

We report that equal yields of [6-1H]-uridine 5'-monophosphate (50%) and [6-2H]-uridine 5'-monophosphate (50%) are obtained from the decarboxylation of orotidine 5'-monophosphate (OMP) catalyzed by orotidine 5'-monophosphate decarboxylase in a solvent of 50/50 (v/v) H2O/D2O. This observation of an unusually small product isotope effect of unity eliminates a proposed mechanism in which proton transfer from Lys-931 to C-6 provides electrophilic push to the loss of CO2 from OMP in a concerted reaction.2,3 It provides evidence that proton transfer from the ammonium cation side-chain of Lys-93 to a vinyl carbanion intermediate is faster than the bond rotation that exchanges the positions of the acidic N-L+ hydrons of this side-chain.
Orotidine 5'-monophosphate decarboxylase (OMPDC) is a remarkable enzyme because it employs no metal ions or other cofactors but yet effects an enormous 1017-fold acceleration of the chemically very difficult decarboxylation of OMP to give uridine 5'-monophosphate (UMP).4,5 It has been shown that a large fraction of the enzymatic rate acceleration results directly from utilization of the intrinsic binding energy of th e remote nonreacting 5'-phosphodianion group of OMP in transition state stabilization.6 The decarboxylation reaction is often proposed to proceed in two steps through a vinyl carbanion intermediate (Scheme 1). However, it has also been suggested that this unstable intermediate might be avoided in a concerted reaction in which decarboxylation and proton transfer to C-6 occur in a single step.2,3
Scheme 1.
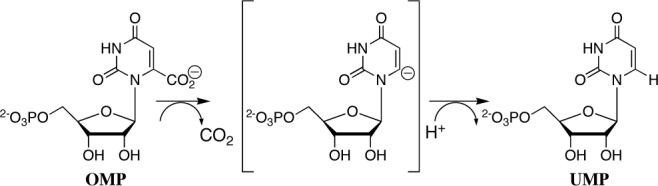
Experimental and computational studies on OMPDC have focused largely on the partly rate-determining and highly unfavorable loss of CO2 from OMP.7-9 There are few data pertaining to the proton transfer to C-6 of the pyrimidine ring. Experimental characterization of this proton transfer step is essential for insight into the existence and lifetime of the putative enzyme-bound vinyl carbanion intermediate.
OMPDC catalyzes incorporation of a hydron from solvent into the UMP product and it has been reported that the decarboxylation of saturating OMP is 30% faster in H2O than in D2O.7 While the origin of this solvent isotope effect on kcat is unclear, it may represent a secondary solvent kinetic isotope effect (SKIE). By contrast, a product isotope effect (PIE) determined in experiments in which H and D in a mixed solvent of H2O/D2O compete for reaction with enzyme-bound OMP to form UMP labeled at C-6 (Scheme 2) would provide insight into the changes in bonding at the transferred hydron that occur on proceeding to the transition state for the product-determining step.10 PIEs are more precise and easier to interpret than SKIEs determined as the ratio of rate constants for reactions in H2O and D2O because: (1) There are no complications from any secondary SKIE when the H- and D-labeled products are formed in the same mixed H2O/D2O solvent. (2) There are no errors due to differences in the conditions for separate reactions in H2O and D2O, such as enzyme concentration, temperature and pL.
Scheme 2.
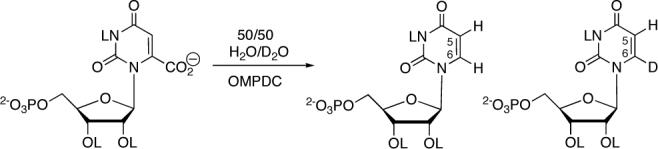
The product distribution for the decarboxylation of OMP catalyzed by OMPDC in 50/50 (v/v) H2O/D2O was determined by 1H NMR spectroscopy at 500 MHz. Figure 1 shows the partial 1H NMR spectrum of UMP obtained from the decarboxylation of OMP (2 mM) catalyzed by OMPDC from S. cerevisiae (C155S mutant, 24 nM, 1 hr, >90% reaction) in 50/50 (v/v) H2O/D2O at pL 7.3 and 25 °C (I = 0.10, NaCl).11,12 The value of PIE = 1.0 was calculated using eq 1, where AH is the integrated area of the doublet due to the C-6 proton of [6-1H]-UMP (7.990 ppm), and AD is the integrated area of the singlet due to the C-5 proton of [6-2H]-UMP (5.865 ppm).13 By comparison, PIEs of 7.3 − 8.1 for proton transfer to ring-substituted aryl vinyl ethers from lyonium ion in 50/50 (v/v) H2O/D2O have been reported recently.10
| (1) |
Figure 1.
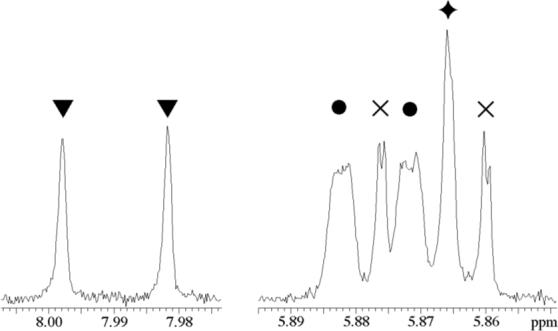
Partial 1H NMR spectrum (500 MHz) of UMP from decarboxylation of OMP (2 mM) catalyzed by OMPDC from S. cerevisiae (24 nM) in 50/50 (v/v) H2O/D2O at pL 7.3 and 25 °C. Key: (▼) Doublet due to the C-6 proton of [6-1H]-UMP; (•) Doublets (not resolved) due to the anomeric protons of [6-1H]-UMP and [6-2H]-UMP; (×) Doublet due to the C-5 proton of [6-1H]-UMP;  Singlet due to the C-5 proton of [6-2H]-UMP.
Singlet due to the C-5 proton of [6-2H]-UMP.
We used similar procedures to determine values of PIE = 1.0 for decarboxylation of OMP (2 mM) catalyzed by OMPDC from both E. coli (40 nM) and M. thermoautotrophicum (40 nM) in 50/50 (v/v) H2O/D2O at pL 7.3 and 25 °C (I = 0.10, NaCl). The essentially identical PIEs determined for OMPDC from different sources is significant, because these enzymes exhibit somewhat different architectures at their active sites.2,9a,14,15
The value of PIE = 1.0 for the OMPDC-catalyzed decarboxylation of OMP in 50/50 (v/v) H2O/D2O shows that the deuterium enrichment of the hydron used to protonate OMP or an intermediate carbanion at the reaction transition state (50%) is the same as that of the 50/50 (v/v) H2O/D2O solvent. The product-determining step is thought to be proton transfer from the NL3+ group of the side-chain of Lys-93 to OMP or to a reaction intermediate (Scheme 3).15 Values of øNL3+ ≈ 1.0 have been reported for the H/D fractionation between L2O and R-NL3+, so that the deuterium enrichment of the NL3+ group of Lys-93 should be similar to that of the solvent L2O.16 Therefore the PIE of 1.0 is essentially equal to the primary kinetic isotope effect for reaction of the H- and D-labeled NL3+ group of Lys-93 to form [6-1H]-UMP and [6-2H]-UMP.
Scheme 3.
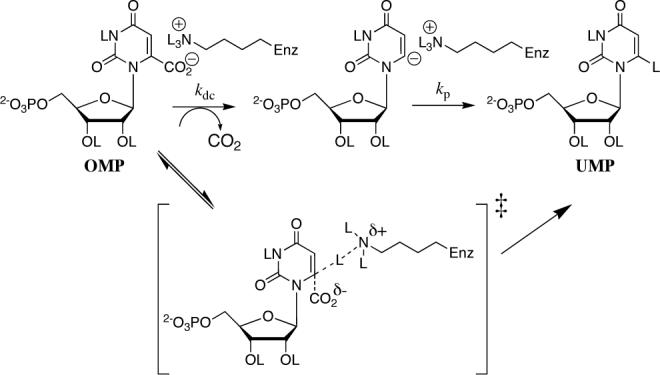
A significant primary product isotope effect is expected for a reaction in which there is movement of the proton in the transition state for the product-determining step,17a and there is no precedent for PIEs as small as 1.0 when carbanion protonation is the product-determining step.17a,18 The observed PIE of 1.0 requires that all of the zero point energy present in the N-L+ bonds of Lys-93 be maintained at the transition state for the step that determines whether the UMP product is labeled at C-6 with H or D. This PIE is not consistent with a mechanism in which proton transfer from Lys-93 to C-6 of OMP provides electrophilic push to the loss of CO2 in a concerted reaction that avoids formation of an unstable vinyl carbanion intermediate (bottom pathway, Scheme 3).2,3,19
We suggest that the essentially statistical yields of [6-1H]-UMP and [6-2H]-UMP from the OMPDC-catalyzed decarboxylation of OMP are established at a step that occurs prior to hydron transfer to a vinyl carbanion intermediate. This could be the decarboxylation step, if an N-L+ bond of Lys-93 is already correctly positioned to deliver a hydron to a vinyl carbanion (kdc, Scheme 3). Alternatively it may be a step that orients an N-L+ bond of Lys-93 into a “reactive position” where hydron transfer to a vinyl carbanion intermediate can occur. In both cases the PIE of 1.0 requires that the chemical step of hydron transfer to the carbanion be faster than any molecular motion that allows its discrimination between reaction with H and D at the NL3+ group of Lys-93.17 We therefore propose that hydron transfer from the side-chain of Lys-93 to a vinyl carbanion intermediate (kp) is faster than any movement that exchanges the positions of the N-L+ hydrons and which would allow the carbanion to select for reaction with H or D.17 In water, the rate constant for such a step is ca. 1011 s−1.20
The X-ray crystal structure of yeast OMPDC complexed with 6-hydroxyuridine 5'-monophosphate shows that the CH2-NH3+ group of Lys-93 is anchored by two hydrogen bonds to the carboxylate groups of Asp-91 and Asp-96 that are proposed to direct the third ammonium hydron of Lys-93 towards the putative vinyl carbanion intermediate.15 These hydrogen bonds should also restrict rotation about the carbon-nitrogen bond of the terminal CH2-NL3+ group of Lys-93 (krot << 1011 s−1). This would favor the observed unselective proton transfer from the remaining free (non-hydrogen-bonded) hydron to a vinyl carbanion intermediate.
Acknowledgement
We acknowledge the National Institutes of Health (GM39754 to JPR and GM65155 to JAG) for generous support of this work.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Residues are numbered according to the sequence for the enzyme from yeast.
- 2.Begley TP, Appleby TC, Ealick SE. Curr. Opin. Struct. Biol. 2000;10:711–718. doi: 10.1016/s0959-440x(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 3.Begley TP, Ealick SE. Curr. Opin. Chem. Biol. 2004;8:508–515. doi: 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Miller BG, Wolfenden R. Annu. Rev. Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 5.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 6.Amyes TL, Richard JP, Tait JJ. J. Am. Chem. Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 7.Ehrlich JI, Hwang C-C, Cook PF, Blanchard JS. J. Am. Chem. Soc. 1999;121:6966–6967. [Google Scholar]
- 8.Rishavy MA, Cleland WW. Biochemistry. 2000;39:4569–4574. doi: 10.1021/bi000376p. [DOI] [PubMed] [Google Scholar]
- 9.a Wu N, Mo Y, Gao J, Pai EF. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gao J, Byun KL, Kluger R. Topics in Current Chemistry. 2004;238:113–136. [Google Scholar]; c Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- 10.Tsang W-Y, Richard JP. J. Am. Chem. Soc. 2007;129:ASAP. doi: 10.1021/ja073679g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.1H NMR spectra were recorded on a Varian Unity Inova-500 spectrometer using a sweep width of 6000 Hz, a 90° pulse angle, an acquisition time of 6 s, a relaxation delay between pulses of 80 s (> 7T1) and with suppression of the water peak. Baselines were subjected to first-order drift correction before integration of the signals.
- 12.Pentz L, Thornton ER. J. Am. Chem. Soc. 1967;89:6931–6938. Reaction mixtures were buffered with 50 mM 3-(N-morpholino)propanesulfonic acid (50% free base). Values of pL were obtained by adding 0.18 to the reading of the pH meter. [Google Scholar]
- 13.Control experiments showed that AH/AD (eq 1) remains constant over a period of ca. 20 hours. The variation in this ratio determined from integration of different NMR spectra is less than 3%.
- 14.Harris P, Poulsen J-CN, Jensen KF, Larsen S. J. Mol. Biol. 2002;318:1019–1029. doi: 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 15.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schowen KB, Schowen RL. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 17.a Thibblin A, Jencks WP. J. Am. Chem. Soc. 1979;101:4963–4973. [Google Scholar]; b Jencks WP. Acc. Chem. Res. 1980;13:161–169. [Google Scholar]
- 18.Fishbein JC, Jencks WP. J. Am. Chem. Soc. 1988;110:5075–5086. [Google Scholar]
- 19.The primary 13C KIE on OMPDC-catalyzed decarboxylation of OMP labeled at the carboxylate carbon decreases from 1.043 for reaction in H2O to 1.034 for the 25% slower reaction in D2O [Ref. 7], which shows that decarboxylation is less rate-determining in D2O than in H2O. This result is not easily rationalized by a mechanism in which proton transfer from Lys-93 to C-6 of OMP is concerted with the loss of CO2 because the change from H2O to D2O should raise the barrier to a reaction in which proton transfer is concerted with loss of CO2, as a result of a normal primary KIE. This would cause the loss of CO2 to become more rate-determining in a multistep enzymatic reaction in D2O and would result in an increase, rather than the observed decrease, in the 13C isotope effect.
- 20.Richard JP, Tsuji Y. J. Am. Chem. Soc. 2000;122:3963–3964. [Google Scholar]