

Abstract
The transition from rolling to firm adhesion is a key step in the adhesion cascade that permits a neutrophil to exit the bloodstream and make its way to a site of inflammation. In this work, we construct an integrated model of neutrophil activation and arrest that combines a biomechanical model of neutrophil adhesion and adhesive dynamics, with fully stochastic signal transduction modeling, in the form of kinetic Monte Carlo simulation within the microvilli. We employ molecular binding parameters gleaned from the literature and from simulation of cell-free rolling mediated by selectin molecules. We create a simplified model of lymphocyte function-associated antigen-1 activation that links P-selectin glycoprotein ligand-1 ligation to integrin activation. The model utilizes an energy profile of various integrin activation states drawn from literature data and permits manipulation of signal diffusivity within the microvillus. Our integrated model recreates neutrophil arrest within physiological timescales, and we demonstrate that increasing signal diffusivity within a microvillus accelerates arrest. If the energy barrier between free unactivated and free activated lymphocyte function-associated antigen-1 increases, the period of rolling before arrest increases. We further demonstrate that, within our model, modification of endothelial ligand surface densities can control arrest. In addition, the relative concentrations of signaling molecules control the fractional activation of the overall signaling pathway and the rolling time to arrest. This work presents the first, to our knowledge, fully stochastic model of neutrophil activation, which, though simplified, can recapitulate significant physiological details of neutrophil arrest yet retains the capacity to incorporate additional information regarding mechanisms of neutrophil signal transduction as they are elucidated.
INTRODUCTION
Recruitment of neutrophils to sites of inflammation begins with the upregulation and presentation of E- and P-selectin on the luminal surfaces of vascular endothelial cells (1). During inflammation, leukocytes start to roll along the endothelium as a result of transient receptor-ligand bonding between the selectins and their glycosylated ligands on the surfaces of leukocytes. Subsequent to selectin-mediated rolling are integrin-mediated firm adhesion, leukocyte extravasation, and chemotaxis to the site of tissue injury.
This work focuses on the transition from rolling to firm adhesion in the neutrophil. In neutrophils, cell arrest is caused by β2 integrin activation, during which the integrin's affinity for intercellular adhesion molecule-1 (ICAM-1), its endothelial ligand, increases as the result of a conformational change. The resting integrin resembles a folded switchblade; upon activation, the α- and β-cytoplasmic domains separate, and the protein swings open into an extended conformation (2,3). The conformational presentation of I-domain, the ICAM-1-binding region in the integrin headpiece, regulates the transition from rolling to firm adhesion (4). I-domain sequestered in the closed or wild-type conformation mediates rolling adhesion; the locked open conformation supports firm adhesion. Recent work suggests that rolling and firm adhesion mediated by lymphocyte function-associated antigen-1 (LFA-1) result from two separate extended conformations that differ in the I-domain affinity for ICAM-1 (5).
Two major hypotheses offer explanations as to how the rolling neutrophil makes the decision to stop. Both hold that the cell samples the endothelial surface through receptor-ligand binding and processes the signals generated by these receptors (6,7). The first hypothesis suggests that ligation of selectins activates a signaling pathway that causes the global activation of β2-integrins. Significant experimental evidence points to the p38 mitogen-activated protein kinase (MAPK) cascade as the signaling pathway involved in this global activation (8–13). Mathematical modeling of the basic MAPK cascade demonstrates the switch-like cooperative kinetics of “ultrasensitivity”, a property well suited for decision making (14). The second hypothesis posits that interactions between endothelium-presented chemokines and specific leukocyte GPCRs release integrins from cytoskeletal restraints, thus promoting mobility and clustering and thereby locally enhancing integrin avidity. This hypothesis also has substantial support (15–17). Our goal is to develop generally applicable methods in which signaling networks can be embedded within mechanically based models of adhesion, regardless of the precise details. In all these cases, activation ultimately flows through the β2-integrins, leading to firm arrest.
Small numbers of molecules at the tips of a spatially restricted population of microvilli participate in the bonding events necessary for rolling and firm adhesion. Thus, the behavior of individual molecules significantly influences overall adhesive behavior. The adhesive dynamics (AD) simulation, a mechanics-based model of leukocyte adhesion developed in our lab, accordingly accounts for individual binding and unbinding events, treating them as stochastic occurrences with bond-length-dependent probabilities (18–20). To capture more accurately the stochasticity inherent in biological processes, we must extend such detailed probabilistic treatment to the activation of individual integrin molecules: the conformational change responsible for activation is thought to be the direct result of a binding event (see below). We employ the kinetic Monte Carlo (KMC) method, in a manner similar to that used to model the immunological synapse (21), to model integrin activation on the microvillus (MV) level. The KMC method treats all molecules in the signaling network, in addition to the receptors at MV tips, as stochastic elements with variable internal states. By defining properties and rules governing the behavior of these elements, we can investigate how stochastic properties of the signaling network affect neutrophil arrest.
Two different β2 integrins on the neutrophil, LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18), mediate the firm adhesion required for extravasation. Although both integrins undergo conformational activation, the surface expression of Mac-1 but not of LFA-1 may rapidly increase in response to chemokine stimulation (22,23). However, the role of LFA-1 in firm adhesion under shear stress is short lived; maintenance of ICAM-1-mediated firm adhesion over several minutes requires Mac-1 (23,24). The duration of our simulations does not extend into times in which Mac-1 is important; so we model only one integrin, LFA-1. Our simulation can be extended easily in the future to account for multiple integrins with different behaviors.
Current opinion holds that three main β2 integrin conformers exist in a dynamic equilibrium whose balance is influenced by the presence of ligand (2). In addition to the inactive and active forms, there is an “intermediate” integrin conformer with a binding affinity between those of the bent (“resting”) and extended (“active”) conformers (2). Experimental evidence indicates that talin, a cytoskeletal protein known to activate αIIbβ3 integrins downstream of calcium-enhanced calpain cleavage (25–27), disrupts the association between the cytoplasmic tails of the αL and β2 subunits and activates the integrin (3). Thus, integrin conversion between states is likely the result of a binding event. Given the small numbers of integrins present at a MV tip, this activation lends itself nicely to modeling as a stochastic process. For simplicity, we consider the conversion from low to high affinity as a one-step process.
Previous AD simulations described the behavior of a neutrophil with two receptor-ligand pairs (i.e., selectin-P-selectin glycoprotein ligand-1 (PSGL-1) and β2-integrin-ICAM-1) in shear flow (28). Adhesion state diagrams showed that the receptor-ligand pairs work synergistically to promote adhesion (28). In other words, a cell with only one receptor type will roll for some range of surface densities beyond which the cell achieves firm adhesion. If the second type of receptor is added to the original cell, the range of first receptor surface densities that mediate rolling is reduced, and the cell achieves firm adhesion at a lower surface density of the initial receptor type. The specific synergism depends upon flow shear rate and integrin properties. Thus for a given cell in one state, we can predict the change in system properties required for the transition to the other state; what remains to be determined is how that transition is affected.
In this work, we embed within AD a KMC model of integrin activation through a plausible signal transduction network. PSGL-1 ligation generates a signal that is transmitted through several intermediates before driving the activation of LFA-1. This simple signal transduction pathway represents the most basic network that can capture the cross talk between selectins and integrins. As the precise details of the signaling pathway emerge, they can be incorporated within the model. Thus we present here a fully stochastic model that integrates signaling and adhesion to simulate integrin-mediated neutrophil arrest.
MODEL DEVELOPMENT
Rolling mechanics: adhesive dynamics
AD (18,20) was modified to account for three receptor-ligand pairs: PSGL-1/E-selectin, resting LFA-1/ICAM-1, and active LFA-1/ICAM-1. A detailed description of AD in which two receptors are involved has already been described by Bhatia and co-workers (28). A model schematic appears in Fig. 1, and a brief description of the algorithm follows.
FIGURE 1.

Schematic diagram of AD. Rigid, cylindrical microvilli are randomly placed on the surface of a sphere. Adhesion molecules are assigned randomly to these microvilli according to a Poisson distribution, and the wall is assumed to be a uniformly reactive planar surface. Length-dependent binding probabilities govern the formation and breakage of bonds. Resting integrins are stochastically activated within a MV-level KMC module. Translational and rotational velocity are calculated from forces and torques due to bonds, gravity, nonspecific surface repulsion, and hydrodynamic flow using a mobility matrix. G = shear rate.
Rigid microvilli are randomly distributed on the rigid, spherical cell surface, and adhesion molecules are placed at their tips according to a Poisson distribution. Flow is in the positive x-direction. To reduce computational demands, motion is restricted to x and z axis translation only and to rotation about the y axis. Microvilli beyond a central hoop-like region will never be able to enter the contact zone and form bonds and are thus excluded from the calculations. At each time step, a contact zone between the cell and flat endothelium is defined, spanning many microvilli, and receptor-ligand pairs within that zone are tested for formation. All preexisting bonds are then tested for breakage. Bonds are modeled as Hookean springs, and the probabilities of breakage and formation are calculated from bond-length-dependent kinetic rates. The reverse reaction rate takes the form of the Bell model (29):
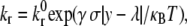 |
(1) |
where 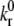 is an intrinsic reverse reaction rate, γ is the reactive compliance, a parameter with units of length describing the bond's sensitivity to force, σ is the bond spring constant, y is the bond length, λ is the unstressed bond length, and κBT is the thermal energy.
is an intrinsic reverse reaction rate, γ is the reactive compliance, a parameter with units of length describing the bond's sensitivity to force, σ is the bond spring constant, y is the bond length, λ is the unstressed bond length, and κBT is the thermal energy.
The forward reaction rate follows from the Boltzmann distribution for affinity (30) and takes the form of
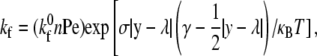 |
(2) |
where 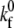 is the intrinsic forward reaction rate, n is the endothelial ligand surface density, and Pe is the Peclet number defined by Pe = (cell radius)(relative velocity)/(lateral diffusivity). The forward rate accounts for the ligand density on the adhesive surface. Furthermore, the effect of relative velocity between the cell and surface is included via Pe to describe the dependence of the forward reaction rate on the encounter rate of binding molecules (31,32). The probability during simulation time step, Δt, of bond formation, Pf, and breakage, Pr, are
is the intrinsic forward reaction rate, n is the endothelial ligand surface density, and Pe is the Peclet number defined by Pe = (cell radius)(relative velocity)/(lateral diffusivity). The forward rate accounts for the ligand density on the adhesive surface. Furthermore, the effect of relative velocity between the cell and surface is included via Pe to describe the dependence of the forward reaction rate on the encounter rate of binding molecules (31,32). The probability during simulation time step, Δt, of bond formation, Pf, and breakage, Pr, are 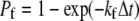 and
and 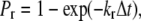 respectively (18,33).
respectively (18,33).
Total forces and torques exerted on the cell due to bonds, hydrodynamic flow, steric repulsion, and gravity are calculated. The repulsion prevents contact of the cell with the surface and is assumed to be a generic short-range force of the form 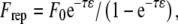 where 1/τ is a length scale, ɛ is the distance between the cell and the surface, and F0 sets the magnitude of the repulsive force (28,32). The equations of motion for a sphere in close contact with a wall have been solved (34,35), and the rotational and translational velocities in each coordinate direction can be calculated from the component forces and torques. The detailed methods for calculation of the cell's motion involve balancing the adhesive forces with the hydrodynamic forces, as described previously (33). The positions of the cell and of the cell-surface receptors are then updated. The fully stochastic activation pathway, described in detail below, is then evaluated. Changes in bond status due to bond formation and breakage within AD are transmitted to the activation pathway.
where 1/τ is a length scale, ɛ is the distance between the cell and the surface, and F0 sets the magnitude of the repulsive force (28,32). The equations of motion for a sphere in close contact with a wall have been solved (34,35), and the rotational and translational velocities in each coordinate direction can be calculated from the component forces and torques. The detailed methods for calculation of the cell's motion involve balancing the adhesive forces with the hydrodynamic forces, as described previously (33). The positions of the cell and of the cell-surface receptors are then updated. The fully stochastic activation pathway, described in detail below, is then evaluated. Changes in bond status due to bond formation and breakage within AD are transmitted to the activation pathway.
Signal transduction: kinetic Monte Carlo method
A schematic of the signal transduction pathway functional within each MV appears in Fig. 2 A. Each MV is modeled as a separate KMC system. The MV is treated as a 1002-rung one-dimensional ladder with LFA-1 and PSGL-1 elements permanently fixed at the bottom rung (MV tip) and MAPK elements permanently fixed at the top rung (MV base). Inward and outward signaling adaptor elements (ADINs and ADOUTs, respectively) are randomly distributed at the start of the simulation and may move along the intermediate ladder rungs via displacements, or hops, of specified size. An MV length of 0.3 μm sets the rungs 0.3 nm apart. With a hop size of 10 rungs (3 nm), an element would require 100 net displacement events in one direction to traverse the entire MV. For the simulation time step of 0.1 μs, a 10-rung hop size corresponds to a one-dimensional diffusion constant of 45 μm2/s. Because the ladder is one dimensional, more than one element may exist on the same rung. ADIN elements carry an activating signal from bound PSGL-1 elements to resting MAPK elements. ADOUT elements carry an activating signal from active MAPK elements to resting LFA-1 elements.
FIGURE 2.

Details of the stochastic integrin activation pathway. (A) Schematic of the integrin activation pathway. At the MV tip, bound PSGL-1 activates ADIN elements (triangles), which diffuse to the MV base and activate a MAPK element (oval). Active MAPK subsequently activates ADOUT elements (rectangles), which diffuse back to the MV tip and convert resting LFA-1 to active LFA-1. (B) Energy profile of multiple LFA-1 internal states. The star marks the energy barrier to free LFA-1 activation achieved through state exchange reactions with ADOUT. (C) Interaction of LFA-1 and ADOUT. Horizontal arrows indicate AD bond formation and breakage. Vertical arrows indicate KMC state exchange reactions. Diagonal arrows indicate KMC association and dissociation events. The star indicates the state exchange reaction corresponding to the barrier similarly marked in (B). Note that in this model, the state exchange reaction for activation of bound LFA-1 is assigned an infinite energy barrier.
Each element represents a protein that may have multiple internal states. In this simple model, the ultrasensitivity of the MAPK cascade is captured by the fact that the MAPK element can be active or inactive. The total numbers of LFA-1 and PSGL-1 elements in each KMC are specified in the initial MV creation within AD. The total numbers of ADIN, ADOUT, and MAPK elements are variable parameters. Receptor binding and unbinding events that occur within AD are reflected in changes in internal states of the corresponding KMC elements.
The energy function for the MV, which accounts for all its molecular components and states, is
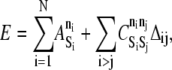 |
(3) |
where N is the number of elements, ni is the internal state of element i, and 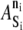 is a species-dependent parameter that describes the energy of the particular internal state.
is a species-dependent parameter that describes the energy of the particular internal state. 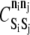 is the binding energy for elements Si and Sj of internal states ni and nj, respectively. The Kronecker delta function, Δij, equals one if the elements are bound and zero otherwise. The integer values of ni for LFA-1 ranges from 0 to 3, inclusive, whereas all other species have binary values, 0 or 1. Free resting LFA-1 and free active LFA-1 (free meaning not bound to ICAM-1 within AD) correspond to ni values of 0 and 1, respectively, whereas bound resting LFA-1 and bound active LFA-1 (bound with respect to ICAM-1) correspond to ni values of 2 and 3, respectively (Fig. 2, B and C). A PSGL-1 element bound (within AD) to E-selectin corresponds to ni = 1.
is the binding energy for elements Si and Sj of internal states ni and nj, respectively. The Kronecker delta function, Δij, equals one if the elements are bound and zero otherwise. The integer values of ni for LFA-1 ranges from 0 to 3, inclusive, whereas all other species have binary values, 0 or 1. Free resting LFA-1 and free active LFA-1 (free meaning not bound to ICAM-1 within AD) correspond to ni values of 0 and 1, respectively, whereas bound resting LFA-1 and bound active LFA-1 (bound with respect to ICAM-1) correspond to ni values of 2 and 3, respectively (Fig. 2, B and C). A PSGL-1 element bound (within AD) to E-selectin corresponds to ni = 1.
KMC simulation starts with the first PSGL-1/E-selectin binding event on each MV and continues for the duration of the MV's residence within the contact zone. When the MV leaves the contact zone, KMC simulation ceases until the MV reenters the contact zone. Restricting KMC calculations to the contact zone saves considerable computation time. Further, the characteristic time an MV spends outside the contact zone is much less than the characteristic time for LFA-1 deactivation.
At each AD time step, each element within the KMC is randomly assigned one of the four permitted KMC moves: displacement, association or dissociation, state exchange, and catalytic reaction. If a displacement move is chosen and the element can be displaced (i.e., it is neither a fixed element nor bound to a fixed element), an upward or downward direction is chosen with equal probability and the element is displaced. If an association or dissociation move is chosen, the element's bonding status is first evaluated. If the element is already bound, it is tested for dissociation. If it is not bound, an element from a neighboring rung (one above or one below) is randomly selected, and formation of a bond between the two elements is tested. Within this one-dimensional model, elements can form bonds with only one other element, and the elements must be exactly one ladder rung apart. Acceptance of bond formation or breakage is based upon the change in MV system energy, ΔE, resulting from the potential event, as well as the barrier for the reaction, 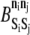 (where
(where 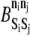 > 0 in the exothermic direction). A random number uniformly distributed between zero and one is compared to
> 0 in the exothermic direction). A random number uniformly distributed between zero and one is compared to 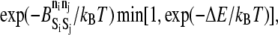 where kB is the Boltzmann constant and T is the temperature. A random number less than this product results in acceptance of the event.
where kB is the Boltzmann constant and T is the temperature. A random number less than this product results in acceptance of the event.
A state exchange involves transfer of energy that alters the state of both participating elements to state n′i, akin to phosphorylation exchange. The elements need not be bound to each other. If a state exchange move is chosen, an element from a neighboring rung is selected at random, and acceptance of the exchange is determined in the same manner as for association and dissociation, with 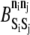 replaced by
replaced by 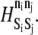 In this model, state exchange reactions are permitted between the pairs ADIN and MAPK, MAPK and ADOUT, and ADOUT and LFA-1. Through these exchanges, energy derived from the extracellular ligation of PSGL-1 is transmitted to the integrin.
In this model, state exchange reactions are permitted between the pairs ADIN and MAPK, MAPK and ADOUT, and ADOUT and LFA-1. Through these exchanges, energy derived from the extracellular ligation of PSGL-1 is transmitted to the integrin.
A catalytic reaction involves two elements yet changes the state of only one of the participating elements. If a catalytic reaction move is chosen, an element from a neighboring rung is selected at random and the move is accepted if a random number is less than 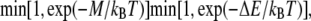 where M is the barrier to the catalytic reaction whose conditions are satisfied. In this model, the sole catalytic event involves an extracellularly bound PSGL-1 element activating or deactivating an ADIN element to which it is bound within the KMC. There is no autocatalysis permitted in this model.
where M is the barrier to the catalytic reaction whose conditions are satisfied. In this model, the sole catalytic event involves an extracellularly bound PSGL-1 element activating or deactivating an ADIN element to which it is bound within the KMC. There is no autocatalysis permitted in this model.
Parameters
Table 1 lists AD system parameters used in these simulations. The cell radius is that of a neutrophil. MV density and length are chosen to match experimental measurements (36). The shear rate of 100 s−1 falls within the range of shear rates at which neutrophil rolling is observed (37) but below the values where significant cell deformation occurs (38).
TABLE 1.
Simulation parameters
| Parameter | Definition | Value | Reference |
|---|---|---|---|
| a | Cell radius | 5 μm | (47) |
| G | Shear rate | 100 s −1 | (32,37) |
| μ | Viscosity | 1 cP | |
| ρ | Fluid density | 1.0 g/cm3 | |
| Δρ | Density difference | 0.05 g/cm3 | |
| ɛw | Wall roughness | 50 nm | (32) |
| σ | Spring constant | 100 dyne/cm | (48) |
| λ | Equilibrium bond length | 70 nm | (49) |
| T | Temperature | 310 K | |
| ρMV | MV density | 5 mv/μm2 | (36) |
| Lmv | MV length | 0.3 μm | (36) |
| nE-sel | E-selectin surface density | 3600 molec/μm2 | (19) |
| nICAM | ICAM-1 surface density | 210 molec/μm2 | (41) |
Molecular binding parameters are shown in Table 2. The Bell model parameters, γ and 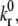 for the E-selectin-PSGL-1 and ICAM-1-resting LFA-1 pairs are taken from pause time distribution analysis and atomic force microscopy measurements, respectively (39,40). The value of
for the E-selectin-PSGL-1 and ICAM-1-resting LFA-1 pairs are taken from pause time distribution analysis and atomic force microscopy measurements, respectively (39,40). The value of 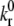 for ICAM-1-active LFA-1 is also taken from Zhang and co-workers (40). To determine two-dimensional
for ICAM-1-active LFA-1 is also taken from Zhang and co-workers (40). To determine two-dimensional 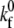 values for the selectin and resting integrin binding pairs, simulations employing the measured Bell model parameters were fit to data from experiments of cell-free rolling mediated by molecular pairs E-selectin and PSGL-1 or ICAM-1 and wild-type LFA-1 I domain (19,41). Measurements of ICAM-1 binding to resting and active LFA-1 in solution indicate that there is a 9000-fold increase in affinity for ligand from the resting to active integrin state (42,43). Using this information along with the previously determined rate constants, we then calculate a
values for the selectin and resting integrin binding pairs, simulations employing the measured Bell model parameters were fit to data from experiments of cell-free rolling mediated by molecular pairs E-selectin and PSGL-1 or ICAM-1 and wild-type LFA-1 I domain (19,41). Measurements of ICAM-1 binding to resting and active LFA-1 in solution indicate that there is a 9000-fold increase in affinity for ligand from the resting to active integrin state (42,43). Using this information along with the previously determined rate constants, we then calculate a 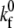 value of 115 μm2/s for the ICAM-1-active LFA-1 bond.
value of 115 μm2/s for the ICAM-1-active LFA-1 bond.
TABLE 2.
Molecular binding parameters
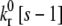
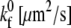
Within the KMC module, the selection of appropriate values for the various internal state energies is important. Unless otherwise specified, the terms bound and free in the following discussion refer to extracellular bonds involving LFA-1/ICAM-1 or PSGL-1/E-selectin pairs. Values of 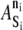 for all KMC elements are listed in Table 3. The resting, free LFA-1 (ni = 0) has a baseline energy of 0 kBT (Fig. 2 B). Scanning electron microscopy of αVβ3 showed that ∼85% of integrins exist in the extended conformation in the presence of Mn2+, an ion thought to mimic integrin conformational changes due to inside out signaling (2). This information yields an estimate of a 2 kBT drop in energy upon activation of a free resting LFA-1. BIAcore measurements of KD values of the resting LFA-1/ICAM-1 and active LFA-1/ICAM-1 interaction provide estimates of energy differences between the bound and free states of resting and active molecules (42). A KD of 1670 μM for resting LFA-1/ICAM-1 interactions corresponds to an approximate 6 kBT drop in energy upon formation of the extracellular bond. A KD of 0.2 μM for active LFA-1/ICAM-1 interactions corresponds to an approximate 15 kBT drop in energy upon formation of the extracellular bond. Thus, a bound resting LFA-1 (ni = 2) has an energy of −6 kBT, and a free active LFA-1 (ni = 1) has an energy of −2 kBT (Fig. 2 B). The bound active LFA-1 (ni = 3) has an energy of −17 kBT, which reflects the −15 kBT change from the free active state (Fig. 2 B). The interactions of LFA-1 with ADOUT and with ICAM-1 (within AD) that underlie these state changes are depicted in detail in Fig. 2 C.
for all KMC elements are listed in Table 3. The resting, free LFA-1 (ni = 0) has a baseline energy of 0 kBT (Fig. 2 B). Scanning electron microscopy of αVβ3 showed that ∼85% of integrins exist in the extended conformation in the presence of Mn2+, an ion thought to mimic integrin conformational changes due to inside out signaling (2). This information yields an estimate of a 2 kBT drop in energy upon activation of a free resting LFA-1. BIAcore measurements of KD values of the resting LFA-1/ICAM-1 and active LFA-1/ICAM-1 interaction provide estimates of energy differences between the bound and free states of resting and active molecules (42). A KD of 1670 μM for resting LFA-1/ICAM-1 interactions corresponds to an approximate 6 kBT drop in energy upon formation of the extracellular bond. A KD of 0.2 μM for active LFA-1/ICAM-1 interactions corresponds to an approximate 15 kBT drop in energy upon formation of the extracellular bond. Thus, a bound resting LFA-1 (ni = 2) has an energy of −6 kBT, and a free active LFA-1 (ni = 1) has an energy of −2 kBT (Fig. 2 B). The bound active LFA-1 (ni = 3) has an energy of −17 kBT, which reflects the −15 kBT change from the free active state (Fig. 2 B). The interactions of LFA-1 with ADOUT and with ICAM-1 (within AD) that underlie these state changes are depicted in detail in Fig. 2 C.
TABLE 3.
KMC elements
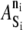
| |||||
|---|---|---|---|---|---|
| Si | Location | n = 0 | 1 | 2 | 3 |
| PSGL-1 | MV tip, fixed | 0 | −2 | na | na |
| LFA-1 | MV tip, fixed | 0 | −2 | −6 | −17 |
| ADIN | Cytosolic | 0 | −2 | na | na |
| ADOUT | Cytosolic | 0 | −2 | na | na |
| MAPK | MV base, fixed | 0 | −2 | na | na |
The energies of pairwise KMC bond formation, 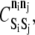 and energy barriers to KMC association and dissociation,
and energy barriers to KMC association and dissociation, 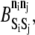 must also be set. These parameters are held at uniform values of −2 kBT and 1 kBT, respectively. The 2 kBT energy drop due to bond formation promotes association between elements in the KMC, and the modest 1 kBT barrier to association/dissociation is not overly obstructive. Values of
must also be set. These parameters are held at uniform values of −2 kBT and 1 kBT, respectively. The 2 kBT energy drop due to bond formation promotes association between elements in the KMC, and the modest 1 kBT barrier to association/dissociation is not overly obstructive. Values of 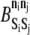 and
and 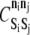 for all permitted pairs of elements are listed in Table 4.
for all permitted pairs of elements are listed in Table 4.
TABLE 4.
Permitted KMC binding pairs: bond energy and barrier to formation
| Si | Sj | ni | nj | 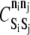 |
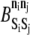 |
|---|---|---|---|---|---|
| PSGL-1 | ADIN | 0 or 1 | 0 or 1 | −2.0 | 1.0 |
| ADIN | MAPK | 1 | 0 | −2.0 | 1.0 |
| ADIN | MAPK | 0 | 1 | −2.0 | 1.0 |
| MAPK | ADOUT | 1 | 0 | −2.0 | 1.0 |
| MAPK | ADOUT | 0 | 1 | −2.0 | 1.0 |
| ADOUT | LFA-1 | 0 | 1 or 3 | −2.0 | 1.0 |
| ADOUT | LFA-1 | 1 | 0 or 2 | −2.0 | 1.0 |
Selection of state exchange energy barriers, 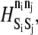 is crucial (Table 5). For state exchanges involving MAPK, we hold the barrier constant at a modest 2.0 kBT to permit uncomplicated transmission of signal. Exchange events involving activation of bound and free LFA-1 are treated differently, however. The energy barrier to activation of free LFA-1 remains unknown, so we permit the energy barrier to the corresponding exchange event to vary among the values 2.0, 6.0, 10.0, and 14.0 kBT. These values correspond to activation rates ranging between 101 and 106 s−1, a reasonable span. For the exchange event that activates bound LFA-1, on the other hand, we set the energy barrier to be very large, effectively preventing this method of integrin activation. We make the reasonable assumption that activation of LFA-1 would require unbinding from ICAM-1 and activation of the free receptor, thus simplifying our model significantly. This assumption can be relaxed in later simulations. The sole catalytic reaction energy barrier, M, involving activation of ADIN by bound PSGL-1, is held at 2.0 kBT.
is crucial (Table 5). For state exchanges involving MAPK, we hold the barrier constant at a modest 2.0 kBT to permit uncomplicated transmission of signal. Exchange events involving activation of bound and free LFA-1 are treated differently, however. The energy barrier to activation of free LFA-1 remains unknown, so we permit the energy barrier to the corresponding exchange event to vary among the values 2.0, 6.0, 10.0, and 14.0 kBT. These values correspond to activation rates ranging between 101 and 106 s−1, a reasonable span. For the exchange event that activates bound LFA-1, on the other hand, we set the energy barrier to be very large, effectively preventing this method of integrin activation. We make the reasonable assumption that activation of LFA-1 would require unbinding from ICAM-1 and activation of the free receptor, thus simplifying our model significantly. This assumption can be relaxed in later simulations. The sole catalytic reaction energy barrier, M, involving activation of ADIN by bound PSGL-1, is held at 2.0 kBT.
TABLE 5.
Permitted KMC exchange reactions
| Si | Sj | ni | nj | n′i | n′j | 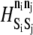 |
|---|---|---|---|---|---|---|
| ADIN | MAPK | 1 | 0 | 0 | 1 | 2.0 |
| MAPK | ADOUT | 1 | 0 | 0 | 1 | 2.0 |
| ADOUT | LFA-1 | 1 | 2 | 0 | 3 | 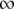 |
| ADOUT | LFA-1 | 1 | 0 | 0 | 1 | Variable |
The diffusivity of the signaling adaptor elements ADIN and ADOUT is varied by setting the elements' hop size. In this work, we explore values of 1, 2, 10, and 100 rungs, corresponding to one-dimensional diffusion constants of 0.45, 1.8, 45, and 4.5 × 103 μm2/s, respectively. These diffusion constants span the range of 5.5–93 μm2/s measured for small solutes in cytoplasm (44).
The numbers of ADIN, ADOUT, and MAPK elements within the KMC module are each fixed at 5 in every MV for all simulated cells unless otherwise noted. These values permit sufficient signaling to bring about cell arrest yet limit the quantity of elements manipulated within the KMC module. For an MV with a radius of 0.1 μm, length of 0.3 μm, and volume of ∼0.01 μm 3, five molecules per MV corresponds to a concentration of ∼1 μM. The effects of increasing the amounts of ADIN, ADOUT, and MAPK beyond these numbers are addressed briefly below.
Computation
Simulations were coded in C++ and run using Dell Precision workstations (Dell, Round Rock, TX) with 2.6 GHz Xeon processors (Intel, Santa Clara, CA). Random number generation employed the C++ version of the Mersenne twister algorithm (45). Simulation output was visualized using MATLAB (Natick, MA). Cell center coordinates and the locations of all MVs were recorded at all times (see Fig. 4). In addition, the activation and bonding states of all receptors on each MV tip were tracked (see Figs. 4 and 5). The total energy function, E, for each MV was recorded (see Fig. 7), so the detailed properties of each MV's KMC module could be followed over time.
FIGURE 4.

Visualization of activating and arresting cell at 0, 2.5, 10, and 20 s. The left-hand column of images represents a side view of a cell rolling to the right. The right-hand column depicts the cell rolling toward the reader. On the surface of the cell, MVs are drawn as individual cylinders in a color corresponding to the degree of LFA-1 activation.
FIGURE 5.

Cell-level simulation output for cell depicted in Fig. 4. (A) Displacement; (B) Cumulative average rolling velocity; (C) Instantaneous velocity; (D) 10-s backward moving average velocity; (E) Number of bound MVs; (F) Total number of PSGL-1 bonds; (G) Total number of active LFA-1 bonds; (H) Total number of resting LFA-1 bonds; and (I) Total number of active LFA-1 molecules.
FIGURE 7.

MV-level simulation output for individual MV from cell depicted in Figs. 4 and 5. (A) Total KMC energy; (B) Number of PSGL-1 molecules (solid line) and bonds (dashed line); (C) Number of active LFA-1 molecules (solid line) and bonds (dashed line); (D) Number of resting LFA-1 molecules (solid line) and bonds (dashed line).
RESULTS
State diagram
Site densities of PSGL-1 and LFA-1 that would permit a transition from rolling to firm adhesion were first determined. For a system without activation, we constructed an adhesion state diagram delineating the boundary between rolling and firm adhesion as a function of receptor density (Fig. 3) for two conditions: a completely unactivated cell whose LFA-1 are all resting and a fully activated cell whose LFA-1 are all in the active state. Firm adhesion was identified when the average rolling velocity over the previous 10 s was <2% of VH, the hydrodynamic velocity. LFA-1 is able to mediate rolling in both resting and active states. As expected, synergistic promotion of adhesion that is characteristic of two-receptor rolling (28)—in which selectins and integrins together promote adhesion beyond what either molecule can do itself—was observed for both fully resting and activated states. Also, as expected, activation of integrins reduces the density of molecules required for firm adhesion. We chose the surface density combination of 3 PSGL-1/MV and 9 LFA-1/MV, marked with a point in Fig. 3, to be used in all simulations in this work. When all LFA-1 are resting, a cell with this density combination rolls at 11.8 ± 1.3 μm/s (SD; n = 4), where as a cell with all its LFA-1 active is firmly adherent.
FIGURE 3.

State diagram on a double logarithmic scale depicting the boundary between rolling and firm adhesion as a function of PSGL-1 and LFA-1 site densities for both resting (dashed curve) and active (solid curve) integrin states. For each integrin state, the boundary represents a 10-s mean velocity of 0.02 VH. The point indicated by the circle corresponds to a cell with receptor densities of 3 PSGL-1 molecule/MV and nine total LFA-1 molecules/MV. Shear rate = 100 s−1. ICAM-1 = 210 #/μm2; E-selectin = 3600 #/μm2.
Total cell response
As the cell rolls along the endothelium, it accumulates signal generated by E-selectin ligation of PSGL-1. Thus, as rolling persists, the cell becomes progressively more activated. This process is depicted in Fig. 4 for an arresting cell with a 2 kBT barrier to free LFA-1 activation and a hop size of 10. As time goes on, more and more MVs become activated, and the degree to which these MVs are activated increases. The number of colored MVs increases with time, and the color of more and more MVs shifts toward the red, or high activation, end of the color scale (Fig. 4).
Global simulation output for this cell appears in Fig. 5. The cell displacement (Fig. 5 A) initially increases with time as it rolls, but the cell later arrests when it becomes more activated. Correspondingly, both the average velocity and 10-s moving average velocity decrease with time (Fig. 5, B and D). The instantaneous velocity (Fig. 5 C) fluctuates until the cell becomes firmly adherent. Periods of lower instantaneous velocity can be associated with periods of higher bonding. For example, the dip in instantaneous velocity at ∼10 s occurs during a peak in the number of active LFA-1 bonds (Fig. 5 G). In fact, the number of active LFA-1 bonds at this time is nearly equal to the number when the cell is arrested. It is likely, then, that the distribution of the bonds in addition to the number contributes to firm adhesion. The number of bound MVs (Fig. 5 E) fluctuates slightly until the cell arrests, whereupon the number of bound MVs remains constant at 2. The numbers of active LFA-1 molecules and bonds (Fig. 5, I and G, respectively) increase with time until reaching maxima as the cell arrests. These numbers do not increase significantly as the cell remains firmly adherent because activating signal can enter the cell only through bound MVs. Because the KMC modules of MVs do not interact, once the bound MVs become fully activated, and no new MVs bind, overall numbers of active LFA-1 bonds and molecules cannot increase. The number of resting LFA-1 bonds remains relatively constant until the cell arrests, at which point the number of resting LFA-1 bonds falls to zero as resting LFA-1 are activated (Fig. 5 H). Note that in this model, LFA-1 cannot activate if it is bound to ICAM-1 due to the large barrier height; any bound resting LFA-1 must first dissociate from ICAM-1 within AD before activating within the KMC and then rebinding ICAM-1 as an active LFA-1.
Microvillus-level output
The behavior of any individual MV can be followed over time. Fig. 6 depicts the activity of KMC elements for a single sample MV with hop size of 100 and 2 kBT barrier to LFA-1 activation. The two bound PSGL-1 (solid asterisks) form bonds (lines) with ADIN elements (triangles), which in turn diffuse and carry the activating signal through MAPK (circles) and ADOUT (squares) elements. LFA-1 (diamonds) activation is indicated by change from open to solid. Note that every interaction between an activated ADIN (solid squares) and a resting LFA-1 does not result in a durable activation. Crossing bond lines exist because the signal transduction model is one dimensional; the KMC elements at any axial (vertical) position are spread out to be resolved visually. The vertical scale has also been altered such that bond lengths spanning only one axial diffusive step are the same as the vertical hop size of ADIN and ADOUT elements (100 axial steps).
FIGURE 6.

KMC activity for a sample MV with continuously bound PSGL-1. Bound PSGL-1 (solid asterisks) and LFA-1 (diamonds) elements are fixed at the MV tip, whereas MAPK (circles) are fixed at the MV base. ADIN (triangles) and ADOUT (squares) diffuse between the base and tip, carrying the activation signal (solid symbols indicate activated state). Lines represent KMC bonds. LFA-1 activation is indicated by solid diamonds.
The behavior relevant to AD binding is displayed in Fig. 7 for one particular MV from the cell depicted in Figs. 4 and 5. This MV is one of the two that remain bound at the end of this particular simulation, in which firm adhesion is achieved. Fig. 7 A shows the value of E, the total energy function of the KMC module, as a function of time. Fig. 7, B—D, shows the changes with time of total and bound numbers of PSGL-1, active LFA-1, and resting LFA-1 molecules, respectively. Troughs in E at 0.25, 4.5, and 12 s correspond, as expected, to bond formation. The number of PSGL-1 molecules remains constant throughout the simulation although bonds form and break. The increases in the number of active LFA-1 molecules match precisely the decreases in the number of resting LFA-1. The number of active LFA-1 molecules does decrease occasionally, reflecting the stochastic nature of integrin activation within this model.
Effect of diffusivity and free integrin activation barrier height
As discussed in Model Development, the two major adjustable parameters in our model are the exchange reaction energy barrier for activation of free LFA-1 and the diffusivity (or hop size) of ADIN and ADOUT. These two parameters allow, respectively, control over the reaction and diffusion limits of the activation pathway. By increasing the barrier height for free LFA-1 activation, we simulate a slower activation reaction. By increasing the signal diffusivity (hop size), we simulate a cell whose MVs facilitate faster transmission of signals along their lengths. Fig. 8 shows how these parameters affect the rolling time to arrest, where arrest is defined as the point at which the 10-s moving average velocity first drops below 2% VH. As expected, increasing the diffusivity (increasing hop size) reduces rolling time before arrest. At large barrier heights, further increasing the barrier to LFA-1 activation prolongs rolling before arrest. When the barrier to LFA-1 activation is small, however, the reaction is fast and other steps in the activation process control the time to arrest.
FIGURE 8.

Rolling time to arrest as a function of ADIN and ADOUT diffusivity (hop size) and energy barrier, 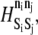 to state exchange reaction activation of free LFA-1. Arrest is defined as the point at which the 10-s moving average velocity first drops below 2% VH. N ≥ 7. Error = SE.
to state exchange reaction activation of free LFA-1. Arrest is defined as the point at which the 10-s moving average velocity first drops below 2% VH. N ≥ 7. Error = SE.
Role of endothelium
Endothelial expression levels of the ligands ICAM-1 and E-selectin represent a major regulatory method for recruitment of neutrophils to sites of inflammation. Within our integrated model, we manipulate the surface densities of ICAM-1 and E-selectin and observe the effects on rolling times before arrest. As expected, increasing the ICAM-1 surface density by 50% (from 210 to 315 molec/μm2) accelerates arrest (Fig. 9). Reducing the surface density of E-selectin by 50% (from 3600 to 1800 molec/μm2) at this higher ICAM-1 surface density prolongs the rolling time before arrest (Fig. 9).
FIGURE 9.

Effect of endothelial surface densities on rolling time to arrest. 3 PSGL-1 molecule/MV and 9 total LFA-1 molecules/MV; hop size = 2; ADIN = ADOUT = MAPK = 5; barrier to free LFA-1 activation = 6 kBT. Shear rate = 100 s−1. N ≥ 7. Error = SE. (* = p < 0.05).
Roles of ADIN, ADOUT, and MAPK
The preceding investigations of signal diffusivity and barrier height to LFA-1 activation were conducted with ADIN, ADOUT, and MAPK = five elements. However, the possibility remains that the amounts of these elements could be rate limiting. Fig. 10 A depicts the effect of increasing numbers of ADIN and ADOUT at constant receptor surface densities, hop size, barrier heights, and number of MAPK. These increases are equivalent to raising the total concentration of signal-carrying molecules (ADIN plus ADOUT) within the MV from 2 μM to 8 and 20 μM. Increasing both ADIN and ADOUT to 20 significantly shortens rolling time before arrest (p < 0.05), but further increase in element numbers to 50 provides no additional acceleration of adhesion. Here, the quantity of ADIN and ADOUT elements saturates the MAPK, and added signal carriers will not result in additional signal transduction.
FIGURE 10.

Effect of number of (A) ADIN and ADOUT and (B) MAPK elements on rolling time to arrest. 3 PSGL-1 molecule/MV and 9 total LFA-1 molecules/MV; hop size = 2; barrier to free LFA-1 activation = 6 kBT. Shear rate = 100 s−1. N ≥ 7. Error = SE. (* = p < 0.05).
The effect of increasing the number of MAPK at constant receptor surface densities, hop size, and barrier heights for two values of ADIN and ADOUT is shown in Fig. 10 B. When the number of signal carriers is high (ADIN = ADOUT = 20), increasing MAPK first accelerates arrest (p < 0.05), suggesting that MAPK = 1 limits the activation throughput. As the number of MAPK increases, the system approaches a 1:1:1 ratio of MAPK/ADIN/ADOUT, and the rolling time before arrest plateaus. When the number of signal carriers is low (ADIN = ADOUT = 5), increasing MAPK prolongs arrest. As the number of MAPK exceeds that of the ADIN or ADOUT, the signal carriers will have an increasing probability of associating with MAPK at the MV base rather than transmitting signal via diffusion. This has the effect of depleting the pool of ADIN and ADOUT, causing an increase in rolling time before arrest.
Tracking the activated fraction of ADIN, ADOUT, and MAPK over the entire cell provides additional information. Fig. 11 A shows this overall activated fraction as a function of time for all simulated cells used to calculate the average values in Fig. 10 A. At a constant MAPK = 5, increasing the numbers of ADIN and ADOUT results in a larger activated fraction of signaling molecules at a given time. This should yield a larger fraction of activated LFA-1 and thus accelerate arrest, as demonstrated in Fig. 10 A. Note that traces for ADIN = ADOUT = 20 and 50 overlap, a finding that is consistent with the saturation depicted in Fig. 10 A. Fig. 11 B depicts the overall activated fraction of signaling molecules as a function of time for all simulated cells corresponding to Fig. 10 B. At constant ADIN = ADOUT = 5, the activated fraction of signaling molecules at a given time decreases as MAPK increases. Thus, a reduced level of activation in the signaling pathway should yield a smaller fraction of activated LFA-1 and prolong rolling before arrest, explaining the findings in Fig. 10 B.
FIGURE 11.

Total cell fraction of activated ADIN, ADOUT, and MAPK for individual simulated cells as a function of time. Each trace ends at the point of the cell's firm arrest. (A) At constant ADIN = ADOUT = 5, traces for MAPK = 1 (black), 5 (blue), 10 (red), 20 (green). (B) At constant MAPK = 5, traces for ADIN = ADOUT = 5 (blue), 20 (black), 50 (red). 3 PSGL-1 molecule/MV and 9 total LFA-1 molecules/MV; hop size = 2; barrier to free LFA-1 activation = 6 kBT. Shear rate = 100 s−1.
DISCUSSION
In this work, we have successfully constructed a fully stochastic simulation of neutrophil activation driven by the probabilistic activation of β2-integrin molecules. Within the AD algorithm, we have embedded a KMC model of a simplified, local LFA-1 activation pathway. It is important to emphasize that the model is mechanically rigorous; as with all AD simulations, we are solving the equations of mechanical energy to determine the adhesive state of the cell (see Model Development). In this case, the advance is that we simultaneously have embedded a signaling network, thus creating a truly integrated model of cell adhesion. Receptor-ligand binding parameters were gleaned from the literature or determined by approximating experimental data with AD simulation. Values of parameters used in the KMC module, such as molecule diffusivity or free energies for LFA-1 in its various states, were also taken from the literature or varied across a range of reasonable values. We have shown that this integrated model can recreate the arrest of a neutrophil on a physiological timescale, in the neighborhood of ∼100 s (46) (see Fig. 8). It is remarkable that we can match the observed timescale for stopping with such a simple recreation of the intracellular signaling network. As more details become available about the kinetics and structure of the signaling network, we will be able to improve the accuracy and predictive potential of our model.
The signaling model presented here is simplified, yet it captures enough detail to allow four separate integrin states, as informed by reports of integrin conformations (4). Thus it permits the manipulation of integrin free energy states and can be further refined as more definitive information regarding integrin conformation emerges. The model also allows us to render the signaling pathway diffusion limited or reaction limited with minimal complexity. Because the methodology presented here is modular, a more sophisticated signaling network, such as one that employs a multiple-subunit GPCR as the chemokine receptor driving integrin activation, could easily be inserted into our integrated model.
Our model excludes a number of processes in an effort to maintain simplicity. First, we omit explicit signal deactivation, whether through inactivation of MAPK or through inactivation of the ADIN or ADOUT signal adaptor elements. It seems reasonable that a physiological activating pathway should have a counteracting deactivating pathway. It is likely that the effect of signal deactivation on neutrophil arrest will depend on the relative timescales of the deactivating event and the entire activating pathway as well as the timescales of MV duration within and outside the contact zone. Second, our model does not permit MAPK to diffuse axially along the MV. Although proteins can be highly associated with the cytoskeleton and relatively immobile within the cell, MAPK diffusion should be considered. The effect of an additional mobile member of the signaling pathway will affect the dynamics of leukocyte arrest and will likely depend, in a diffusion-limited system, on the relative diffusivities of all mobile signal carriers. Third, our model's assumptions prevent activation of LFA-1 when it is bound to ICAM-1. It has been suggested that LFA-1, when tethering the neutrophil to endothelium through its bond with ICAM-1, may undergo mechanoactivation (4), and it would seem reasonable to introduce a force dependence to the energy barrier to this event within the KMC module if future experimental evidence confirms this. Such a pathway would likely result in accelerated neutrophil arrest or permit firm adhesion at conditions of higher hydrodynamic shear forces.
Although the model includes changes in LFA-1 binding kinetics and strength, our model does not consider changes in LFA-1 length (LFA-1/ICAM-1 bond length) upon activation. The current structural models of integrin activation suggest that the major conformational rearrangement, in which the N-terminal headpiece is freed from the C-terminal, membrane-proximal domain, results in an extended, longer protein (3,4). Our integrated model could be modified easily to include any or all of the above mentioned details, as we will likely pursue in future work. In particular, we are likely to explore how GPCRs may also activate integrins and how the cross talk between G-protein and MAP-kinase signaling pathways would dynamically control the docking of leukocytes.
In this work, we have shown, for the first time to our knowledge, how a stochastic simulation of adhesion and signaling can be coupled. The basic paradigm we describe here—of integrating signaling and mechanical models of cell function—should ultimately prove useful for models of complex, mechanically based behavior, such as spreading, motility, and contraction as well as adhesion. The result of this work is that we are able to predict timescales for leukocyte activation that are reasonable and consistent with that seen in experiments. The model strikes a balance between detail and tractability. We have incorporated the minimal functional details for both the adhesion and signaling cascades to give rise to leukocyte stopping while restricting ourselves to real, identifiable cellular componentry that can be altered through molecular biology or knock-down assays. (For example, we showed how changes in MAPK might affect adhesion dynamics.) The goal of our work is to develop plausible, testable models of cell decision making with just the right number of components—however large that needs to be—to capture the indicated biology. As details into adhesion and signaling pathways emerge, we will improve our assays accordingly.
Acknowledgments
The authors are grateful for support from the National Institutes of Health (HL18208 and HL87353).
Editor: Jennifer Linderman.
References
- 1.Ley, K. 2003. The role of selectins in inflammation and disease. Trends Mol. Med. 9:263–268. [DOI] [PubMed] [Google Scholar]
- 2.Takagi, J., B. M. Petre, T. Walz, and T. A. Springer. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110:599–611. [DOI] [PubMed] [Google Scholar]
- 3.Kim, M., C. V. Carman, and T. A. Springer. 2003. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 301:1720–1725. [DOI] [PubMed] [Google Scholar]
- 4.Salas, A., M. Shimaoka, S. Q. Chen, C. V. Carman, and T. Springer. 2002. Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 277:50255–50262. [DOI] [PubMed] [Google Scholar]
- 5.Salas, A., M. Shimaoka, A. N. Kogan, C. Harwood, U. H. von Andrian, and T. A. Springer. 2004. Rolling adhesion through an extended conformation of integrin alpha(L)beta(2) and relation to alpha I and beta I-like domain interaction. Immunity. 20:393–406. [DOI] [PubMed] [Google Scholar]
- 6.Alon, R., V. Grabovsky, and S. Feigelson. 2003. Chemokine induction of integrin adhesiveness on rolling and arrested leukocytes local signaling events or global stepwise activation? Microcirculation. 10:297–311. [DOI] [PubMed] [Google Scholar]
- 7.Ley, K. 2002. Integration of inflammatory signals by rolling neutrophils. Immunol. Rev. 186:8–18. [DOI] [PubMed] [Google Scholar]
- 8.Hidari, K., A. S. Weyrich, G. A. Zimmerman, and R. P. McEver. 1997. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J. Biol. Chem. 272:28750–28756. [DOI] [PubMed] [Google Scholar]
- 9.Evangelista, V., S. Manarini, R. Sideri, S. Rotondo, N. Martelli, A. Piccoli, L. Totani, P. Piccardoni, D. Vestweber, G. de Gaetano, and C. Cerletti. 1999. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. 93:876–885. [PubMed] [Google Scholar]
- 10.Blanks, J. E., T. Moll, R. Eytner, and D. Vestweber. 1998. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta(2)-integrin mediated cell attachment to ICAM-1. Eur. J. Immunol. 28:433–443. [DOI] [PubMed] [Google Scholar]
- 11.Smolen, J. E., T. K. Petersen, C. Koch, S. J. O'Keefe, W. A. Hanlon, S. Seo, D. Pearson, M. C. Fossett, and S. I. Simon. 2000. L-selectin signaling of neutrophil adhesion and degranulation involves p38 mitogen-activated protein kinase. J. Biol. Chem. 275:15876–15884. [DOI] [PubMed] [Google Scholar]
- 12.Simon, S. I., Y. Hu, D. Vestweber, and C. W. Smith. 2000. Neutrophil tethering on E-selectin activates beta(2) integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J. Immunol. 164:4348–4358. [DOI] [PubMed] [Google Scholar]
- 13.Hentzen, E., D. McDonough, L. McIntire, C. W. Smith, H. L. Goldsmith, and S. I. Simon. 2002. Hydrodynamic shear and tethering through E-selectin signals phosphorylation of p38 MAP kinase and adhesion of human neutrophils. Ann. Biomed. Eng. 30:987–1001. [DOI] [PubMed] [Google Scholar]
- 14.Huang, C. Y. F., and J. E. Ferrell. 1996. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA. 93:10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiVietro, J. A., M. J. Smith, B. R. E. Smith, L. Petruzzelli, R. S. Larson, and M. L. Lawrence. 2001. IL-8 triggers progressive activation of neutrophil rolling in vitro on P-selectin and intercellular adhesion molecule-1. J. Immunol. 167:4017–4025. [DOI] [PubMed] [Google Scholar]
- 16.Lum, A. F. H., C. E. Green, G. R. Lee, D. E. Staunton, and S. I. Simon. 2002. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J. Biol. Chem. 277:20660–20670. [DOI] [PubMed] [Google Scholar]
- 17.Smith, M. L., T. S. Olson, and K. Ley. 2004. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J. Exp. Med. 200:935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer, D. A., and S. M. Apte. 1992. Simulation of cell rolling and adhesion on surfaces in shear flow: general results and analysis of selectin-mediated neutrophil adhesion. Biophys. J. 63:35–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang, K. C., and D. A. Hammer. 2000. Adhesive dynamics simulations of sialyl-Lewis(x)/E-selectin-mediated rolling in a cell-free system. Biophys. J. 79:1891–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang, K. C., D. F. J. Tees, and D. A. Hammer. 2000. The state diagram for cell adhesion under flow: leukocyte rolling and firm adhesion. Proc. Natl. Acad. Sci. USA. 97:11262–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, K. H., A. R. Dinner, C. Tu, G. Campi, S. Raychaudhuri, R. Varma, T. N. Sims, W. R. Burack, H. Wu, O. Kanagawa, M. Markiewicz, P. M. Allen, M. L. Dustin, A. K. Chakraborty, and A. S. Shaw. 2003. The immunological synapse balances T cell receptor signaling and degradation. Science. 302:1218–1222. [DOI] [PubMed] [Google Scholar]
- 22.Tandon, R., R. I. Sha'afi, and R. S. Thrall. 2000. Neutrophil beta 2-integrin upregulation is blocked by a p38 MAP kinase inhibitor. Biochem. Biophys. Res. Commun. 270:858–862. [DOI] [PubMed] [Google Scholar]
- 23.Ding, Z. M., J. E. Babensee, S. I. Simon, H. F. Lu, J. L. Perrard, D. C. Bullard, X. Y. Dai, S. K. Bromley, M. L. Dustin, M. L. Entman, C. W. Smith, and C. M. Ballantyne. 1999. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163:5029–5038. [PubMed] [Google Scholar]
- 24.Hentzen, E. R., S. Neelamegham, G. S. Kansas, J. A. Benanti, L. V. McIntire, C. W. Smith, and S. I. Simon. 2000. Sequential binding of CD11a/CD18 and CD11b/CD18 defines neutrophil capture and stable adhesion to intercellular adhesion molecule-1. Blood. 95:911–920. [PubMed] [Google Scholar]
- 25.Yan, B., D. A. Calderwood, B. Yaspan, and M. H. Ginsberg. 2001. Calpain cleavage promotes talin binding to the beta(3) integrin cytoplasmic domain. J. Biol. Chem. 276:28164–28170. [DOI] [PubMed] [Google Scholar]
- 26.Vinogradova, O., A. Velyvis, A. Velyviene, B. Hu, T. A. Haas, E. F. Plow, and J. Qin. 2002. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 110:587–597. [DOI] [PubMed] [Google Scholar]
- 27.Calderwood, D. A., B. X. Yan, J. M. de Pereda, B. G. Alvarez, Y. Fujioka, R. C. Liddington, and M. H. Ginsberg. 2002. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 277:21749–21758. [DOI] [PubMed] [Google Scholar]
- 28.Bhatia, S. K., M. R. King, and D. A. Hammer. 2003. The state diagram for cell adhesion mediated by two receptors. Biophys. J. 84:2671–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell, G. I. 1978. Models for specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- 30.Dembo, M., D. C. Torney, K. Saxman, and D. Hammer. 1988. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B. Biol. Sci. 234:55–83. [DOI] [PubMed] [Google Scholar]
- 31.Chang, K. C., and D. A. Hammer. 1999. The forward rate of binding of surface-tethered reactants: effect of relative motion between two surfaces. Biophys. J. 76:1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King, M. R., and D. A. Hammer. 2001. Multiparticle adhesive dynamics. Interactions between stably rolling cells. Biophys. J. 81:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang, K. C., and D. A. Hammer. 1996. Influence of direction and type of applied force on the detachment of macromolecularly-bound particles from surfaces. Langmuir. 12:2271–2282. [Google Scholar]
- 34.Goldman, A. J., R. G. Cox, and H. Brenner. 1967. Slow viscous motion of a sphere parallel to a plane wall. I. Motion through a quiescent fluid. Chem. Eng. Sci. 22:637–652. [Google Scholar]
- 35.Goldman, A. J., R. G. Cox, and H. Brenner. 1967. Slow viscous motion of a sphere parallel to a plane wall. 2. Couette flow. Chem. Eng. Sci. 22:653–660. [Google Scholar]
- 36.Shao, J. Y., H. P. Ting-Beall, and R. M. Hochmuth. 1998. Static and dynamic lengths of neutrophil microvilli. Proc. Natl. Acad. Sci. USA. 95:6797–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawrence, M. B., and T. A. Springer. 1991. Leukocytes roll on a selectin at physiological flow-rates—distinction from and prerequisite for adhesion through integrins. Cell. 65:859–873. [DOI] [PubMed] [Google Scholar]
- 38.Lei, X., M. R. Lawrence, and C. Dong. 1999. Influence of cell deformation on leukocyte rolling adhesion in shear flow. J. Biomech. Eng. 121:636–643. [DOI] [PubMed] [Google Scholar]
- 39.Smith, M. J., E. L. Berg, and M. B. Lawrence. 1999. A direct comparison of selectin-mediated transient, adhesive events using high temporal resolution. Biophys. J. 77:3371–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang, X. H., E. Wojcikiewicz, and V. T. Moy. 2002. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys. J. 83:2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eniola, A. O., E. F. Krasik, L. A. Smith, G. Song, and D. A. Hammer. 2005. I-domain of lymphocyte function-associated antigen-1 mediates rolling of polystyrene particles on ICAM-1 under flow. Biophys. J. 89:3577–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimaoka, M., C. F. Lu, R. T. Palframan, U. H. von Andrian, A. McCormack, J. Takagi, and T. A. Springer. 2001. Reversibly locking a protein fold in an active conformation with a disulfide bond: integrin alpha L I domains with high affinity and antagonist activity in vivo. Proc. Natl. Acad. Sci. USA. 98:6009–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimaoka, M., C. F. Lu, A. Salas, T. Xiao, J. Takagi, and T. A. Springer. 2002. Stabilizing the integrin alpha M inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc. Natl. Acad. Sci. USA. 99:16737–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arrio-Dupont, M., G. Foucault, M. Vacher, P. F. Devaux, and S. Cribier. 2000. Translational diffusion of globular proteins in the cytoplasm of cultured muscle cells. Biophys. J. 78:901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsumoto, M., and T. Nishimura. 1998. Mersenne twister: a 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans. Model. Comput. Simul. 8:3–30. [Google Scholar]
- 46.Kunkel, E. J., J. L. Dunne, and K. Ley. 2000. Leukocyte arrest during cytokine-dependent inflammation in vivo. J. Immunol. 164:3301–3308. [DOI] [PubMed] [Google Scholar]
- 47.Brunk, D. K., D. J. Goetz, and D. A. Hammer. 1996. Sialyl Lewis(x)/E-selectin-mediate rolling in a cell-free system. Biophys. J. 71:2902–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morozov, V. N., and T. Y. Morozova. 1990. What does a protein molecule look like? Comments Mol. Cell. Biophys. 6:249–270. [Google Scholar]
- 49.Patel, K. D., M. U. Nollert, and R. P. McEver. 1995. P-selectin must extend a sufficient length from the plasma membrane to mediate rolling of neutrophils. J. Cell Biol. 131:1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]