

Abstract
Snail family proteins are key regulators of epithelial-mesenchymal transition, but their role in endothelial-to-mesenchymal transition (EMT) is less well studied. We show that Slug, a Snail family member, is expressed by a subset of endothelial cells as well as mesenchymal cells of the atrioventricular canal and outflow tract during cardiac cushion morphogenesis. Slug deficiency results in impaired cellularization of the cardiac cushion at embryonic day (E)–9.5 but is compensated by increased Snail expression at E10.5, which restores cardiac cushion EMT. We further demonstrate that Slug, but not Snail, is directly up-regulated by Notch in endothelial cells and that Slug expression is required for Notch-mediated repression of the vascular endothelial cadherin promoter and for promoting migration of transformed endothelial cells. In contrast, transforming growth factor β (TGF-β) induces Snail but not Slug. Interestingly, activation of Notch in the context of TGF-β stimulation results in synergistic up-regulation of Snail in endothelial cells. Collectively, our data suggest that combined expression of Slug and Snail is required for EMT in cardiac cushion morphogenesis.
Introduction
Epithelial-mesenchymal transition is the process by which epithelial cells undergo phenotypic and morphological reorganization. Epithelial-mesenchymal transition is essential during embryogenesis for the formation of many tissues, including the formation of the mesoderm, the migration of neural crest cells, and the development of the heart valves and septa (Hay, 2005). Endothelial-to-mesenchymal transition (EMT) is a specific form of epithelial-mesenchymal transition that is initiated at embryonic day (E)–9.5 in the atrioventricular (AV) canal and E10.5 in the outflow tract (OFT) cardiac cushions, the two sites of EMT in the developing heart (Camenisch et al., 2002a). This process generates cells that contribute to the connective tissue of the valves and septa of the adult heart (Eisenberg and Markwald, 1995).
Recent studies have demonstrated a critical role of the Notch signaling pathway during cardiac EMT, and disruption of this pathway has been implicated in the pathogenesis of various cardiovascular diseases (Iso et al., 2003; Niessen and Karsan, 2007). In the mouse, targeted deletion of Notch1 or its key nuclear partner CSL (CBF1/Suppressor of Hairless/Lag-1) results in cardiac cushion EMT defects (Oka et al., 1995; Timmerman et al., 2004). Further, targeted deletion of the downstream Notch/CSL effector Hey2 or double-deficiency of Hey1 and Hey2 or Hey1 and HeyL results in various congenital heart anomalies including cardiac cushion defects (Donovan et al., 2002; Fischer et al., 2004, 2007). In humans, mutations at the Notch1 locus are associated with bicuspid aortic valve disease as well as mitral valve anomalies and tetralogy of Fallot (Garg et al., 2005). Further, patients with mutations of the Notch ligand Jagged1 develop Alagille syndrome, a polymalformative disorder which includes cardiac cushion defects (Li et al., 1997; Oda et al., 1997; Eldadah et al., 2001).
TGF-β–related pathways have also been shown to be essential for proper heart development through their role in regulating EMT (Azhar et al., 2003). Of particular interest, BMP2 and TGF-β2 are expressed by the AV canal cushion myocardium (Dickson et al., 1993; Zhang and Bradley, 1996). BMP2-deficient mice die before cardiac cushion development (Zhang and Bradley, 1996); however, deficiency of the BMP2 receptor Alk2 results in AV canal EMT defects (Wang et al., 2005). TGF-β2–deficient mouse embryos do not display obvious cardiac cushion EMT defects, although later remodeling of the AV canal cushion is impaired (Dickson et al., 1993; Sanford et al., 1997; Bartram et al., 2001; Molin et al., 2002, 2003). However, using an ex vivo AV canal explant assay, TGF-β2–blocking antibodies or blocking antibodies for its coreceptor TβRIII inhibit AV canal EMT, suggesting redundancy of this pathway in vivo (Brown et al., 1999; Camenisch et al., 2002a). These data and others have established a clear role of TGF-β–related pathways during mammalian cardiac cushion development.
The Snail family members Snail (also known as Snai1) and Slug (also known as Snai2) encode zinc finger–containing transcriptional repressors that trigger EMT during embryonic development and tumor progression, in part by regulating expression of junctional proteins, most notably E-cadherin (Nieto, 2002). In the mouse, Snail has been shown to be expressed in the cardiac cushions from E9.5 onwards (Timmerman et al., 2004). Mice deficient for Snail die at E7.5, before cardiac development, and display defects in mesoderm formation (Carver et al., 2001). Conditional deletion of Snail after E8 results in lethality by E9.5, partially because of severe cardiovascular defects, but before the initiation of cardiac cushion EMT (Murray and Gridley, 2006). In the mouse, Slug is expressed in the cardiac cushions at E13.5, and mice deficient for Slug are viable but are growth retarded and display defects in pigmentation and hematopoiesis (Jiang et al., 1998; Inoue et al., 2002). To date, there is no direct evidence demonstrating the requirement for any Snail family member during mammalian heart development.
In this paper, we demonstrate that Slug is first expressed by a subset of endothelial cells as well as mesenchymal cells of the AV canal at E9.5, at the initiation of EMT. In keeping with a requirement for Slug during the initiation of cardiac EMT, the AV canal cushions show markedly reduced cellularization at E9.5, which normalizes by E10.5. Concordant with the in vivo findings, AV canal explant assays demonstrate that EMT in Slug-deficient embryos is impaired at E9.5 but not E10.5, as EMT in Slug-deficient embryos is rescued by an increase in Snail expression by E10.5. Accordingly, abolishing both Slug and Snail expression results in EMT defects at E10.5. In contrast to a previous study, we show that Notch signaling, through CSL, directly regulates the Slug promoter, resulting in the up-regulation of Slug, but not Snail, in endothelial cells (Timmerman et al., 2004). We further show that Slug directly binds and represses the vascular endothelial cadherin (VE-cadherin) promoter. Slug also promotes increased migration toward PDGF. In contrast, TGF-β2 and BMP2 induce Snail expression but minimal Slug expression. However, Notch synergistically induces Snail in concert with TGF-β2. Our data demonstrate that Notch-induced expression of Slug plays an important role in the initiation of EMT in the heart but that increased Snail compensates for the lack of Slug in Slug-targeted embryos as cardiac cushion morphogenesis progresses.
Results
Activation of the Notch pathway induces Slug but not Snail in endothelial cells
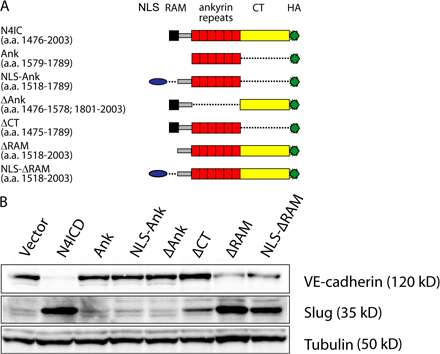
It has previously been suggested that EMT initiated by Notch may proceed through the induction of Snail; however, the degenerate primers used in that study amplify both Snail and Slug (Timmerman et al., 2004). To clarify which Snail family members are regulated by Notch signaling, we activated Notch in endothelial cells by ectopically expressing the Notch ligands Jagged1 or Dll4 or the constitutively active form of Notch1 or Notch4 (Notch1ICD and Notch4ICD, respectively), all of which are expressed in the cardiac cushion (Loomes et al., 1999; Noseda et al., 2004). Activated Notch up-regulated Slug, but not Snail, in all endothelial cells tested, as demonstrated by RT-PCR (Fig. 1 A), quantitative RT-PCR (qRT-PCR; Fig. 1 B), and immunoblotting (Fig. 1, C and D; and see Fig. 6 B). As a positive control, we confirmed that the known Notch targets Hey1, Hey2, and HeyL were induced by NotchICD (Fig. 1, A and B). Additional experiments with NotchICD deletion constructs revealed that the Ankyrin repeats of Notch are required for induction of Slug (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1). These findings indicate that Notch activation induces Slug but not Snail expression in endothelial cells.
Figure 1.

Expression of Slug, but not Snail, is induced by Notch activation. (A) Analysis of mRNA expression by semi–qRT-PCR in human mammary epithelial cells (HMEC) and human umbilical vein epithelial cells (HUVEC) expressing constitutively active Notch1 (Notch1ICD) or Notch4 (Notch4ICD). (B) Analysis of mRNA expression by qRT-PCR in HMEC expressing Notch1ICD. Results are normalized to the vector control (n = 3). *, P < 0.05. Error bars show SEM. (C) Immunoblots for Slug and Snail in HMEC, HUVEC, and human aortic endothelial cells (HAEC) transduced with Notch1ICD, Notch4ICD, or the empty vector. (D) HMEC expressing Jagged1 or Dll4 were cocultured with parental HMEC and immunoblotting for Slug was performed.
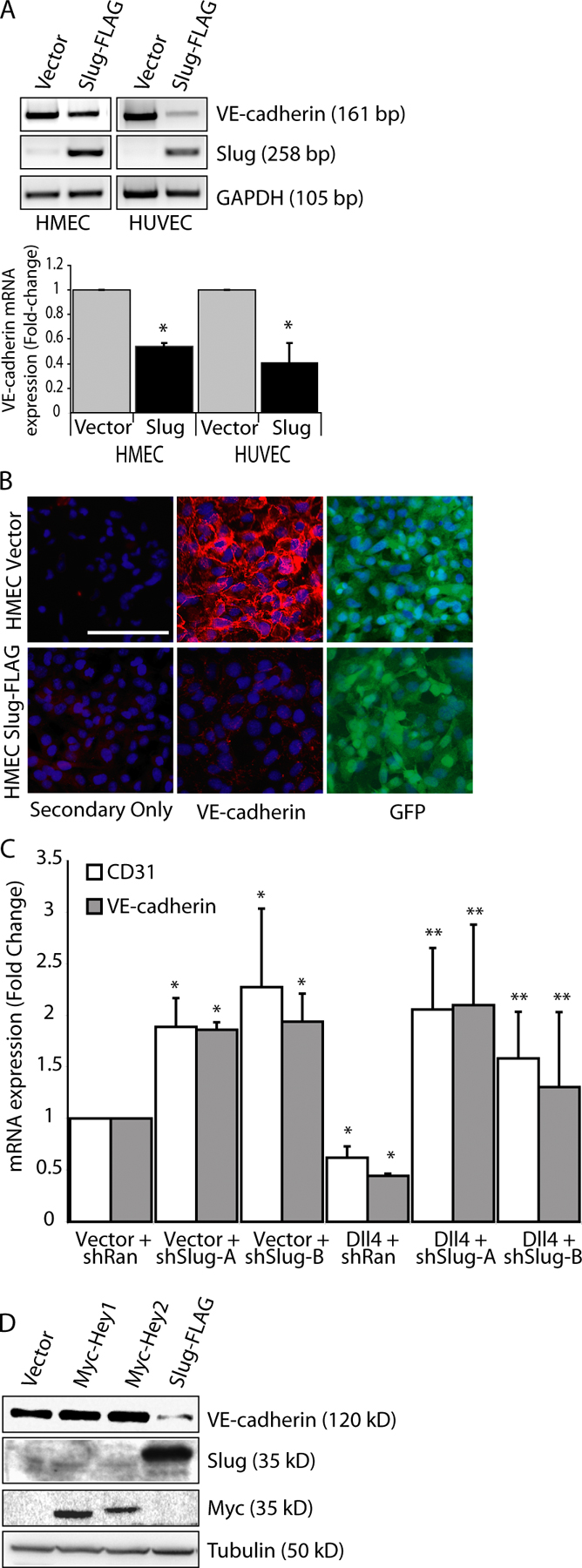
Figure 6.

Induction of Snail by TGF-β2 is synergistically enhanced in Dll4-activated endothelial cells. (A) qRT-PCR for Snail mRNA in vector- or Dll4-activated HMEC treated with 2.5 ng/ml TGF-β2 for the indicated times (n = 3). *, P < 0.05. (B) Immunoblots for Snail and Slug in vector- or Dll4-activated HMEC treated with 2.5 ng/ml TGF-β2. (C) qRT-PCR for Snail, Slug, Hey1, and Smad7 mRNA in vector- or Dll4-activated HMEC treated with 10 μM DMSO or DAPT for 16 h followed by treatment with 2.5 ng/ml TGF-β2 for 3 h (n = 3). *, P < 0.05. (D) qRT-PCR for Snail, Slug, Hey1, and Smad7 mRNA in vector- or Dll4-activated HMEC treated with 20 or 50 ng/ml BMP2 (n = 3). Error bars show SEM.
Slug is expressed in a subset of endothelial cells and the mesenchymal cells of the AV canal, OFT, and valves of the embryonic mouse and human heart
It has been reported that Slug mRNA is not expressed at E9.5 in the cardiac cushion (Timmerman et al., 2004), which suggests that it is dispensable for cardiac EMT. Because Notch activation has been shown to be critical for EMT during cardiac cushion development (Noseda et al., 2004; Timmerman et al., 2004), we were interested in defining the expression of Slug during the period of cellularization of the mammalian cardiac cushions. We thus examined Slug-lacZ mice, which have the lacZ gene inserted into the Slug locus with concomitant deletion of the zinc finger DNA binding motifs. Expression of lacZ in this model has been shown to faithfully match expression of Slug mRNA in all tissues analyzed, as determined by in situ hybridization (Jiang et al., 1998). β-galactosidase staining of E9.5–11.5 embryos showed that Slug-expressing cells were abundant in the heart (Fig. 2 A) with expression within a subset of endothelial cells as well as the mesenchymal cells of the AV canal and OFT at E9.5, with increasing expression over E10.5 and 11.5 (Fig. 2 B). Detailed analysis of Slug expression around E9.5 revealed that Slug expression is induced in the AV canal at the 25–28-somite stage at the initiation of EMT (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1). Immunofluorescent staining for Slug protein at E11.5 confirmed expression in the cardiac cushion mesenchyme and a subset of endothelial cells that costained for CD31 (Fig. 2 C, arrowheads).
Figure 2.

Slug expression during cardiac cushion development. (A) β-galactosidase staining (representing Slug expression) of whole-mounted hearts from Slug-lacZ+/− embryos from E9.5 to 11.5. Arrows point to the AV canal or OFT at E11.5. Bars, 250 μm. (B) Sections through the AV canal and OFT of β-galactosidase–stained Slug-lacZ+/− hearts from E9.5 to 11.5. Bars, 100 μm. (C) Immunofluorescence staining for Slug (red) and CD31 (green) in E11.5 embryonic mouse hearts. Arrowheads point to cells coexpressing Slug and CD31. Bars, 25 μm. (D) In situ hybridization for Snail and Slug in a 65-d human embryonic heart. Arrows point to the mitral and tricuspid valves, arrowheads indicate the interatrial septum, and the asterisk marks the AV septum. A higher magnification image of the heart valve is shown in the right panel of each analysis. Bars, 1 mm.
It has been suggested that EMT continues to take place to allow valvular remodeling later in development as well as in the adult (Armstrong and Bischoff, 2004). To confirm a role for Snail family members in the human heart, we examined expression of Snail and Slug in embryonic human hearts at various developmental stages (days 52–78 of gestation) with similar results at various stages. Fig. 2 D shows that Snail and Slug are both expressed in the tricuspid and mitral valves, the AV septum, and the interatrial septum in a 65-d human heart. This staining pattern is similar to Slug expression in later stages of mouse heart development (Oram et al., 2003). Higher magnification images revealed that the mesenchymal cells of the valves, as well as endothelial cells at the root of the valves, express Snail and Slug (Fig. 2 D), suggesting a role for Snail family members in human cardiac cushion development and remodeling.
Slug is necessary for EMT in the cardiac cushions
To determine whether targeted disruption of the Slug gene has a functional effect on cardiac cushion development, the AV canal of E9.5 embryos were placed on collagen gels to monitor EMT ex vivo, as previously described (Camenisch et al., 2002a; Chang et al., 2004). Occasional endothelial cell outgrowths occur proximal to 100 μm of the AV canal explant. Therefore, only the morphologically distinct mesenchymal cells distal to 100 μm from the AV canal were quantitated to determine the degree of EMT. Homozygous Slug-LacZ mutants behave as Slug−/− (Slug-deficient) animals (Jiang et al., 1998; Inoue et al., 2002), and Slug−/− AV canal explants had significantly reduced migration and invasion compared with Slug+/− or wild-type controls at E9.5 (Fig. 3, A and B). Of the few Slug−/− cells that did migrate, many had a rounded morphology, and were not able to differentiate into spindled mesenchymal cells. Analysis of β-galactosidase activity in Slug+/− AV explants revealed Slug expression in the migrating cells as well as the proximal cardiac endothelial cells (Fig. 3 C). Interestingly, the majority of β-galactosidase staining was seen in rounded cells, which is consistent with morphology of cells that are intermediate between endothelial and mesenchymal phenotype, as previously described (Camenisch et al., 2002b).
Figure 3.

Slug−/− embryos display defects in AV canal EMT at E9.5. (A) Phase contrast (left) and DAPI (right) images of AV canal explants from wild-type and Slug−/− embryos. Bars, 250 μm. (B) Quantitative analysis of EMT in AV canal explants from E9.5 wild-type (wt), Slug+/−, and Slug−/− embryos after 48 h in culture. Results represent the distance of a positive pixel (DAPI-stained nucleus) to the closest point of the AV canal normalized to the area of the AV canal tissue. *, P < 0.05. (C) Slug expression in the AV canal explant assay as visualized by β-galactosidase staining of wild-type and Slug-lacZ+/− AV explants. The rounded morphology of most of the LacZ+ cells is shown on the right. The black square indicates the region of higher magnification shown to the right. Bars, 50 μm. (D) Representative sections of wild-type and Slug−/− hearts counterstained with Nuclear Fast Red used for analysis in E. Dotted blue lines highlight the superior and inferior AV cushions. Bars, 50 μm. (E) Quantitation of the cellularity of the superior and inferior cushions in E9.5 wild-type (wt; n = 3) and Slug−/− (n = 3) embryos. Error bars show SEM. (F) BrdU analysis on the percentage of proliferating cells in wild-type (n = 4) and Slug−/− (n = 6) AV canal cardiac cushions (total), the AV canal endocardium (Endo), and AV canal mesenchymal cells (Mesen; 10–15 sections per embryo). Error bars show SEM. (G) Vector- or Slug-transduced HMEC were subjected to an endothelial wounding assay. Bars represent the distance migrated after 24 h (n = 4). *, P < 0.05. Error bars show SD. (H) Vector- or Slug-transduced HMEC were evaluated in a modified Boyden chamber assay with 20 ng/ml PDGF-BB present in the lower chamber. Bars represent the total number of cells migrated after 4 h (n = 6). *, P < 0.05. Error bars show SD.
To confirm that cushion cellularization was impaired in Slug-deficient embryos in vivo, E9.5 hearts were serially sectioned (between 7 and 20 sections for each heart) and the number of cushion cells was quantitated in every section. At E9.5, Slug−/− embryos had significantly fewer mesenchymal cells in the cardiac cushions compared with wild-type controls (Fig. 3, D and E). However, by E10.5 there was no difference in cellularity of the cardiac cushions, and a defect in AV canal EMT ex vivo was not evident (Fig. S2). These findings implicate Slug in the early activation and migration of endothelial cells during cushion EMT with potential compensation by other factors later in cardiac cushion development. To investigate the reason for the normalization of cardiac cushion cellularization by E10.5, the degree of cardiac cushion apoptosis and proliferation was evaluated. AV canal endocardial and mesenchymal cell proliferation was measured by BrdU incorporation and no difference in S-phase entry was noted at E9.5 (unpublished data). However, analysis at E10.25 revealed that there is an increase in BrdU incorporation in both the endocardium and mesenchyme in Slug−/− embryos (Fig. 3 F). Although the increase in BrdU incorporation was small, a greater pool of endocardial cells able to undergo EMT combined with increased mesenchymal proliferation may be sufficient to normalize cushion cellularity by E10.5. In contrast, quantitation of active caspase-3 to enumerate the numbers of cells undergoing apoptosis did not reveal much cell death (< 1%) in either wild-type or Slug−/− AV canals, with no difference between the two groups (unpublished data).
To determine whether Slug is sufficient to promote a motile phenotype in endothelial cells, endothelial cells were transduced with vector or Slug, and an in vitro wound healing (scratch) assay was performed. The scratch assay revealed increased migration of Slug-expressing endothelial cells as early as 4 h and up to 24 h after wounding of the endothelial monolayer, resulting in Slug-expressing cells migrating almost twice as far after 24 h (Fig. 3 G). PDGF has been shown to be expressed in the cardiac cushions during EMT (Van Den Akker et al., 2005). Using a modified Boyden chamber assay with 20 ng/ml PDGF-BB present in the lower chamber, we found that Slug-expressing endothelial cells showed significantly increased directed migration toward PDGF-BB (Fig. 3 H). Thus, Slug expression in the endothelium appears to be sufficient for endothelial motility and directed migration.
Slug represses endothelial phenotype
Given our findings demonstrating the requirement of Slug in cardiac EMT, we sought to examine the role that Slug plays in modulating the endothelial phenotype. Enforced expression of Slug repressed expression of key endothelial genes such as VE-cadherin, CD31, and Tie2 as determined by qRT-PCR, immunoblotting, and immunofluorescence (Fig. 4, A and B; and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1). However, in contrast to activated Notch, Slug did not induce the mesenchymal markers smooth muscle α-actin and h1-calponin (Fig. 4 B). These findings suggest that Slug expression promotes the initial phases of EMT associated with the loss of endothelial phenotype but is not sufficient to complete the transition into a mesenchymal cell that is mediated by Notch activation.
Figure 4.

Slug represses the endothelial phenotype and directly regulates the VE-cadherin promoter. (A) Analysis of endothelial marker expression by qRT-PCR in Slug-transduced HMEC (n = 3). *, P < 0.05. (B) Immunoblots for endothelial and mesenchymal markers in empty vector–, NotchICD-, and Slug-expressing HMEC. (C) EMSA using in vitro–translated luciferase (Luc) or Slug-FLAG protein and 32P-labeled double-stranded oligonucleotides for an E-box cis element (−97) or two putative Slug E2-box motifs (−306 and −379) in the human VE-cadherin promoter. Supershift assays with anti–FLAG-M2 or IgG control antibodies and competition assays with 50× wild-type (wt) or mutant probes are also shown. (D) Promoter activity in endothelial cells cotransfected with vector or Slug plasmids and wild-type, E2-box mutant, or E-box mutant mouse VE-cadherin promoter-luciferase constructs (n = 4; each experiment performed in triplicate). *, P < 0.05. Error bars show SEM.
VE-cadherin is a key endothelial adherens junction protein that is required for maintenance of endothelial homeostasis and that must be down-regulated before endothelial remodeling (Crosby et al., 2005). We thus determined whether Slug was capable of directly repressing VE-cadherin. Promoter analysis of human VE-cadherin identified two putative Slug binding E2-box (CACCTG) motifs 5′ to the transcriptional start site (TSS), located at −306 to −311 and −379 to −384 (Prandini et al., 2005). As demonstrated by electrophoretic mobility shift assays (EMSA), Slug was capable of binding both E2-box motifs, but was unable to bind a CAGCTG E-box element located at −97 to −102 in the human VE-cadherin promoter (Fig. 4 C). Of the three cis elements tested in the human VE-cadherin promoter, only the −379 to −384 E2-box and the −97 to −102 E-box motifs are conserved in the mouse VE-cadherin promoter. Consistent with the EMSA result, mutation of the E2-box, but not the E-box motif, significantly reduced the ability of Slug to repress the mouse VE-cadherin promoter as measured by luciferase assays (Fig. 4 D). Thus, VE-cadherin transcription is directly repressed by Slug binding to the E2-box promoter elements in endothelial cells.
Notch acts through CSL to induce Slug and repress the endothelial phenotype
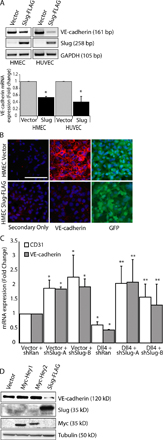
We next determined whether Notch induces Slug through the canonical CSL-dependent pathway or the less well-defined CSL-independent route (Ramain et al., 2001). Dll4-mediated induction of Slug mRNA and protein was dramatically reduced when CSL was knocked down using either of two lentiviral-delivered short hairpin RNA (shRNA) constructs, which target distinct regions of CSL (Fig. 5, A–C). As expected, induction of the Notch target HeyL was also abolished by CSL knockdown (Fig. 5 B). In addition, the ability of Notch activation to down-regulate the endothelial markers VE-cadherin and CD31 (Fig. 5 C) was abrogated when CSL was knocked down. We also targeted Slug using two distinct lentiviral-delivered shRNAs and found that the ability of Dll4-activated Notch to down-regulate VE-cadherin and CD31 was also reversed by Slug knockdown (Fig. 5 C and Fig. S3), thus demonstrating the requirement of CSL-mediated induction of Slug for Notch-mediated EMT. Furthermore, activation of CSL using a constitutively active CSL mutant (CSL-VP16; MacKenzie et al., 2004) demonstrated that CSL activation alone was sufficient to up-regulate Slug expression as well as the Notch target HeyL (Fig. 5 D). However, enforced expression of the Notch targets Hey1 or Hey2, which have been implicated in cardiac EMT, did not up-regulate Slug or repress VE-cadherin (Fig. S3). Together, these findings indicate that Notch, via CSL, directly up-regulates Slug expression and that Slug is the Notch target responsible for repressing VE-cadherin expression.
Figure 5.

Notch signaling regulates Slug expression through a CSL-dependent pathway. (A) qRT-PCR analysis demonstrating efficient knockdown of CSL in HMEC with two different shRNAs targeting CSL (shCSL) compared with a random control sequence (shRan). (B) qRT-PCR of Slug and HeyL in vector- or Dll4-activated HMEC transduced with shCSL constructs (n = 3). *, P < 0.05 vector shRandom versus HA-D114 shRandom; **, P < 0.05 HA-D114 shRandom versus HA-D114 shCSL-A or shCSL-B. (C) Immunoblotting for Slug, VE-cadherin, and CD31 in vector- or Dll4-activated HMEC transduced with shCSL or shSlug constructs. (D) qRT-PCR of vector- or CSL-VP16–expressing HMEC for Slug and HeyL (n = 3). *, P < 0.05. (E) PCR after ChIP with anti–FLAG-M2 antibody on HMEC-expressing vector (vec) or FLAG-CSL (CSL) to demonstrate CSL binding to the human Slug promoter. The negative (-ve) control represents PCR of the ZNF3 promoter after ChIP using FLAG-M2. (F) EMSA using nuclear lysates collected from vector- or FLAG-CSL–expressing HMEC and 32P-labeled double-stranded oligonucleotides spanning each of the two CSL binding sites in the human Slug promoter. Supershift assays with anti–FLAG-M2 or IgG control antibodies, and competition assays with 50× wild-type (wt) or mutant probes are also shown. Error bars show SEM.
Analysis of the human and mouse Slug promoters (−2,000 to +100 relative to the TSS) identified six putative CSL binding sites ((C/T)(A/G)TG(A/G/T)GA(A/G/T)) in the human and two putative CSL binding sites in the mouse. Of the six putative binding sites in the human Slug promoter, two were further investigated based on conservation of the CSL binding sites in the mouse Slug promoter. The first binding site (TATGGGAA) is located at −846 to −853, whereas the second binding site (TGTGGGAA) is located at −1,679 to −1,686 upstream of the TSS. Using chromatin immunoprecipitation (ChIP) followed by PCR with primers flanking the CSL elements, we found that CSL was capable of binding both CSL consensus motifs in the Slug promoter (Fig. 5 E). In contrast, PCR of the same ChIP DNA did not enrich the ZNF3 promoter, which lacks a putative CSL binding site. EMSA of nuclear lysates harvested from FLAG-CSL–expressing endothelial cells confirmed that CSL is capable of binding both consensus elements present in the human Slug promoter (Fig. 5 F). Collectively, these data demonstrate that Slug is a direct target of Notch through a CSL-dependent pathway and that Slug induction is required for Notch-mediated repression of the endothelial phenotype.
Notch and TGF-β act synergistically to induce Snail
Components of the TGF-β pathway have been shown to be required for EMT and for the regulation of Snail family genes during heart development (Romano and Runyan, 2000; Camenisch et al., 2002a; Wang et al., 2005). Additionally, the Notch and TGF-β pathways have been shown to coregulate target gene expression in various cell types (Blokzijl et al., 2003; Zavadil et al., 2004). To investigate the relationship between the Notch and TGF-β pathways and Snail family member expression, endothelial cells cocultured with vector- or Dll4-transduced cells were treated with 2.5 ng/ml TGF-β2 or 20 or 50 ng/ml BMP2. TGF-β2 stimulation induced maximal induction of Snail mRNA and protein expression after 2 h of treatment in vector-transduced cells, followed by rapid down-regulation (Fig. 6, A and B). Although Dll4 stimulation alone did not induce Snail, combined activation of the Notch and TGF-β pathways resulted in a synergistic increase of Snail mRNA levels and maintenance of expression for at least 8 h after stimulation with TGF-β2 (Fig. 6 A). Protein expression of Snail peaked slightly later (4 h) and was sustained at a much higher level in the context of Dll4 and TGF-β2 costimulation compared with TGF-β2 stimulation alone (Fig. 6 B). In contrast, there was minimal induction of Slug by TGF-β2, whereas Notch activation alone dramatically up-regulated Slug (Fig. 6, B and C). Costimulation by Dll4 and TGF-β2 did not increase the level of Slug induction over that seen with Dll4 alone (Fig. 6, B and C).
To investigate the role of Notch activation in TGF-β2–mediated induction of Snail, we used the γ-secretase inhibitor DAPT to block ligand-activated Notch signaling. TGF-β2 treatment dramatically up-regulated the expression of Snail, but the addition of DAPT did not affect the ability of TGF-β2 to induce Snail, which is consistent with Notch-independent induction (Fig. 6 C). In the context of combined Notch and TGF-β2 activation, the synergistic up-regulation of Snail expression was reduced by DAPT to the level seen by TGF-β2 stimulation alone (Fig. 6 C). TGF-β2 had no effect on Slug levels, and the addition of DAPT abrogated Slug induction by Dll4, suggesting complete dependence on Notch activation for Slug up-regulation (Fig. 6 C). Similar results were observed with TGF-β1 treatment (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1). As expected, stimulation of endothelial cells with TGF-β2 induced expression of Smad7, a TGF-β target gene, to similar levels in control and Notch-activated cells (Fig. 6 C). Addition of DAPT appeared to block the ability of TGF-β2 to induce Smad7, but the results were variable and did not reach statistical significance for TGF-β2 or TGF-β1 (Fig. 6 C and Fig. S4), suggesting a minimal role for Notch activation in TGF-β–induced Smad7 induction. Hey1 expression was induced by Notch signaling and TGF-β2 and was dependent on active Notch signaling (Fig. 6 C). Similar to what has been described for BMP4/6, Hey1 was also synergistically induced to very high levels by TGF-β2 and Dll4 (Fig. 6 C; Dahlqvist et al., 2003; Itoh et al., 2004).
Similar to TGF-β2, when endothelial cells were stimulated with BMP2 there was dramatic up-regulation of Snail expression, minimal up-regulation of Slug, and synergistic activation of Hey1 in Notch activated cells (Fig. 6 D). However, unlike TGF-β2, combined activation of the Notch pathway and BMP2 stimulation did not synergistically up-regulate Snail expression (Fig. 6 D). This suggests that a Smad3-dependent process is involved in the synergistic activation of Snail expression by the Notch and TGF-β pathways. These findings clearly confirm that Slug is a direct target of Notch and that Snail is not but that Snail is synergistically induced when Notch activation is superimposed on TGF-β stimulation.
Snail and Slug cooperatively induce cardiac EMT
Given that the cardiac EMT defect seen in Slug−/− mice at E9.5 was reduced by E10.5 (Fig. 3, A and B; and Fig. S2), we sought to determine whether Snail was compensating for the absence of Slug in vivo. qRT-PCR analysis of wild-type and Slug−/− hearts was conducted for Snail and Slug. Slug expression increased from E9.5 to 11.5 in the wild-type heart and its expression was abolished in the Slug−/− hearts. In contrast, Snail expression did not increase from E9.5 to 11.5 in the wild-type hearts. However, in the Slug−/− hearts Snail was up-regulated 3.6-fold by E11.5 (Fig. 7 A). In situ hybridization at E10.5 and 11.5 revealed Snail expression in the AV canal and OFT in both wild-type and Slug−/− hearts, with increased expression in the Slug−/− embryos (Fig. 7 B), suggesting that the region of Snail expression is not expanded but, rather, that the cells normally expressing Snail do so at a higher level in the Slug−/− hearts.
Figure 7.

Increased Snail expression compensates for Slug deficiency. (A) qRT-PCR analysis for Snail and Slug of whole hearts isolated from E9.5, 10.5, and 11.5 wild-type (wt) and Slug−/−embryos (n = 3). *, P < 0.05. Error bars show SEM. (B) In situ hybridization for Snail in E10.5 and 11.5 wild-type (wt) and Slug−/− hearts (n = 3; five embryos in each replicate). Bars, 250 μm. (C) Quantitation of EMT in AV canal explants from E9.5 wild-type (wt) or Slug+/− and Slug−/− embryos treated with 5 ng/ml TGF-β2 or vehicle (UT). (D) Quantitation of EMT in AV canal explants from E10.5 wild-type (wt) or Slug+/− and Slug−/− embryos transduced with shRandom or shSnail constructs.
We next determined whether the TGF-β pathway, potentially through Snail, could compensate for Slug deficiency at E9.5. Treatment of E9.5 AV canal explants with 5 ng/ml TGF-β2 completely rescued the EMT defect seen in E9.5 Slug−/− embryos (Fig. 7 C). Given the increase in Snail expression noted at E10.5 and 11.5 in Slug−/− hearts (Fig. 7, A and B), the ability of Snail to compensate for the absence of Slug after E9.5 was directly assessed using a lentiviral-delivered shRNA to knock down Snail expression in Slug−/− E10.5 AV canal explants. Knockdown of Snail in wild-type or heterozygote Slug AV explants did not result in a decrease in the number or distance of migrating cells at E10.5 (Fig. 7 D). In contrast, knockdown of Snail in Slug−/− AV explants resulted in a significant reduction in the number of migrating/invading cells (Fig. 7 D). The degree of EMT is reduced in E10.5 AV canals compared with E9.5 AV canals, which is consistent with previous data (Dor et al., 2001; Chang et al., 2004). These data support the redundancy of Slug and Snail during the later stages of EMT in the cardiac cushions and suggest that parallel activation by the Notch and TGF-β–BMP pathways is required to maintain the appropriate level of expression of Snail family members in order for cushion development to proceed.
Discussion
The Notch signaling pathway has been found to be a key regulator of cardiac cushion EMT and has been implicated in the pathogenesis of various cardiovascular diseases (Niessen and Karsan, 2007). TGF-β–related pathways have also been shown to be essential for proper heart development through their role in regulating EMT (Azhar et al., 2003). Thus, there are clear requirements for both the Notch and TGF-β–related pathways during mammalian cardiac cushion development. However, there is limited detail of how these pathways function and interact with each other during cardiac development. Our findings suggest cooperation between the Notch and TGF-β–BMP pathways during cardiac EMT, through the coordinate regulation of a group of genes such as the Snail family of transcription factors. In contrast to a previous study, we demonstrate that the transcriptional repressor Slug, but not Snail, is a direct target of the Notch pathway (Timmerman et al., 2004). Conversely, activation of the TGF-β pathway dramatically up-regulates the expression of Snail but not Slug, and Slug is not required for TGF-β–mediated EMT. Importantly we reveal synergistic up-regulation of Snail expression by the Notch and TGF-β pathways, despite the fact that Snail is not a direct target of Notch. This synergistic activation of TGF-β–induced Snail by Notch is consistent with the decrease in Snail expression seen in CSL-deficient embryos (Timmerman et al., 2004).
Interestingly, endothelial-specific gene targeting of the BMP receptor Alk2 results in cardiac cushion defects that are associated with a decrease in the expression of Snail, but not Hey2 or Slug, in the AV canal (Wang et al., 2005). In contrast, Notch-mediated EMT is cell autonomous and TGF-β independent (Noseda et al., 2004). Collectively, these findings support the data presented herein that Slug is a direct target of the Notch pathway and that Snail is a target of TGF-β–related pathways.
We show for the first time that Slug is expressed in a subset of cardiac endothelial cells and the mesenchyme of the AV canal and OFT from the onset of EMT at E9.5 and is essential for initiating cardiac cushion cellularization. We further demonstrate that Slug binds and represses the VE-cadherin promoter, inducing a motile phenotype. Taken with the defect in AV canal EMT at E9.5, the ability of Slug to bind and repress the VE-cadherin promoter and induce migration suggests that the activation phase of EMT in the endocardium is impaired by loss of Slug.
Using an AV canal explant model, we demonstrate that Slug-deficient hearts have a specific defect in cardiac cushion EMT at E9.5 but not at E10.5. In Slug-deficient hearts at E10.5, cardiac EMT is compensated for by a relative increase in Snail expression. Accordingly, inducing Snail expression by treatment with TGF-β2 at E9.5 rescues the EMT defect in Slug-deficient mice. Conversely, abolishing both Slug and Snail expression results in EMT defects at E10.5. Consistent with a requirement for both Snail and Slug during cardiac EMT, both members are expressed during mouse and human heart development with similar localization in a subset of endothelial cells and the mesenchymal cells of the AV canal and OFT. It is of interest that deletion of Slug results in up-regulation of Snail. This finding suggests that Slug may act to repress Snail, either by directly targeting the Snail promoter or through the repression of elements of the TGF-β–related or Notch pathways. Consistent with the latter hypothesis, we have seen that both Hey2 and Smad7 are up-regulated in Slug-deficient hearts at E11.5 (unpublished data). Additionally, it has been demonstrated that Slug does not affect Snail-promoter activity (Peiro et al., 2006), which we have verified (unpublished data). Our findings are concordant with a recent study showing that Snail heterozygosity increases the penetrance of palate defects in Slug-deficient mice, suggesting that Snail also compensates for Slug deficiency during palate development (Murray et al., 2007). In addition, the finding that there is increased Slug expression in the developing palate in Snail-deficient embryos (Murray et al., 2007) suggests reciprocal regulation of gene expression between Slug and Snail.
The interaction between the Notch and TGF-β pathways likely occurs at multiple levels and may be context-dependent. Targeting of CSL results in reduced TGF-β2 and its receptor TBRII in the mouse heart (Timmerman et al., 2004). In contrast, the Notch-ligand Jagged1 has been shown to be induced by TGF-β at the onset of EMT in epithelial cells (Zavadil et al., 2004). Despite our evidence showing cooperation of the TGF-β and Notch pathways in cardiac cushion development, several studies have suggested that constitutively active NotchICD inhibits TGF-β signaling through the sequestration of Smad3 or the coactivator p300 (Masuda et al., 2005; Sun et al., 2005). However, overexpression of NotchICD may have resulted in artifactual sequestering of TGF-β signaling components. Alternatively, the outcome of Notch–TGF-β cross talk may be dependent on the context. Indeed, in mouse embryonic endothelial cells, BMP signaling synergizes with NotchICD through a ternary interaction between Smad5, NotchICD, and p/CAF (Itoh et al., 2004).
Combining the findings in this paper with published data, one can propose a model where endothelial Notch activation induces Slug and release of TGF-β and BMPs from the cushion myocardium activates the cardiac endothelium to up-regulate Snail, which is enhanced in Notch-activated cells. Based on the Slug-deficient hearts and RNAi studies in the AV explants, a minimal total dose of Snail/Slug is required in order for EMT to be initiated at E9.5, with a diminishing requirement at E10.5 as cushion development proceeds.
Materials and methods
Reagents
The mouse monoclonal antibody against the FLAG epitope (M2), mouse anti–h1-calponin, and mouse anti-tubulin were purchased from Sigma-Aldrich. Goat anti–VE-cadherin (C-19), goat anti-CD31 (C-20), and goat anti-Slug (G-18) were obtained from Santa Cruz Biotechnology Inc. Rabbit anti–Snail antibody was obtained from Abcam. Mouse anti–VE-cadherin (TEA1/31) was obtained from Beckman Coulter. Rabbit anti–α–smooth muscle actin antibody was obtained from Thermo Fisher Scientific.
Cell culture and gene transfer
The HMEC-1 human microvascular endothelial cell line, HUVEC, and human aortic endothelial cells were obtained and cultured as previously described (Noseda et al., 2004). Endothelial cells were transduced using the retroviral vectors pLNCX, pLNC-Slug-FLAG, pLNC-FLAG-CSL, MIY, MIY-Slug-FLAG, MIY-Notch4IC-HA, MIY-Notch1IC, and MIY-CSL-VP16 as previously described (Karsan et al., 1996). pcDNA3-Slug-FLAG cDNA was a gift from E.R. Fearon (The University of Michigan Heath Systems, Ann Arbor, MI).
RNA collection and RT-PCR
RNA was isolated and cDNA was made as previously described (Noseda et al., 2004). PCR was performed on a PCR cycler (PTC-200 [Bio-Rad Laboratories] or 7900HT [Applied Biosystems]) with primers listed in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1).
Luciferase reporter assay
8 × 104 HMEC was plated 24 h before transfection in 24-well plates. HMEC were transfected using SuperFect (QIAGEN) reagent, with 0.3125 μg of total plasmid DNA as per the manufacturer's recommendations. Each well was transfected with 0.3 μg of the VE-cadherin promoter plasmid or mutant VE-cadherin promoter constructs, 5 ng pcDNA3 or pcDNA3-Slug-FLAG, and 7.5 ng pRL-CMV (Promega). Luminescence was measured on a Lumat LB 9507 (EG&G Berthold) 24 h after transfection using dual luciferase reporter assays according to the manufacturer's recommendations (Promega).
EMSA
For Slug EMSA assays, in vitro–translated (TNT; Promega) Slug-FLAG or control luciferase protein was preincubated with FLAG-M2 antibody overnight at 4°C in 12 mM Hepes, pH 7.9, 4 mM Tris, pH 7.9, 133 mM KCl, 10% Glycerol, and 2 μg PolydI-dC binding buffer. 50-fold excess nonradioactive duplex oligos were preincubated for 15 min on ice, and then 150,000-cpm 32P-labeled double-stranded oligo nucleotides were added and incubated for 30 min at room temperature. Binding reactions were run on 5% Tris-borate EDTA gels and exposed to a phosphorimager plate for 12–16 h. For CSL EMSA assays, nuclear lysates were collected from FLAG-CSL–overexpressing HMEC cells. Binding reaction and detection were the same as used for Slug-FLAG EMSA assays.
ChIP
HMEC were transduced with pLNCX or pLNC-FLAG-CSL, and ChIP assay was performed as previously described (Noseda et al., 2006). ChIP DNA was amplified for the ZNF3 promoter or the two CSL binding sites in the human Slug promoter using primers listed in Table S1.
Mice and AV explant assay analysis
Slug-lacZ mice were provided by T. Gridley (Jackson Laboratories, Bar Harbor, ME). Slug-lacZ+/− mice were crossed to C57BL/6J mice for embryo collection. Embryos were assayed for β-galactosidase activity in situ using published protocols (Nagy et al., 2003). AV canal explants were performed as previously described (Camenisch et al., 2002a). Explants were cultured for 48 h and analyzed for the number and distance of migrating cells.
RNA interference
shRNAs targeting human CSL, Snail, and Slug were cloned into the HpaI–XhoI sites of the pLentilox3.7 vector (gift from L. Van Parijs, Massachusetts Institute of Technology, Cambridge, MA; Table S1). Constructs were sequence verified and validated for efficient knockdown.
Collection of human tissues
Human embryonic hearts were collected, after institutionally approved protocols and informed consent, at the Children's and Women's Health Sciences Centre (Vancouver, Canada). Tissue was fixed in 4% PFA overnight, embedded in OCT, and cryosectioned.
In situ hybridization
In situ hybridization was performed as previously described (Wilkinson, 1992). Mouse Snail probe (−55 to +454) was cloned into pBluescript. Human Snail and human Slug probes were comprised of the entire ORF cloned into pCDNA3.
BrdU analysis
Slug-lacZ+/− male and female mice were crossed and pregnant females were injected with 1,500 mg/ml BrdU (Sigma-Aldrich) 2 h before killing. Embryos were collected, paraffin embedded, and sectioned (6 μm) onto Histobond slides (Marienfeld Laboratory Glassware). Slides were boiled for 30 min in 0.1M citrate buffer, rinsed in water, and then denatured in 2N HCl for 45 min at 37°C. Slides were then rinsed in PBS, and BrdU staining was performed using mouse anti-BrdU (BU33; Sigma-Aldrich) and goat anti–mouse Alexa 488 (Invitrogen).
Online supplemental material
Fig. S1 shows that the ankyrin repeats of NotchICD are required for induction of Slug expression. Fig. S2 shows the Slug-lacZ expression in the 18 to 29 somite stage heart and that the AV canal EMT defect observed in Slug-deficient embryos at E9.5 is no longer present at E10.5. Fig. S3 shows that Slug represses endothelial cell phenotype in HMEC and HUVEC, that the knockdown of Slug expression in Notch-activated cells restores VE-cadherin and CD31 expression, and that the ectopic expression of Hey1 or Hey2 does not induce Slug expression or repress VE-cadherin expression. Fig. S4 shows that the induction of Snail by TGF-β1 is synergistically enhanced in Notch-activated endothelial cells. Table S1 is a list of primers used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710067/DC1.
Supplementary Material
Acknowledgments
We would like to thank Megan Abbott for help with the animal studies and Jennifer Baker and Alastair Kyle for help with the AV canal data analysis and immunohistochemistry studies.
This work was supported by grants from the Canadian Institutes of Health Research (MOP 64354), the Heart and Stroke Foundation of British Columbia and the Yukon, Genome Canada, and Genome British Columbia. K. Niessen and L. Chang are supported by Doctoral Research Awards and Y. Fu by a Postdoctoral Award from the Michael Smith Foundation for Health Research. A. Karsan and P.A. Hoodless are Senior Scholars of the Michael Smith Foundation for Health Research, and P.A. Hoodless is a Canadian Institutes of Health Research New Investigator.
Abbreviations used in this paper: AV, atrioventricular; ChIP, chromatin immunoprecipitation; E, embryonic day; EMSA, electrophoretic mobility shift assay; EMT, endothelial-to-mesenchymal transformation; HMEC, human mammary epithelial cell; HUVEC, human umbilical vein epithelial cell; OFT, outflow tract; qRT-PCR, quantitative RT-PCR; shRNA, short hairpin RNA; TSS, transcriptional start site; VE-cadherin, vascular endothelial cadherin.
References
- Armstrong, E.J., and J. Bischoff. 2004. Heart valve development: endothelial cell signaling and differentiation. Circ. Res. 95:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar, M., J. Schultz Jel, I. Grupp, G.W. Dorn II, P. Meneton, D.G. Molin, A.C. Gittenberger-de Groot, and T. Doetschman. 2003. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 14:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartram, U., D.G. Molin, L.J. Wisse, A. Mohamad, L.P. Sanford, T. Doetschman, C.P. Speer, R.E. Poelmann, and A.C. Gittenberger-de Groot. 2001. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 103:2745–2752. [DOI] [PubMed] [Google Scholar]
- Blokzijl, A., C. Dahlqvist, E. Reissmann, A. Falk, A. Moliner, U. Lendahl, and C.F. Ibanez. 2003. Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 163:723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C.B., A.S. Boyer, R.B. Runyan, and J.V. Barnett. 1999. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 283:2080–2082. [DOI] [PubMed] [Google Scholar]
- Camenisch, T.D., D.G. Molin, A. Person, R.B. Runyan, A.C. Gittenberger-de Groot, J.A. McDonald, and S.E. Klewer. 2002. a. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 248:170–181. [DOI] [PubMed] [Google Scholar]
- Camenisch, T.D., J.A. Schroeder, J. Bradley, S.E. Klewer, and J.A. McDonald. 2002. b. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat. Med. 8:850–855. [DOI] [PubMed] [Google Scholar]
- Carver, E.A., R. Jiang, Y. Lan, K.F. Oram, and T. Gridley. 2001. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 21:8184–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.P., J.R. Neilson, J.H. Bayle, J.E. Gestwicki, A. Kuo, K. Stankunas, I.A. Graef, and G.R. Crabtree. 2004. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 118:649–663. [DOI] [PubMed] [Google Scholar]
- Crosby, C.V., P.A. Fleming, W.S. Argraves, M. Corada, L. Zanetta, E. Dejana, and C.J. Drake. 2005. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood. 105:2771–2776. [DOI] [PubMed] [Google Scholar]
- Dahlqvist, C., A. Blokzijl, G. Chapman, A. Falk, K. Dannaeus, C.F. Ibanez, and U. Lendahl. 2003. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 130:6089–6099. [DOI] [PubMed] [Google Scholar]
- Dickson, M.C., H.G. Slager, E. Duffie, C.L. Mummery, and R.J. Akhurst. 1993. RNA and protein localisations of TGF beta 2 in the early mouse embryo suggest an involvement in cardiac development. Development. 117:625–639. [DOI] [PubMed] [Google Scholar]
- Donovan, J., A. Kordylewska, Y.N. Jan, and M.F. Utset. 2002. Tetralogy of Fallot and other congenital heart defects in Hey2 mutant mice. Curr. Biol. 12:1605–1610. [DOI] [PubMed] [Google Scholar]
- Dor, Y., T.D. Camenisch, A. Itin, G.I. Fishman, J.A. McDonald, P. Carmeliet, and E. Keshet. 2001. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 128:1531–1538. [DOI] [PubMed] [Google Scholar]
- Eisenberg, L.M., and R.R. Markwald. 1995. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 77:1–6. [DOI] [PubMed] [Google Scholar]
- Eldadah, Z.A., A. Hamosh, N.J. Biery, R.A. Montgomery, M. Duke, R. Elkins, and H.C. Dietz. 2001. Familial tetralogy of fallot caused by mutation in the jagged1 gene. Hum. Mol. Genet. 10:163–169. [DOI] [PubMed] [Google Scholar]
- Fischer, A., N. Schumacher, M. Maier, M. Sendtner, and M. Gessler. 2004. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 18:901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, A., C. Steidl, T.U. Wagner, E. Lang, P.M. Jakob, P. Friedl, K.P. Knobeloch, and M. Gessler. 2007. Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ. Res. 100:856–863. [DOI] [PubMed] [Google Scholar]
- Garg, V., A.N. Muth, J.F. Ransom, M.K. Schluterman, R. Barnes, I.N. King, P.D. Grossfeld, and D. Srivastava. 2005. Mutations in NOTCH1 cause aortic valve disease. Nature. 437:270–274. [DOI] [PubMed] [Google Scholar]
- Hay, E.D. 2005. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 233:706–720. [DOI] [PubMed] [Google Scholar]
- Inoue, A., M.G. Seidel, W. Wu, S. Kamizono, A.A. Ferrando, R.T. Bronson, H. Iwasaki, K. Akashi, A. Morimoto, J.K. Hitzler, et al. 2002. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell. 2:279–288. [DOI] [PubMed] [Google Scholar]
- Iso, T., Y. Hamamori, and L. Kedes. 2003. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 23:543–553. [DOI] [PubMed] [Google Scholar]
- Itoh, F., S. Itoh, M.J. Goumans, G. Valdimarsdottir, T. Iso, G.P. Dotto, Y. Hamamori, L. Kedes, M. Kato, and P. ten Dijke Pt. 2004. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 23:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, R., Y. Lan, C.R. Norton, J.P. Sundberg, and T. Gridley. 1998. The Slug gene is not essential for mesoderm or neural crest development in mice. Dev. Biol. 198:277–285. [PubMed] [Google Scholar]
- Karsan, A., E. Yee, and J.M. Harlan. 1996. Endothelial cell death induced by tumor necrosis factor-alpha is inhibited by the Bcl-2 family member, A1. J. Biol. Chem. 271:27201–27204. [DOI] [PubMed] [Google Scholar]
- Li, L., I.D. Krantz, Y. Deng, A. Genin, A.B. Banta, C.C. Collins, M. Qi, B.J. Trask, W.L. Kuo, J. Cochran, et al. 1997. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 16:243–251. [DOI] [PubMed] [Google Scholar]
- Loomes, K.M., L.A. Underkoffler, J. Morabito, S. Gottlieb, D.A. Piccoli, N.B. Spinner, H.S. Baldwin, and R.J. Oakey. 1999. The expression of Jagged1 in the developing mammalian heart correlates with cardiovascular disease in Alagille syndrome. Hum. Mol. Genet. 8:2443–2449. [DOI] [PubMed] [Google Scholar]
- MacKenzie, F., P. Duriez, F. Wong, M. Noseda, and A. Karsan. 2004. Notch4 inhibits endothelial apoptosis via RBP-Jkappa-dependent and -independent pathways. J. Biol. Chem. 279:11657–11663. [DOI] [PubMed] [Google Scholar]
- Masuda, S., K. Kumano, K. Shimizu, Y. Imai, M. Kurokawa, S. Ogawa, M. Miyagishi, K. Taira, H. Hirai, and S. Chiba. 2005. Notch1 oncoprotein antagonizes TGF-beta/Smad-mediated cell growth suppression via sequestration of coactivator p300. Cancer Sci. 96:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin, D.G., U. Bartram, K. Van der Heiden, L. Van Iperen, C.P. Speer, B.P. Hierck, R.E. Poelmann, and A.C. Gittenberger-de-Groot. 2003. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev. Dyn. 227:431–444. [DOI] [PubMed] [Google Scholar]
- Molin, D.G., M.C. DeRuiter, L.J. Wisse, M. Azhar, T. Doetschman, R.E. Poelmann, and A.C. Gittenberger-de Groot. 2002. Altered apoptosis pattern during pharyngeal arch artery remodelling is associated with aortic arch malformations in Tgfbeta2 knock-out mice. Cardiovasc. Res. 56:312–322. [DOI] [PubMed] [Google Scholar]
- Murray, S.A., and T. Gridley. 2006. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. USA. 103:10300–10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, S.A., K.F. Oram, and T. Gridley. 2007. Multiple functions of Snail family genes during palate development in mice. Development. 134:1789–1797. [DOI] [PubMed] [Google Scholar]
- Nagy, A.G., M. Gertsenstein, K. Vintersten, and R. Behringer. 2003. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press. 779 pp.
- Niessen, K., and A. Karsan. 2007. Notch signaling in the developing cardiovascular system. Am. J. Physiol. Cell Physiol. 293:C1–C11. [DOI] [PubMed] [Google Scholar]
- Nieto, M.A. 2002. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3:155–166. [DOI] [PubMed] [Google Scholar]
- Noseda, M., G. McLean, K. Niessen, L. Chang, I. Pollet, R. Montpetit, R. Shahidi, K. Dorovini-Zis, L. Li, B. Beckstead, et al. 2004. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 94:910–917. [DOI] [PubMed] [Google Scholar]
- Noseda, M., Y. Fu, K. Niessen, F. Wong, L. Chang, G. McLean, and A. Karsan. 2006. Smooth Muscle {alpha }-Actin Is a Direct Target of Notch/CSL. Circ. Res. 98:1468–1470. [DOI] [PubMed] [Google Scholar]
- Oda, T., A.G. Elkahloun, B.L. Pike, K. Okajima, I.D. Krantz, A. Genin, D.A. Piccoli, P.S. Meltzer, N.B. Spinner, F.S. Collins, and S.C. Chandrasekharappa. 1997. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 16:235–242. [DOI] [PubMed] [Google Scholar]
- Oka, C., T. Nakano, A. Wakeham, J.L. de la Pompa, C. Mori, T. Sakai, S. Okazaki, M. Kawaichi, K. Shiota, T.W. Mak, and T. Honjo. 1995. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 121:3291–3301. [DOI] [PubMed] [Google Scholar]
- Oram, K.F., E.A. Carver, and T. Gridley. 2003. Slug expression during organogenesis in mice. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 271:189–191. [DOI] [PubMed] [Google Scholar]
- Peiro, S., M. Escriva, I. Puig, M.J. Barbera, N. Dave, N. Herranz, M.J. Larriba, M. Takkunen, C. Franci, A. Munoz, et al. 2006. Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res. 34:2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandini, M.H., I. Dreher, S. Bouillot, S. Benkerri, T. Moll, and P. Huber. 2005. The human VE-cadherin promoter is subjected to organ-specific regulation and is activated in tumour angiogenesis. Oncogene. 24:2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramain, P., K. Khechumian, L. Seugnet, N. Arbogast, C. Ackermann, and P. Heitzler. 2001. Novel Notch alleles reveal a Deltex-dependent pathway repressing neural fate. Curr. Biol. 11:1729–1738. [DOI] [PubMed] [Google Scholar]
- Romano, L.A., and R.B. Runyan. 2000. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Dev. Biol. 223:91–102. [DOI] [PubMed] [Google Scholar]
- Sanford, L.P., I. Ormsby, A.C. Gittenberger-de Groot, H. Sariola, R. Friedman, G.P. Boivin, E.L. Cardell, and T. Doetschman. 1997. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 124:2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y., W. Lowther, K. Kato, C. Bianco, N. Kenney, L. Strizzi, D. Raafat, M. Hirota, N.I. Khan, S. Bargo, et al. 2005. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene. 24:5365–5374. [DOI] [PubMed] [Google Scholar]
- Timmerman, L.A., J. Grego-Bessa, A. Raya, E. Bertran, J.M. Perez-Pomares, J. Diez, S. Aranda, S. Palomo, F. McCormick, J.C. Izpisua-Belmonte, and J.L. de la Pompa. 2004. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 18:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Akker, N.M., H. Lie-Venema, S. Maas, I. Eralp, M.C. DeRuiter, R.E. Poelmann, and A.C. Gittenberger-De Groot. 2005. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev. Dyn. 233:1579–1588. [DOI] [PubMed] [Google Scholar]
- Wang, J., S. Sridurongrit, M. Dudas, P. Thomas, A. Nagy, M.D. Schneider, J.A. Epstein, and V. Kaartinen. 2005. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev. Biol. 286:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, D.G. 1992. In situ hybridization: a practical approach. Oxford University Press, New York. 224 pp.
- Zavadil, J., L. Cermak, N. Soto-Nieves, and E.P. Bottinger. 2004. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23:1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., and A. Bradley. 1996. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 122:2977–2986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}