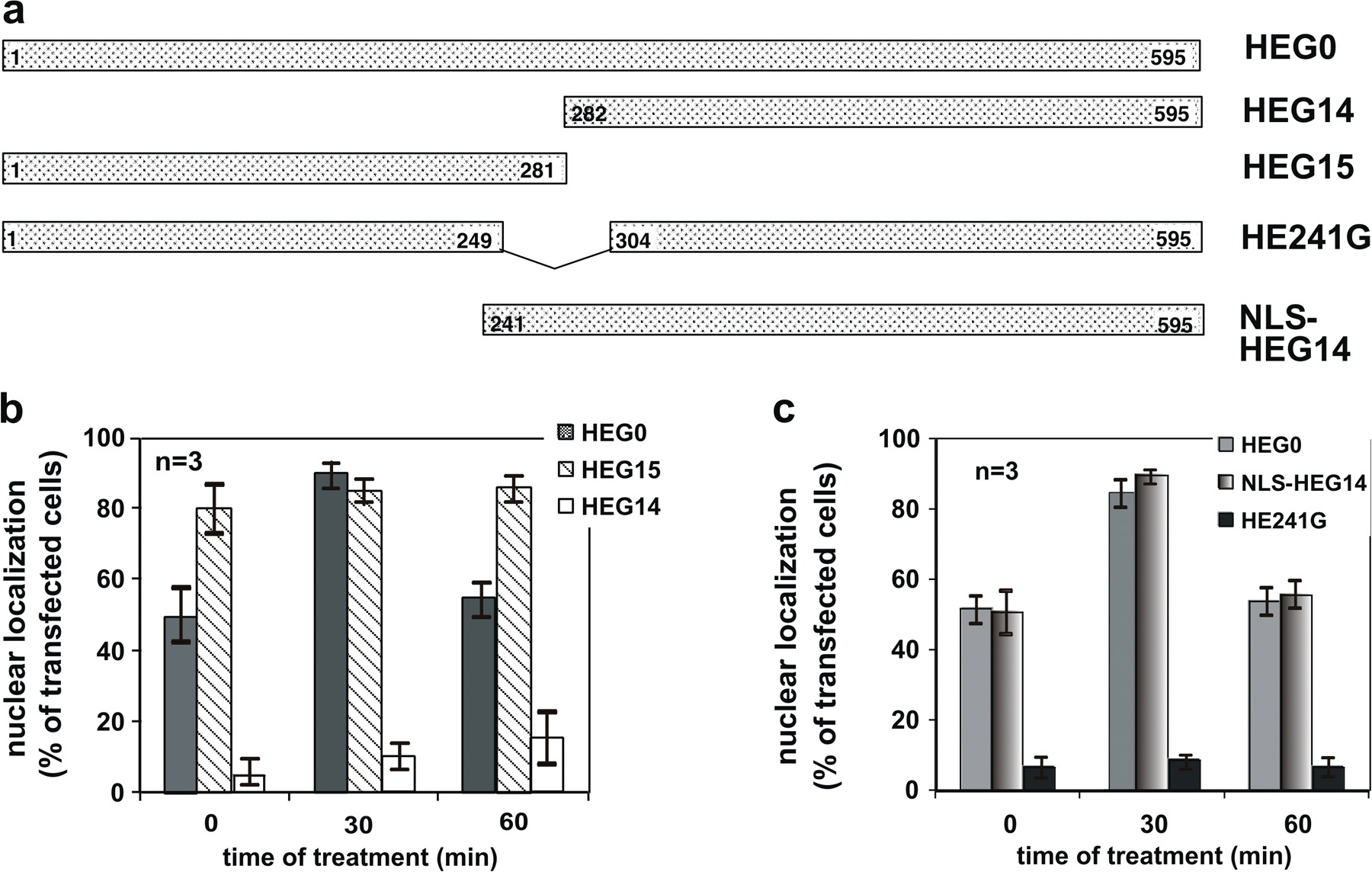
Identification of an ERα nuclear export sequence
Two ERα mutants, HEG15 and HEG14, were subcloned into pEGFP. HEG15 (Δ282–595 HEG0) contains highly conserved NLSs (2 and 3; Guiochon-Mantel et al., 1991). It binds DNA but is unable to bind estradiol (Ylikomi et al., 1992). In turn, HEG14 (Δ1–281) contains NLS1. It binds the hormone but does not bind DNA (Ylikomi et al., 1992). The resulting chimeras, GFP-HEG15 and GFP-HEG14, were transiently transfected into quiescent MCF-7 cells. Consistent with previous findings (Ylikomi et al., 1992), results in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200712125/DC1) show that neither GFP-HEG15 nor GFP-HEG14 shuttle between the nucleus and the cytoplasm. Irrespective of the hormonal treatment, the first mutant mostly resides in the nuclear compartment. Although GFP-HEG14 contains the NLS1 sequence and binds the hormone, it does not enter nuclei even after estradiol addition. From these data, we speculated that the hormone-binding domain localized in the HEG14 mutant as well as NLSs (2 and 3) of ERα are both required for the estradiol-induced nuclear import of the receptor. We next subcloned HE241G into pEGFP. This mutant contains the hormone-binding domain and lacks the three NLSs. It is, in fact, prevalently localized in the cytoplasm regardless of estradiol treatment. The behavior of this mutant confirms that in the absence of NLS, the hormone-binding domain is not sufficient to induce nuclear translocation of the receptor. Because HEG15 is localized in the nuclear compartment and its localization is unaffected by hormonal treatment of cells, we reasoned that the addition of NLSs to HEG14 might restore the estradiol-induced import of ERα. Analysis of transfected cells with the resulting chimera, GFP-NLS/HEG14, showed not only nuclear import but also export similar to GFP-wtERα in response to estradiol treatment. These findings collected in Fig. S2 point to the presence of nuclear export sequences in the NLS–HEG14 construct, which is made up of the NLSs and the hormone-binding domain of ERα.
It has been found that NLSs of the PgR are responsible for its nuclear export (Guiochon-Mantel et al., 1994). Therefore, we studied whether NLSs are involved in ERα export using in vivo export assay (Henderson and Eleftheriou, 2000). In this assay, nuclear export sequences are identified by their ability to restore export activity of the NES-deficient REV1.4-GFP (Rev mutant) to levels similar to those observed with the wild-type (wt) pREV-GFP or the REV1.4-GFP NES (Rev positive control), in which the NES is the canonical export sequence of the REV protein. The NLS sequences of ERα were subcloned into the Rev mutant and expressed in MCF-7 cells. After transfection, cells were incubated in the absence or presence of actinomycin D because it causes cytoplasmic accumulation of the putative NES-containing REV protein by preventing nuclear import of REV (Henderson, 2000). Irrespective of experimental conditions, the Rev mutant NLS localizes in nuclei of MCF-7 cells (Fig. 2, a and b). In the same experiment, the Rev-positive control completely shifted to the cytoplasm in the presence of actinomycin D, whereas the Rev mutant NLS sequences from the ERα showed nuclear, sometimes nucleolar, staining. These data indicate that the NLS sequences of ERα are inactive in this export assay.
Figure 2.

The ERα 444–456 sequence restores export activity of the NES-deficient REV1.4-GFP. Growing MCF-7 cells were used. (a and b) Cells were transfected with the indicated constructs. After transfection, the cells were left untreated (no drug) or treated with actinomycin D (ActD) at 5 μg/ml, alone or together with 5 ng/ml LMB. The subcellular distribution of GFP proteins was determined by fluorescence microscopy, and cells that fell into the category of exclusively nuclear fluorescence were scored. Data are expressed as a percentage of transfected cells, with mean values taken from at least three experiments. For each experiment, at least 600 cells were scored. (b) Images of one experiment in panel a. (c) The wt ERα 444–456 sequence (NES ERα wt) as well as its mutated version (NES ERα mutant). The putative NES-ERα sequence is indicated by the underlined amino acids, which were substituted with alanine residues in the mutant sequence. The NES-ERα wt as well as the NES-ERα mutant subcloned into the Rev mutant were transfected (d and e) in growing MCF-7 cells. After transfection, the cells were left untreated (−) or treated with 5 μg/ml Act D (+). The percentage of cells with nuclear GFP protein was determined by fluorescence microscopy and graphically shown in panel d. Data were derived from at least 600 scored cells. The results of several independent experiments were averaged. (a and d) Means and SEM are shown. n represents the number of experiments. (e) Images of one experiment in panel d. Bars, 5 μm
We then subcloned different sequences of ERα containing leucine residues into a Rev mutant. These constructs were transfected into MCF-7 cells and then analyzed for their ability to restore the nuclear export of the Rev mutant. Using this assay, we verified that ERα residues 427–456 induced a nucleocytoplasm redistribution of Rev mutant in cells treated with actinomycin D (Fig. 2, a and b). LMB treatment blocked the export activity of the 427–456 ERα sequence (Fig. 2, a and b). Regardless of the experimental conditions, the leucine-rich 296–335 ERα sequence was unable to drive the Rev mutant to the cytoplasm (Fig. 2, a and b). These data show that the 427–456 region is involved in the export of ERα through CRM1/exportin binding.
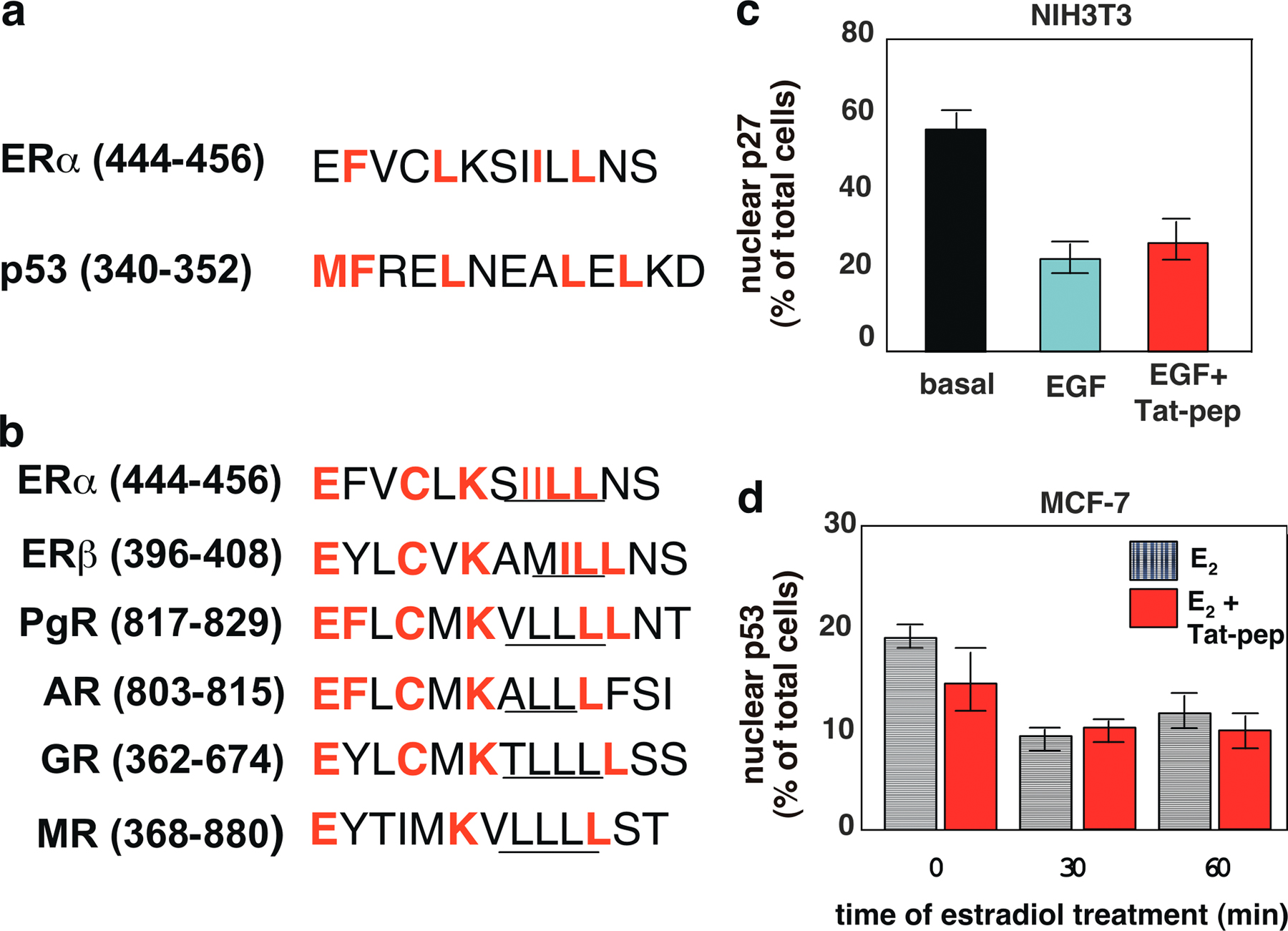
A thorough sequence analysis showed homology between the 444–456 amino acids of ERα and the conserved leucine-rich and REV-like NES of p53 (residues 340–351; Stommel et al., 1999). A comparison of the homologous amino acids across the family of steroid receptors revealed a high level of sequence homology with ERβ, PgR, AR, and GR. The alignment between the sequences is shown in Fig. S3 (a and b, available at http://www.jcb.org/cgi/content/full/jcb.200712125/DC1).
The possibility that residues 444–456 of the ERα sequence contain the NES-ERα was further investigated. The 444–456 ERα sequence were subcloned into the Rev mutant, as they were with a mutated version in which residues 444–456 were mutated from IILL to alanines (Fig. 2 c). The ERα wt sequence (NES-ERα wt) was able to shift the Rev mutant into the cytoplasm of MCF-7 cells, whereas the mutated form of ERα (NES-ERα mutant) failed to do so (Fig. 2, d and e). This difference is even more evident in the presence of actinomycin D.
These results show that a small sequence of the ERα hormone-binding domain contains a CRM1-mediated ERα NES.
A peptide mimicking the 444–456 ERα sequence sequesters the receptor in the nuclear compartment and inhibits estradiol-induced S phase entry in breast cancer cells
Small cationic peptides (also called protein transduction domains) derived from nucleic acid–binding proteins, such as HIV-Tat protein, deliver a myriad of peptides into animal models and represent a biological and potentially therapeutic tool for targeting proteins into the cells (Joliot and Prochiantz, 2004). To trap ERα in the nuclear compartment of cells, we synthesized a Tat-conjugated peptide construct corresponding to the residues 444–456 of ERα (Fig. 3 a). Confocal microscopy analysis revealed in preliminary experiments that the carboxyfluorescein-conjugated peptide translocated across the plasma membrane and, within 30 min, accumulated in nuclei of MCF-7 cells (unpublished data). This peptide displaced the estradiol-induced interaction between recombinant CRM1 and ERα (Fig. 3 b) and blocked the 60-min estradiol-induced nuclear export of GFP-wtERα, thus sequestering the receptor in nuclei (Fig. 3, c and d). The Tat alone did not affect both the ERα–CRM1 interaction (Fig. 3 b) as well as the trafficking of GFP-wtERα (unpublished data). Furthermore, the Tat-conjugated peptide did not affect p27 nuclear export or p53 subcellular localization (Fig. S3, c and d), which indicates that it specifically interferes in ERα localization.
Figure 3.

A peptide mimicking the ERα 444–456 sequence displaces the CRM1–ERα interaction, sequesters the receptor into nuclei, and inhibits S phase entry in estradiol-treated MCF-7 cells. (a) The amino acidic sequence of the Tat-conjugated ERα (444–456) peptide. (b) 35S-labeled HA-CRM1 was incubated with recombinant ERα from baculovirus in the absence or presence of 10 nM estradiol alone or together with a 200-fold excess of the Tat-conjugated peptide (Tat-pep). A 200-fold excess of a nonspecific peptide (Tat) was used as a control. CRM1 was immunoprecipitated with the anti-HA mAb, and proteins in immunocomplexes were revealed by fluorography (top) and immunoblotting with the anti-ERα antibody (bottom). (c) Quiescent MCF-7 cells transfected with GFP-HEG0 were incubated for 1 h with the Tat-conjugated peptide (Tat pep). Thereafter, the cells were left untreated or treated for the indicated times with 10 nM estradiol. The proportion of cells with nuclear GFP-wtERα protein was determined by fluorescence microscopy and graphically shown. For each experiment, at least 200 cells were scored. The results of several independent experiments were averaged. n represents the number of experiments. (d) Images of one experiment in panel c. This shows the localization of GFP-wtERα in MCF-7 cells treated for 60 min with estradiol in the absence or presence of Tat-pep. Bar, 5 μm. (e) MCF-7 cells on coverslips were made quiescent or serum starved (maintained for 24 h in 0.5% FCS). The cells were incubated for 24 h with 10 nM estradiol or 20% serum in the absence or presence of the Tat-conjugated ERα (444–456) peptide (Tat-pep) or Tat alone. After in vivo pulse with BrdU, DNA synthesis was analyzed and BrdU incorporation was expressed as a percentage of total nuclei. For each experiment, at least 200 cells were scored. The results of several independent experiments were averaged. n represents the number of experiments. (f) Quiescent MCF-7 cells were stimulated with 10 nM estradiol for 24 h, and the Tat peptide was added after the addition of the hormone at the time points indicated in the figure. After an in vivo pulse with BrdU, DNA synthesis was analyzed and expressed as a percentage of BrdU incorporation inhibition. For each experiment, at least 300 cells were scored. The results of two independent experiments were averaged. (c, e, and f) Means and SEM are shown.
The extranuclear activity of ERα triggers hormone-dependent DNA synthesis (Castoria et al., 1999), and ERα sequestration in the cytoplasm increases its nongenomic actions and drives neoplastic transformation (Kumar et al., 2002). Fig. 3 e shows that the Tat-conjugated peptide reduced estradiol-induced DNA synthesis in MCF-7 cells by 60%. A negligible effect was observed in cells treated with Tat alone. Furthermore, the Tat peptide did not interfere in serum-induced DNA synthesis in MCF-7 cells (Fig. 3 e), which indicates that the peptide specifically interferes in estradiol action. Addition of the Tat-conjugated peptide at different times after estradiol stimulation shows that after 60 min, when ERα is almost completely exported from nuclei, the peptide does not affect S phase entry in MCF-7 cells (Fig. 3 f), thus reinforcing the view that the estradiol-induced nuclear export of ERα plays a role in DNA synthesis.
ERα NES mutants do not exit nuclei and fail to mediate estradiol-induced DNA synthesis
Based on our findings (Fig. 2), two mutants of GFP-wtERα were prepared by site-directed mutagenesis to identify the NES-ERα sequence and analyze the biological effects induced by these mutations. Fig. 4 a shows a schematic representation of the GFP-wtERα NES and its mutated versions, which were transiently transfected into MCF-7 cells. Estradiol treatment induced nuclear export of GFP-wtERα, which contains the wild-type human ERα. In contrast, both mutants, GFP-ERα 4A and GFP-ERα IL, were unable to be exported from nuclei upon hormonal stimulation (Fig. 4 b). Images in Fig. 4 c show the diffuse, sometimes extranuclear localization of GFP-wtERα in MCF-7 cells stimulated for 60 min with estradiol. Under these conditions, a nuclear/nucleolar localization of both GFP-ERα 4A and GFP-ERα IL was detected.
Figure 4.

Mutations of NES-ERα impair nuclear export of full-length ERα and prevent S phase entry in MCF-7 cells stimulated by estradiol. (a) The wild-type NES-ERα endowed in the amino acid 444–456 sequence is shown in bold. Mutated amino acids are underlined. The NES mutants, GFP-HEG4A and GFP-HEGIL, were prepared as described in Materials and methods. (b) Quiescent MCF-7 cells were transfected with the indicated plasmids and then left unstimulated or stimulated with 10 nM estradiol for the indicated times. The percentage of cells with nuclear GFP protein was determined by fluorescence microscopy and shown graphically. For each experiment, at least 150 cells were scored. The results of different independent experiments were averaged; n represents the number of experiments. (c) Images of one experiment in panel b that shows the intracellular distribution of GFP-wtERα, GFP-ERα 4A, or GFP-ERα IL in MCF-7 cells treated for 1 h with estradiol. Bar, 5 μm. (d–f) Growing NIH3T3 fibroblasts were used. The Western blot in panel d shows that NIH3T3 fibroblasts are ERα negative. In the graph in panel d, cells were transfected with an ERE-Luc construct along with the indicated plasmids. After transfection, the cells were made quiescent and then left unstimulated or stimulated with 10 nM estradiol. Luciferase activity was assayed, normalized using β-gal as an internal control, and expressed as fold induction. (e) Cells were transfected with the indicated plasmids and made quiescent. The cells were left unstimulated or stimulated for 5 min with 10 nM estradiol. Lysates were analyzed for AKT activation using the anti–P-Ser-473 antibody (bottom). The nitrocellulose filters were then reprobed with anti-AKT antibody (top). (f) Cells on coverslips were transfected with the indicated plasmids and made quiescent. The cells were left unstimulated or stimulated for 18 h with 10 nM estradiol or 20% serum. In cells transfected with GFP-wtERα, the inhibitory action of Tat peptide on estradiol-induced BrdU incorporation was analyzed by including this compound (at 1 μM) to the cell medium (bars with asterisks). After in vivo pulse with BrdU, DNA synthesis was analyzed by immunofluorescence. In transfected cells, BrdU incorporation was calculated by the formula (percentage of BrdU-positive cells = [number of transfected-positive cells/number of transfected cells] × 100) and compared with BrdU incorporation of untransfected cells from the same coverslips. For each plasmid, data were derived from at least 500 scored cells. The results of several independent experiments were averaged; n represents the number of experiments. (b, d, and f) Means and SEM are shown.
We then verified the ability of the GFP-ERα IL mutant to activate estradiol-induced gene transcription. To this end, we used ERα-negative NIH3T3 fibroblasts (Castoria et al., 1999, 2003). The Western blot in Fig. 4 d (inset) confirms that NIH3T3 fibroblasts are indeed ERα negative. The cells were then transiently transfected with GFP-wtERα, GFP-ERα IL, or GFP alone. The ERE-Luc reporter gene was cotransfected and its activity assayed. Fig. 4 d shows that the constructs GFP-wtERα and GFP-ERα IL are equally efficient in activating gene transcription upon estradiol stimulation of the cells, with a sixfold induction of ERE-Luc activity. Transcriptional activation was almost undetectable in unstimulated cells or in cells expressing GFP alone.
In another set of experiments, we assessed the ability of GFP-wtERα and GFP-ERα IL to mediate estradiol-induced AKT activation as well as DNA synthesis when ectopically expressed in ERα-negative NIH3T3 fibroblasts. We first verified that GFP-wtERα and its export mutant GFP-ERα IL both activate AKT upon estradiol stimulation of NIH3T3 cells (Fig. 4 e). Although able to activate the nongenomic pathway usually engaged by estradiol to transmit its mitogenic signal, the mutant GFP-ERα IL failed to induce S phase entry in fibroblasts treated with estradiol (Fig. 4 f). The mutant did not affect S phase entry induced by the receptor-independent serum stimulation (Fig. 4 f). In contrast, the GFP-wtERα induced a robust BrdU incorporation in hormone-treated cells (Fig. 4 f). As observed in MCF-7 cells (Fig. 3 e), addition of Tat-conjugated peptide inhibited the estradiol-induced S phase entry in fibroblasts transfected with GFP-wtERα. Here again, the peptide did not interfere in DNA synthesis induced by serum stimulation of the same cells (Fig. 4 f, asterisks).
These experiments demonstrate that the leucine-rich sequence 444–456 of ERα contains a functional NES. Mutations in the core (IILL) of this sequence impair the estradiol-induced nuclear export of ERα and DNA synthesis without affecting the signal transduction pathway or gene transcription regulation by estradiol.
Estradiol regulates FKHR/ERα export
Estradiol activation of PI3K is required to drive MCF-7 cells into S phase (Castoria et al., 2001). Activation of the PI3K–AKT pathway by estradiol regulates ERα nuclear export (Fig. 1), and FKHR nuclear export depends on its phosphorylation by AKT (Biggs et al., 1999). In addition, previous studies reported that an estradiol-dependent interaction between ERα and FKHR occurs in vitro (Schuur et al., 2001). Thus, we hypothesized a role for FKHR in both estradiol-regulated ERα nuclear export and cell cycle arrest mediated by the NES-ERα mutant. To analyze the role of FKHR in estradiol-induced DNA synthesis and ERα nuclear export, quiescent MCF-7 cells were transiently transfected with wt FKHR (GFP-FKHR wt) or a mutant containing a triple alanine substitution (GFP-FKHR AAA) that localizes in nuclei and blocks FKHR phosphorylation by AKT, thereby inducing G1 arrest of cells (Nakamura et al., 2000). Fig. 5 a shows that expression of this mutant reduced estradiol-triggered BrdU incorporation in MCF-7 cells, whereas expression of GFP-FKHR wt did not. We then used confocal microscopy to analyze the role of FKHR in the estradiol-regulated subcellular distribution of ERα. Although ectopically expressed GFP-FKHR wt or GFP alone did not modify the 60-min estradiol-induced nuclear export of ERα, the mutant GFP-FKHR AAA sequestered ERα in the nuclear compartment at that time (Fig. 5, b and d). Conversely, overexpression of the tagged NES-ERα mutant, Myc-ERα IL, resulted in retention of GFP-FKHR wt in the nuclear compartment 60 min after estradiol treatment of MCF-7 cells (Fig. 5, c and d). Thus, estradiol simultaneously regulates ERα- and FKHR-associated nuclear export.
Figure 5.

FKHR nuclear export: regulation by estradiol and a role in hormone-induced DNA synthesis in MCF-7 cells. Quiescent MCF-7 cells on coverslips were used. (a) Cells were transfected with the indicated plasmids then left unstimulated or stimulated for 24 h with 10 nM estradiol. After in vivo pulse with BrdU, DNA synthesis was analyzed by immunofluorescence and BrdU incorporation was calculated as in Fig. 4. (b) Cells were transfected with the indicated plasmids then left unstimulated or stimulated with 10 nM estradiol for the indicated times. Endogenous ERα localization as well as expression of GFP, GFP-FKHR wt, or GFP-FKHR AAA mutant was monitored by confocal microscopy. Cells that fell into the category of exclusively ERα nuclear fluorescence were scored, and data were expressed as a percentage of transfected cells. (c) Cells were cotransfected with the indicated plasmids then left unstimulated or stimulated with 10 nM estradiol for 60 min. Localization of GFP-FKHR wt, Myc-HEG0, or Myc-HEGIL mutant was monitored by confocal microscopy. Cells that fell into the category of exclusively FKHR nuclear fluorescence were scored and the data were expressed as a percentage of cotransfected cells. For each experiment in panels a–c, data were derived from at least 500 scored cells. The results of several independent experiments were averaged; n represents the number of experiments. (d) Images from one experiment in b or c are shown. They represent the staining of endogenous ERα (red) in MCF-7 cells expressing GFP-FKHR wt (left, green) or the mutant, GFP-FKHR AAA (middle, green), and treated for 60 min with estradiol. (right) The staining of Myc-tagged NES-ERα mutant (red) in MCF-7 cells coexpressing GFP-FKHR wt (green) and treated for 60 min with estradiol. Merged images are also shown on the bottom. Bar, 5 μm. (e) The cells were cotransfected with ERα siRNA (ERα siRNA) or nontargeting siRNA (nt siRNA) and GFP-FKHR wt. The cells were then left unstimulated or stimulated with 10 nM estradiol for the indicated times. GFP-FKHR wt localization was monitored by confocal microscopy. Cells that fell into the category of exclusively GFP-FKHR wt nuclear fluorescence were scored and data were expressed as a percentage of transfected cells. Data were derived from at least 200 scored cells. The results of two independent experiments were averaged. The blot in panel e confirms the silencing of ERα in MCF-7 cells transfected with ERα siRNA (top). The bottom shows the blot of loading proteins revealed using the anti-tubulin antibody. (a, b, c, and e) Means and SEM are shown.
We then verified that silencing of ERα by siRNA retains GFP-FKHR wt in the nuclear compartment of 60-min hormonal-treated MCF-7 cells (Fig. 5 e). These findings confirm that, in ERα-positive breast cancer cells, FKHR moves from nucleus to the cytoplasm by a mechanism depending on ERα.
Finally, we observed by Western blot analysis that estradiol stimulation of quiescent MCF-7 cells rapidly induces FKHR phosphorylation in Ser256 (Fig. 6 a). To complement colocalization experiments, we next analyzed the interaction between ERα and FKHR in coimmunoprecipitation experiments. Data in Fig. 6 b show that 30-min hormonal stimulation increases the association of FKHR wt with ERα in MCF-7 cells (Fig. 6 b, left). Whatever the experimental condition, the mutant FKHR AAA does not coimmunoprecipitate with ERα (Fig. 6 b, right), therefore implicating that phosphorylation of FKHR induced by estradiol is required for its association with the receptor.
Figure 6.

Estradiol-induced FKHR activation and Erα–FKHR complex assembly in MCF-7 cells. Model of ERα nuclear export. Quiescent MCF-7 cells were used. (a) Cells were left unstimulated or stimulated for 5 min with 10 nM estradiol in the absence or presence of LY294002 (at 5 μM; Qbiogene). Lysates were analyzed for FKHR phosphorylation using the anti–P-Ser-256–FKHR antibody (bottom). The nitrocellulose filter was then reprobed with anti-FKHR antibody (top). (b) The cells were transfected with the indicated plasmids (FKHR wt or FKHR AAA) then left unstimulated or stimulated for 30 min with 10 nM estradiol. Lysates were immunoprecipitated with anti-ERα antibody. Immunocomplexes were immunoblotted using the antibodies against the indicated proteins. (c) The model of ERα nuclear export is depicted. Estradiol stimulates the PI3K–AKT pathway, leading to FKHR phosphorylation and triggering the functionally associated export of FKHR–ERα. This export results in stimulation of DNA synthesis.
These findings led us to hypothesize the model of ERα export and its interplay with FKHR depicted in Fig. 6 c.
Discussion
Nucleocytoplasmic shuttling of proteins plays a critical role in cell function (for review see Kau and Silver, 2003). Most of the ERα is localized in nuclei of hormone target cells (Stenoien et al., 2001), and its best-known function as a ligand-activated transcription factor requires nuclear localization (Mangelsdorf et al., 1995). Much evidence, however, has demonstrated rapid, extranuclear action of ERα (for review see Migliaccio et al., 2007a). Ligand stimulation of different cell types recruits different signaling effectors to ERα or ERβ, which in turn leads to signal transduction pathway activation (Migliaccio et al., 2000; Castoria et al., 2001). A transcriptionally inactive ERα mutant, permanently residing in the cytoplasm, mediates S phase entry triggered by estradiol activation of the Src–Ras–ERK and PI3K–AKT pathways (Castoria et al., 1999, 2004). These findings suggest a proliferative function for this extranuclear receptor that is independent of its transcriptional activity. This view is supported by the observation that a classical mouse AR, which is expressed at a very low level in NIH3T3 fibroblasts, does not enter nuclei and does not activate gene transcription in androgen-stimulated cells. Nevertheless, it recruits several signaling effectors that control hormone-induced S phase entry and cytoskeleton changes in fibroblasts (Castoria et al., 2003). Rat uterine stromal cells, although they express low levels of transcriptionally incompetent PgR, respond to progestin with active proliferation (Vallejo et al., 2005). In addition, the extranuclear cross talk between ERα–AR and the EGF receptor regulates EGF-elicited responses such as actin changes and DNA synthesis in breast and prostate cancer cells (Migliaccio et al., 2005). Direct evidence of the proliferative role of the ERα–AR complex association with Src in mammary and prostate cancer cell xenografts has been recently described (Migliaccio et al., 2007b; Varricchio et al., 2007). These and other similar findings point to the critical role of steroid receptor extranuclear localization in steroid hormone or growth factor action.
In this study, we analyzed the nucleocytoplasmic shuttling of ERα in MCF-7 cells, focusing on its export from the nucleus. We observed that estradiol induces CRM1-dependent nuclear export of ERα. Thus, identification of the ERα sequence responsible for its nuclear export was initially performed by a nuclear export assay (Henderson and Eleftheriou, 2000). Previous studies found that the outward movement of PgR from the nuclear compartment is mediated by its NLSs (Guiochon-Mantel et al., 1994). We then assayed the ability of ERα NLSs to shift the REV mutant into the cytoplasm. Our data show that NLSs are not involved in the nuclear export of ERα. It has also been hypothesized that the nuclear exit of ERα is regulated by its phosphorylation on Thr311 (Lee and Bai, 2002). This residue is present in a putative leucine-rich ERα NES (305–322 of ERα), which shares homology with the p53 N-terminal NES (Zhang and Xiong, 2001). We found that the leucine-rich 296–335 ERα sequence does not drive the REV mutant into the cytoplasm. Using the same approach, we observed that the leucine-rich 444–456 sequence of ERα contains a functional NES, which is sensitive to LMB treatment. A Tat-conjugated peptide mimicking this amino acid sequence displaces the in vitro interaction between ERα and CRM1. These findings support the conclusion that the 444–456 sequence contains a putative NES. Mutations in the core of this sequence impair estradiol-induced nuclear export of full-length ERα in MCF-7 cells. The 444–456 sequence shows homology with the conserved leucine-rich and REV-like NES of p53 (340–351; Fig. S3 a; Stommel et al., 1999). Remarkably, the 444–456 region of ERα is conserved in other steroid receptors, such as ERβ, PgR, AR, GR, and MR (Fig. S3 b), which suggests that such a sequence is responsible for the CRM1-dependent nuclear export of most steroid receptors. This view is in agreement with the inhibition of nuclear export of different steroid receptors by LMB treatment described by several groups under different experimental conditions (Savory et al., 1999; Prufer and Barsony, 2002; Maruvada et al., 2003).
ERα localization was also modulated by a Tat-conjugated peptide mimicking the ERα 444–456 sequence. It specifically traps ERα in the nuclear compartment of MCF-7 cells and reduces the estradiol-induced S phase entry in these cells, thus confirming that ERα nuclear export plays a key role in DNA synthesis of target cells. This view is reinforced by the finding that addition of the peptide to the cell medium 1 h after hormonal stimulation, when ERα nuclear export is almost complete, does not affect estradiol-induced S phase entry in MCF-7 cells. Experiments with NIH3T3 fibroblasts show that an NES-ERα mutant fails to induce S phase entry in these cells, whereas it activates gene transcription. These findings further point to the role of ERα nuclear export in cell cycle progression modulated by estrogens in breast cancer cells.
The function of many proteins is regulated by their subcellular localization. These proteins include cell cycle regulators and transcription factors, such as nuclear factor κB, p53, and mammalian members of the FHKR transcription factors (for review see Kau and Silver, 2003). A triple alanine mutant of FKHR localizes in the nucleus and induces G1 arrest of cells (Nakamura et al., 2000; Birkenkamp and Coffer, 2003). Consistent with the hypothesis that permanent localization of transcription factors in the nucleus can stop the cell cycle, it has been shown that estradiol stimulation of Pak-1 and ERα promotes cell survival by inducing phosphorylation and nuclear exclusion of Foxo1 in breast cancer cells (Mazumdar and Kumar, 2003). Relevant to the observed association between estradiol-regulated export of ERα and FKHR, an estradiol-dependent interaction between ERα and FKHR has been observed using a yeast two-hybrid screen and in vitro pull-down experiments (Schuur et al., 2001). Extension of these investigations to other nuclear receptor family members has shown that, depending on the receptor type, FKHR represents a bifunctional intermediary protein acting as either a coactivator or corepressor of these receptors (Zhao et al., 2001).
Estradiol stimulation of MCF-7 cells triggers DNA synthesis through PI3K- and Src-dependent pathway activation, which causes an increase in cyclin D1 transcription and p27 nuclear release (Castoria et al., 2001, 2004). We observe here that estradiol-activated AKT phosphorylates FKHR, thus triggering the FKHR–ERα association and facilitating the nuclear export of the complex. By removing the inhibitory action of FKHR, this event triggers cell cycle progression. Increased FKHR–Ser-256 phosphorylation by estradiol activation of AKT has been recently described in neuronal cells (Won et al., 2006). Association of FKHR with ERα, as indicated by their colocalization and coimmunoprecipitation, might favor FKHR nuclear exit by masking its NLS. A similar interaction with FKHR has been described for the 14-3-3 protein (for review see Van Der Heide et al., 2004).
Our present results provide further insight on regulation of ERα intracellular trafficking and demonstrate the key role of estradiol rapid action in modulating this process. We have identified for the first time a functional NES of ERα. Targeting of this motif by synthetic peptides or site-directed mutagenesis modulates ERα subcellular localization and impairs estradiol-induced S phase entry in target cells. The model of ERα nuclear export is depicted in Fig. 6 c.
Our findings highlight the biological functions of this receptor and offer a powerful tool to modify the intracellular distribution of ERα. A strategy of trapping ERα in nuclear compartment using peptides derived from the NES-ERα might be a potential approach to the therapy of human breast cancer.
Materials and methods
Constructs
The constitutive active Ran Q69L was in the pQE plasmid was a gift of I.W. Mattaj (European Molecular Biology Laboratory, Heidelberg, Germany; Izaurralde et al., 1997). The pCDNA/HA-CRM1 (Alt et al., 2000) was digested with KpnI and BamH1 and subcloned into pSG5. cDNA encoding the wt of hERα (HEG0) was in the pSG5 expression vector was provided by P. Chambon (Institut de Génétique et de Biologie Moléculaire et Cellulaire [Centre National de la Recherche Scientifique/Institut National de la Santé et de la Recherche Médicale], Université Louis Pasteur, Illkirch, France; Tora et al., 1989). It was subcloned into EcoRI-pEGFP (C2; Clontech Laboratories, Inc.) or EcoRI-pSG5Myc as described previously (Castoria et al., 2004). The ERα mutants HEG14, HEG15, and HEG241 in pSG5 were provided by P. Chambon (Tora et al., 1989; Ylikomi et al., 1992) were digested with EcoRI and ligated into the pEGFP plasmid (C2). pEGFP-NLS/HEG14 was obtained by digestion of pEGFP-HEG0 with MspI and BamHI and subcloned into SmaI–BamHI pEGFP. The site-directed mutations of ERα were introduced into pSG5-HEG0 using PCR. The pSG5-HEG4A (Ile 451 and 452 changed with Ala; Leu 453 and 454 changed with Ala) and pSG5-HEGIL (Ile 451 changed with Ala; Leu 454 changed with Ala) mutants were generated. The cDNAs coding for the ERα mutants (HEG4A and HEGIL; ERα 4A and ERα IL) were subcloned into EcoRI-pEGFP. All junctions were verified by DNA sequencing. The dominant-negative form of p85α (Δp85α) in pSG5 was provided by J. Downward (Cancer Research UK London Research Institute, England, UK; Dhand et al., 1994). The Myc-His–tagged dominant-negative form of AKT (K179M) in the pUSEAmp plasmid was obtained from United Biotech Inc. The Myc-tagged pSG5 was prepared as described previously (Castoria et al., 2004). FKHR wt or mutant (FKHR AAA) were in pcDNA-GFP (Addgene). For the export assay, the putative export sequences of ERα were cloned into the BamHI and AgeI sites of pREV(1.4)-GFP and inserted between the REV and GFP coding sequence (EGFP-N1; Clontech Laboratories, Inc.). To insert the putative export ERα sequence into pREV(1.4)-GFP (Rev mutant), a PCR strategy was adopted for the following fragments: NLS sequence (241–306 aa) was amplified by forward primer 5′-GACTGGATCCAATGATGAAAGGTGGGATACGAAAAGACCG-3′ and reverse primer 5′-GCGACCGGTGGCAGGCTGTTCTTCTTAGAGCGTTTGATCA-3′; 286 sequence (286–311 aa) by forward primer 5′-GATCGGATCCAATGAGAGCTGCCAACCTTTGGCCAAGCCCG-3′ and reverse primer 5′-GCGACCGGTGGGGCCGTCAGGGACAAGGCCAGGCTGTTCT-3′; 286 sequence (286–382 aa) by reverse primer 5′-GCGACCGGTGGGGCACATTCTAGAAGGTGGACCTGATCATG-3′; 296 sequence (296–335 aa) by forward primer 5′-GATCGGATCCAATGATCAAACGCTCTAAGAAGAAC-3′ and reverse primer 5′-GCGACCGGTGGGGGTCTGGTAGGATCATACTCGGAATAGA-3′; 316 sequence (316–335 aa) by forward primer 5′-GATCGGATCCAAGTGCCTTGTTGGATGCTGAGCCCCCCAT-3′; 341 sequence (341–361 aa) by forward primer 5′GATCGGATCCATCGATGATGGGCTTACTGACCAACCTGGC-3′ and reverse primer 5′-GCGACCGGTGGCGCCCAGTTGATCATGTGAACCAGCTCCC-3′; 368 sequence (368–394 aa) by forward primer 5′-GATCGGATCCAGTGGATTTGACCCTCCATGATCAGGTCCA-3′ and reverse primer 5′-GCGACCGGTGGGCGCCAGACGAGACCAATCATCAGGATCT-3′; 381 sequence (381–412 aa) by forward primer 5′-GATCGGATCCATGTGCCTGGCTAGAGATCCTGATGATTGGT-3′ and reverse primer 5′-GCGACCGGTGGGTTCCTGTCCAAGAGCAAGTTAGGAGCAA-3′; 395 sequence (395–412 aa) by forward primer 5′GATCGGATCCATCCATGGAGCACCCAGTGAAGCTACTGTT-3′; 427 sequence (427–457 aa) by forward primer 5′-GATCGGATCCAATGCTGCTGGCTACATCATCTCGGTTCCG-3′ and reverse primer 5′-GCGACCGGTGGTCCAGAATTAAGCAAAATAATAGATTTGAGGCACAC-3′; 381–457 sequence; and 458–552 sequence by forward primer 5′-GATCGGATCCAGTGTACACATTTCTGTCCAGCACCCTGAAGTCT-3′ and reverse primer 5′-GCGACCGGTGGGGGCGCATGTAGGCGGTGGGCGTCCAGCATCTC-3′. All these fragments were amplified using Platinum Pfx (Invitrogen) according to the manufacturer's instructions. PCR products were purified, digested with BamHI–AgeI, and finally subcloned into pREV(1.4)-GFP (Rev mutant). The ERα 444–456 sequence was subcloned in the same vector as a short fragment generated by annealing of specific oligonucleotides. The same strategy was used to generate the ERα 444–456 mutant. All putative NES-ERαs were verified by sequencing to confirm the exact reading frame between REV and GFP.
Cell culture, transfection, and siRNA
Human breast cancer–derived MCF-7 cells and low-passage mouse embryo NIH3T3 fibroblasts were grown and made quiescent as described previously (Castoria et al., 1999, 2003). Unless otherwise stated, quiescent MCF-7 cells on coverslips were transfected with 1 μg of each purified plasmid using the SuperFect reagent (QIAGEN). Silencing of ERα was silenced by siRNA as described previously (Migliaccio et al., 2005), and purified GFP-FKHR wt was cotransfected at 0.5 μg. Nontargeting siRNA was used as a control, according to the same study (Migliaccio et al., 2005). After transfection, the cells were incubated at 37°C for 24 h and then used for the indicated experiments.
Nuclear export and transactivation assays
The nuclear export assay was performed into growing MCF-7 cells as described previously (Henderson and Eleftheriou, 2000). In brief, each putative NES-ERα was subcloned into the REV (1.4)-GFP (Rev mutant, provided by B. Henderson, Westmead Institute for Cancer Research, University of Sydney and Westmead Millennium Institute at Westmead Hospital, Westmead, Australia) and then transfected (at 2 μg) by SuperFect into growing cells. After 18 h, the cells were incubated with cycloheximide (at 15 μg/ml; EMD) and left untreated or treated for 8 h with actinomycin D (at 5 μg/ml; EMD). When indicated, LMB (at 5 ng/ml; EMD) was added 30 min before the addition of actinomycin D. Growing NIH3T3 fibroblasts were plated on Petri dishes. They were then transfected by SuperFect using 2 μg of either pEGFP, pEGFP-HEG0, or pEGFP-HEGIL. For transactivation assay, 2 μg of pGL2 ERE-Luc (Bonapace et al., 1996) provided by L. Altucci (University of Naples Federico II, Naples, Italy) were cotransfected. 12 h after transfection, cells were made quiescent and left untreated or treated with 10 nM estradiol for 16 h. Lysates were prepared and the luciferase activity was measured using a luciferase assay system (Promega). The results were corrected using CH110-expressed β-galactosidase activity (GE Healthcare).
DNA synthesis analysis and peptides
Quiescent MCF-7 or NIH3T3 cells on coverslips were left unstimulated or stimulated for 18 h with the indicated compounds. Estradiol was used at 10 nM and serum was added at 20%. The Tat-peptide conjugated to the 444–456 amino acid sequence of ERα (Tat-pep) as well as the Tat control peptide were used at 1 μM. The peptides were N-terminal acetylated and C-terminal amidated. After a 6-h pulse with 100 μM BrdU (Sigma-Aldrich), BrdU incorporation was analyzed as described previously (Castoria et al., 1999) using the mouse monoclonal anti-BrdU antibody (clone BU-1 from GE Healthcare). Diluted (1:200 in PBS) Texas red–conjugated goat anti–mouse antibody (Jackson ImmunoResearch Laboratories) was used to detect the primary antibody.
Immunofluorescence and confocal microscopy
Cells on coverslips were fixed for 10 min with paraformaldehyde (3% wt/vol in PBS), washed with PBS, and then analyzed by fluorescence microscopy for the subcellular localization of GFP-wtERα, its mutants, as well as putative NES-ERα constructs. Similar analysis was performed to detect GFP-FKHR wt or its mutant GFP-FKHR AAA. In all other experiments, the cells on coverslips were fixed and permeabilized as described previously (Castoria et al., 1999). Endogenous ERα was visualized as described previously (Castoria et al., 1999) using the A314 mouse mAb or the H222 rat mAb. Mouse antibody was detected using diluted (1:200 in PBS) Texas red–conjugated goat anti–mouse antibodies (EMD). Diluted (1:200 in PBS containing 0.2% BSA) Texas red–conjugated AffiniPure anti–rat IgG (Jackson ImmunoResearch Laboratories) was used to detect the H222 rat monoclonal antibody. The dominant-negative Δp85α and Myc-His–tagged dominant-negative AKT were detected using diluted (1:100 in PBS) either anti-p85α or anti-Myc tag (both from United Biotech Inc.) rabbit polyclonal antibodies. Diluted (1:200 in PBS containing 0.2% BSA) anti–rabbit fluorescein-conjugated antibodies (Jackson ImmunoResearch Laboratories) were added (Castoria et al., 2001). The Myc-tagged pSG5 or wt ERα or ERα IL were detected as described previously (Castoria et al., 2004) using the mouse anti-Myc tag monoclonal antibody (Clontech Laboratories, Inc.). Diluted (1:200 in PBS) Texas red–conjugated goat anti–mouse antibody (Jackson ImmunoResearch Laboratories) was used as a secondary antibody. Endogenous p27 was visualized as described previously (Castoria et al., 2004) using the anti-p27 mAb (BD Biosciences). Endogenous p53 was visualized using the using the mouse monoclonal anti-p53 antibody (D0-1; Santa Cruz Biotechnology, Inc.). Diluted (1:200 in PBS) anti–mouse fluorescein isothiocyanate-conjugated antibodies (EMD) were added to detect the primary mouse antibodies. Coverslips were finally stained with Hoechst 33258, inverted, and mounted in Mowiol (EMD). Fields were analyzed with a fluorescent microscope (DMBL; Leica) using HCX PL Fluotar 40×, HCX PL Apo 63× oil, and HCX PL Fluotar 100× oil objectives (Leica). Images were captured using a DC200 or DC480 camera (Leica) and acquired using IMI1000 or FW4000 software (Leica). For confocal microscopy, fields were analyzed and acquired using a confocal microscope (LSM510; Carl Zeiss, Inc.) using a Plan-Neofluar 40× immersion objective (Carl Zeiss, Inc.). Images were magnified two times.
Purified proteins and in vitro protein–protein interactions
Ran Q69L expressed in JM109 bacteria was purified using the Ni-NTA agarose (QIAGEN) and recombinant human ERα (2800 pmol/ml; Invitrogen). S35-labeled HA-CRM1 was produced in rabbit reticulocyte lysate (Promega), and protein–protein interaction was performed as described previously (Migliaccio et al., 2000). Immunoprecipitation was performed using the rabbit polyclonal anti-ERα Ab (G-20 1 μg/ml; Santa Cruz Biotechnology, Inc.) or the mouse monoclonal anti-HA antibody (at 2 μg/ml; Covance). Eluted proteins were analyzed by immunoblotting or fluorography.
Lysates, immunoprecipitation, and immunoblotting
Lysates were prepared as described previously (Migliaccio et al., 2000), and ERα was immunoprecipitated according to the same protocol. Rabbit polyclonal anti-ERα antibody (G-20; Santa Cruz Biotechnology, Inc.) was used to detect ERα. P-Ser473-AKT and AKT from cell lysates were detected as described previously (Castoria et al., 2001). P-Ser256-FKHR and FKHR were revealed by Western blotting using the appropriate antibodies (Cell Signaling Technology). Expression and production of Ran Q69L was verified using the mouse monoclonal anti-His tag antibody (QIAGEN). When indicated, ERα and the chimera GFP-ERα were detected using either the rat monoclonal (H222; Abbott Labs) anti-ERα antibody or the mouse monoclonal anti-GFP antibody (Clontech Laboratories, Inc.). Immunoreactive proteins were revealed by the ECL detection system (GE Healthcare).
Online supplemental material
Fig. S1 shows the cytoplasmic localization of endogenous ERα in indirect IF staining of untreated or estradiol-treated MCF-7 cells. Corresponding cell lysates do not show significant change in ERα or GFP-ERα levels. The figure also presents images from experiments shown in the paper as well as estradiol activation of Akt in MCF-7 cells. Fig. S2 shows that the NLS–HEG14 construct, which is made up of the nuclear localization signals and the hormone binding domain of ERα contains the NES-ERα. Fig. S3 shows the homology between the 444–456 amino acids of ERα and the conserved leucine-rich and REV-like NES of p53. It also shows that the 444–456 sequence of ERα is highly conserved among the members of steroid receptor family. The specificity of Tat-conjugated peptide is also presented. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200712125/DC1.
Supplementary Material
Acknowledgments
We thank L. Altucci, P. Chambon, A. Diehl, J. Downward, B. Henderson, and I. Mattaj for plasmids and reagents.
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (National and Regional grants) and Ministero dell'Università e della Ricerca Scientifica (PRIN 2006; grant Nos. 2006065483 and 2006062798_002). We declare that we do not have competing financial interests.
M. Lombardi and G. Castoria contributed equally to this paper.
Abbreviations used in this paper: AR, androgen receptor; CRM1, chromosome region maintenance 1; ER, estradiol receptor; FKHR, forkhead in rhabdomyosarcoma; LMB, leptomycin B; NES, nuclear export signal; PgR, progesterone receptor; PI3K, phosphatidylinositol-3-kinase; wt, wild type.
References
- Abbondanza, C., V. Rossi, A. Roscigno, L. Gallo, A. Belsito, G. Piluso, N. Medici, V. Nigro, A.M. Molinari, B. Moncharmont, and G.A. Puca. 1998. Interaction of vault particles with estrogen receptor in the MCF-7 breast cancer cell. J. Cell Biol. 141:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt, J.R., J.L. Cleveland, M. Hannink, and J.A. Diehl. 2000. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes. Dev. 14:3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askjaer, P., A. Bachi, M. Wilm, F.R. Bischoff, D.L. Weeks, V. Ogniewski, M. Ohno, C. Niehrs, J. Kjems, I.W. Mattaj, and M. Fornerod. 1999. Ran-regulated interactions of CRM1 with nucleoporins and a shuttling DEAD-Box helicase. Mol. Cell. Biol. 19:6276–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs, W.H. III, J. Meisenhelder, T. Hunter, W.K. Cavenee, and K.C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 96:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenkamp, K.U., and P.J. Coffer. 2003. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem. Soc. Trans. 31:292–297. [DOI] [PubMed] [Google Scholar]
- Bischoff, F.R., C. Klebe, J. Kretschemer, A. Wiittinghofer, and H. Ponstingl. 1994. RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc. Natl. Acad. Sci. USA. 91:2587–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonapace, I.M., R. Addeo, L. Altucci, L. Cicatiello, M. Bifulco, C. Laezza, S. Salzano, V. Sica, F. Bresciani, and A. Weisz. 1996. 17 beta-estradiol overcomes a G1 block induced by HMG-CoA reductase inhibitors and fosters cell cycle progression without inducing ERK-1 and -2 MAP kinases activation. Oncogene. 12:753–763. [PubMed] [Google Scholar]
- Castoria, G., M.V. Barone, M. Di Domenico, A. Bilancio, D. Ametrano, A. Migliaccio, and F. Auricchio. 1999. Non-transcriptional action of estrogen and progestin triggers DNA synthesis. EMBO J. 18:2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria, G., A. Migliaccio, A. Bilancio, M. Di Domenico, A. de Falco, M. Lombardi, R. Fiorentino, L. Varricchio, M.V. Barone, and F. Auricchio. 2001. PI3-kinase in concert with Src promotes the S-phase entry of estradiol-stimulated MCF-7 cells. EMBO J. 20:6050–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria, G., M. Lombardi, M.V. Barone, A. Bilancio, M. Di Domenico, D. Bottero, F. Vitale, A. Migliaccio, and F. Auricchio. 2003. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J. Cell Biol. 161:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria, G., A. Migliaccio, M. Di Domenico, M. Lombardi, A. de Falco, L. Varricchio, A. Bilancio, M.V. Barone, and F. Auricchio. 2004. Role of atypical PKC in estradiol-triggered G1/S progression of MCF-7 cells. Mol. Cell. Biol. 24:7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvois, S., R. White, and M.G. Parker. 1993. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 106:1377–1388. [DOI] [PubMed] [Google Scholar]
- DeFranco, D.B. 2001. Nuclear export: DNA-binding domains find a surprising partner. Curr. Biol. 11:R1036–R1037. [DOI] [PubMed] [Google Scholar]
- Dhand, R., K. Hara, I. Hiles, B. Bax, I. Gout, G. Panayotou, M.J. Fry, K. Yonezawa, M. Kasuga, and M.D. Waterfield. 1994. PI3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod, M., M. Ohno, M. Yoshida, and I.W. Mattaj. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 90:1051–1060. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., S. Asano, T. Nakamura, M. Adachi, M. Yoshida, M. Yanagida, and E. Nishida. 1997. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 390:308–311. [DOI] [PubMed] [Google Scholar]
- Guiochon-Mantel, A., P. Lescop, S. Christin-Maitre, H. Loosfelt, M. Perrot-Applanat, and E. Milgrom. 1991. Nucleocytoplasmic shuttling of the progesterone receptor. EMBO J. 10:3851–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiochon-Mantel, A., K. Delabre, P. Lescop, and E. Milgrom. 1994. Nuclear localization signals also mediate the outward movement of proteins from the nucleus. Proc. Natl. Acad. Sci. USA. 91:7179–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururaj, A.E., S.K. Rayala, R.K. Vadlamudi, and R. Kumar. 2006. Novel mechanisms of resistance to endocrine therapy: genomic and nongenomic considerations. Clin. Cancer Res. 12:1001s–1007s. [DOI] [PubMed] [Google Scholar]
- Henderson, B.R. 2000. Nuclear-cytoplasmic shuttling of APC regulates b-catenin subcellular localization and turnover. Nat. Cell Biol. 2:653–660. [DOI] [PubMed] [Google Scholar]
- Henderson, B.R., and A. Eleftheriou. 2000. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 256:213–224. [DOI] [PubMed] [Google Scholar]
- Izaurralde, E., U. Kutay, C. von Kobbe, I.W. Mattaj, and D. Gorlich. 1997. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 16:6535–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joliot, A., and A. Prochiantz. 2004. Transduction peptides: from technology to physiology. Nat. Cell Biol. 6:189–196. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen, B.S., J.F. Elliston, F.J. Jr Monsma, P.A. Springer, Y.S. Ziegler, and G.L. Greene. 1987. Structural analysis of covalently labeled estrogen receptors by limited proteolysis and monoclonal antibody reactivity. Biochemistry. 26:2364–2373. [DOI] [PubMed] [Google Scholar]
- Kau, T.R., and P.A. Silver. 2003. Nuclear transport as a target for cell growth. Drug Discov. Today. 8:78–85. [DOI] [PubMed] [Google Scholar]
- Kumar, R., R.A. Wang, A. Mazumdar, A.H. Talukder, M. Mandal, Z. Yang, R. Bagheri-Yarmand, A. Sahin, G. Hortobagyi, L. Adams, et al. 2002. A naturally occurring MTA1 variant sequesters oestrogen receptor-a in the cytoplasm. Nature. 418:654–657. [DOI] [PubMed] [Google Scholar]
- Kudo, N., B. Wolff, T. Sekimoto, E.P. Schreiner, Y. Yoneda, M. Yanagida, S. Horinouchi, and M. Yoshida. 1998. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242:540–547. [DOI] [PubMed] [Google Scholar]
- Lee, H., and W. Bai. 2002. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol. Cell. Biol. 22:5835–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., and D.B. De Franco. 2000. Protracted nuclear export of glucocorticoid receptor limits its turnover and does not require the exportine1/CRM-directed nuclear export pathway. Mol. Endocrinol. 14:40–51. [DOI] [PubMed] [Google Scholar]
- Liu, J.L., Z. Mao, T.A. LaFortune, M.M. Alonso, G.E. Gallick, J. Fueyo, and W.K. Yung. 2007. Cell cycle-dependent nuclear export of phosphatase and tensin homologue tumor suppressor is regulated by the phosphoinositide-3-kinase signaling cascade. Cancer Res. 67:11054–11063. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf, D.J., C. Thummel, M. Beato, P. Herrlich, G. Schütz, K. Umesono, B. Blumberg, P. Kastner, M. Mark, P. Chambon, and R.M. Evans. 1995. The nuclear receptor superfamily: the second decade. Cell. 83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruvada, P., C.T. Baumann, G.L. Hager, and P.M. Yen. 2003. Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J. Biol. Chem. 278:12425–12432. [DOI] [PubMed] [Google Scholar]
- Mazumdar, A., and R. Kumar. 2003. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 535:6–10. [DOI] [PubMed] [Google Scholar]
- Migliaccio, A., G. Castoria, M. Di Domenico, A. de Falco, A. Bilancio, M. Lombardi, M.V. Barone, D. Ametrano, M.S. Zannini, C. Abbondanza, and F. Auricchio. 2000. Steroid-induced androgen receptor-oestradiol receptor b2Src complex triggers prostate cancer cell proliferation. EMBO J. 19:5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio, A., M. Di Domenico, G. Castoria, M. Nanayakkara, M. Lombardi, A. de Falco, A. Bilancio, L. Varricchio, A. Ciociola, and F. Auricchio. 2005. Steroid receptor regulation of EGF signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. 65:10585–10593. [DOI] [PubMed] [Google Scholar]
- Migliaccio, A., G. Castoria, and F. Auricchio. 2007. a. Src-dependent signalling pathway regulation by sex-steroid hormones: Therapeutic implications. Int. J. Biochem. Cell Biol. 39:1343–1348. [DOI] [PubMed] [Google Scholar]
- Migliaccio, A., L. Varricchio, A. De Falco, G. Castoria, C. Arra, H. Yamaguchi, A. Ciociola, M. Lombardi, R. Di Stasio, A. Barbieri, et al. 2007. b. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene. 26:6619–6629. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., S. Ramaswamy, F. Vazquez, S. Signoretti, M. Loda, and W.R. Sellers. 2000. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 20:8969–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg, E.A. 1997. Nucleocytoplasmic transport: signals, mechanism and regulation. Nature. 386:779–787. [DOI] [PubMed] [Google Scholar]
- Prufer, K., and J. Barsony. 2002. Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol. Endocrinol. 16:1738–1751. [DOI] [PubMed] [Google Scholar]
- Saporita, A.J., Q. Zhang, N. Navai, Z. Dincer, J. Hahn, Z. Cai, and Z. Wang. 2003. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J. Biol. Chem. 278:41998–42005. [DOI] [PubMed] [Google Scholar]
- Savory, J.G., B. Hsu, I.R. Laquian, W. Giffin, T. Reich, R.J. Hache, and Y.A. Lefebre. 1999. Discrimination between NL1 and NL2 mediated nuclear localization of the glucocorticoid receptor. Mol. Cell. Biol. 19:1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuur, E.R., A.V. Loktev, M. Sharma, Z. Sun, R.A. Roth, and R.J. Weigel. 2001. Ligand-dependent interaction of estrogen receptor-alpha with members of the forkhead transcription factor family. J. Biol. Chem. 276:33554–33560. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J., W. Godolphin, L.A. Jones, J.A. Holt, S.G. Wong, D.E. Keith, W.J. Levin, S.G. Stuart, J. Udove, and A. Ullrich. 1989. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 244:707–712. [DOI] [PubMed] [Google Scholar]
- Stenoien, D.L., K. Patel, M.G. Mancini, M. Dutertre, C.L. Smith, B.W. O'Malley, and M.A. Mancini. 2001. FRAP reveals that mobility of oestrogen receptor-alpha is ligand-and proteasome-dependent. Nat. Cell Biol. 3:15–23. [DOI] [PubMed] [Google Scholar]
- Stommel, J.M., N.D. Marchenko, G.S. Jimenez, U.M. Moll, T.J. Hope, and G.M. Wahl. 1999. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18:1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tora, L., A. Mullick, D. Metzger, M. Ponglikitmongkol, I. Park, and P. Chambon. 1989. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 8:1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi, R.K., Y. Lavrovsky, S.C. Ahn, S.C. Song, B. Chatterjee, and A.K. Roy. 2000. Dynamics of intracellular movement and nucleocytoplasmic recycling of the ligand-activated androgen receptor in living cells. Mol. Endocrinol. 14:1162–1174. [DOI] [PubMed] [Google Scholar]
- Vadlamudi, R.K., B. Manavathi, S. Balasenthil, S.S. Nair, Z. Yang, A.A. Sahin, and R. Kumar. 2005. Functional implications of altered subcellular localization of PELP1 in breast cancer cells. Cancer Res. 65:7724–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo, G., C. Ballaré, J.L. Barañao, M. Beato, and P. Saragüeta. 2005. Progestin activation of nongenomic pathways via cross talk of progesterone receptor with estrogen receptor β induces proliferation of endometrial stromal cells. Mol. Endocrinol. 19:3023–3037. [DOI] [PubMed] [Google Scholar]
- Van Der Heide, L.P., M.F. Hoekman, and M.P. Smidt. 2004. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 380:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varricchio, L., A. Migliaccio, G. Castoria, H. Yamaguchi, A. de Falco, M. Di Domenico, P. Giovannelli, W. Farrar, E. Appella, and F. Auricchio. 2007. Inhibition of estradiol receptor/Src association and cell growth by an estradiol receptor alpha tyrosine-phosphorylated peptide. Mol. Cancer Res. 5:1213–1221. [DOI] [PubMed] [Google Scholar]
- Vicent, G.P., C. Ballaré, A.S. Nacht, J. Clausell, A. Subtil-Rodríguez, I. Quiles, A. Jordan, and M. Beato. 2006. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol. Cell. 24:367–381. [DOI] [PubMed] [Google Scholar]
- Won, C.K., H.H. Ji, and P.O. Koh. 2006. Estradiol prevents the focal cerebral ischemic injury-induced decrease of forkhead transcription factors phosphorylation. Neurosci. Lett. 398:39–43. [DOI] [PubMed] [Google Scholar]
- Ylikomi, T., M.T. Bocquel, M. Berry, H. Gronemeyer, and P. Chambon. 1992. Cooperation of proto-signals for nuclear accumulation of estrogen and progesterone receptors. EMBO J. 11:3681–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., and Y. Xiong. 2001. A p-53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 292:1910–1915. [DOI] [PubMed] [Google Scholar]
- Zhao, H.H., R.E. Herrera, E. Coronado-Heinsohn, M.C. Yang, J.H. Ludes-Meyers, K.J. Seybold-Tilson, Z. Nawaz, D. Yee, F.G. Barr, S.G. Diab, et al. 2001. Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J. Biol. Chem. 276:27907–27912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}