

Abstract
The function of a protein generally is determined by its three-dimensional (3D) structure. Thus, it would be useful to know the 3D structure of the thousands of protein sequences that are emerging from the many genome projects. To this end, fold assignment, comparative protein structure modeling, and model evaluation were automated completely. As an illustration, the method was applied to the proteins in the Saccharomyces cerevisiae (baker’s yeast) genome. It resulted in all-atom 3D models for substantial segments of 1,071 (17%) of the yeast proteins, only 40 of which have had their 3D structure determined experimentally. Of the 1,071 modeled yeast proteins, 236 were related clearly to a protein of known structure for the first time; 41 of these previously have not been characterized at all.
Keywords: comparative protein structure modeling/computer analysis of genome sequences
Despite great progress in biology, there is a need to describe and understand the function of many proteins in more detail than has been achieved so far. Although protein function is best determined experimentally (1), it sometimes can be predicted by matching the sequence of a protein with proteins of known function (1–3). This is possible because similar protein sequences tend to have similar functions, although exceptions also occur (4). The success and utility of computational assignment of protein function recently has increased dramatically because of the many genome sequencing projects (5). For example, sequence matching of the proteins encoded by the Saccharomyces cerevisiae (baker’s yeast) genome (6) has resulted in assignment of 58% of the yeast proteins into 11 functional classes with 93 subclasses (http://www.mips.-biochem.mpg.de/mips/yeast/index.html). One way to add to sequence-based predictions of function would be to determine or predict the three-dimensional (3D) structures of proteins. The 3D structure of a protein generally provides more information about its function than its sequence because interactions of a protein with other molecules are determined by amino acid residues that are close in space but are frequently distant in sequence. In addition, because evolution tends to conserve function and function depends more directly on structure than on sequence, structure is more conserved in evolution than sequence (7). The net result is that patterns in space are frequently more recognizable than patterns in sequence. For example, several mouse mast cell proteases have a conserved surface region of positively charged residues that binds proteoglycans (8). This region is not easily recognizable in the sequence because the constituting residues occur at variable and sequentially nonlocal positions that form a binding site only when the protease is fully folded. Approximately 7,500 protein structures have been determined experimentally by x-ray crystallography and nuclear magnetic resonance spectroscopy (ref. 9; http://www.pdb.bnl.gov), while there are over 325,000 entries in the GenPept sequence database alone (ref. 10; ftp://ncbi.nlm.nih.gov/genbank/genpept.fsa). To bridge this increasingly large gap between the numbers of known protein sequences and structures, we calculated useful all-atom 3D models for a significant fraction of the translated ORFs in the yeast genome (6). Specifically, we show how to automate modeling of thousands of proteins and how to predict the overall accuracy of the models with a high degree of certainty. We also discuss new ways of using a large number of protein models and point out several unexpected similarities between previously uncharacterized yeast ORFs and proteins of known structure.
MATERIALS AND METHODS
Protein Structure Modeling Method.
Comparative protein structure modeling (11, 12) was the method chosen for this study. The reasons were that, of all protein structure prediction methods, comparative modeling results in the most accurate, detailed, and explicit models of protein structure. This maximizes their usefulness in applications such as interpretation of the existing functional data, design of ligands, and construction of mutants and chimeric proteins for testing new functional hypotheses (11). Comparative protein structure modeling of a target sequence consists of (i) identification of known structures related to the target sequence (templates), (ii) alignment of the templates with the target sequence, (iii) building a model based on the alignment, and (iv) evaluation of the model. This flowchart has been implemented in a UNIX perl script that calls the appropriate programs for the individual tasks, each of which is described in more detail below. Program clustor was used to distribute efficiently smaller jobs on many workstations, without having to adapt the individual programs for parallel execution (http://www.activetools.com). All of the alignments and models are available on Internet at http://guitar.rockefeller.edu, as is our program modeller used for sequence–structure alignment, model building, and model evaluation. The models are also accessible through the Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces/).
Template Search.
To find template structures for modeling of the translated ORF sequences, each of the 6,218 ORFs from yeast (Saccharomyces Genome Database) was compared with each of the 2,045 potential templates corresponding to the protein chains representative of the PDB (March, 1997). The representative protein chains had at most 95% sequence identity to each other or had length difference of at least 30 residues or 30%; they were also the highest quality structures within each group. Although a small fraction of the yeast ORFs (<7%) is likely to be incorrect (3), this is not a serious limitation because an ORF that matches a known protein structure is likely to correspond to a real protein. The matching was done by the program align, which implements the local dynamic programming method with a new gap penalty function and has a search sensitivity higher than that of blast (13). Each ORF–PDB matching was run with the default gap penalty parameters first. A match was considered significant or insignificant if the alignment score was >22 or <19 nats, respectively, where the nat is a unit for measuring significance of a match (14). All of the pairs with intermediate matches with scores between 19 and 22 nats were realigned by using 600 combinations of the gap penalty parameters. The match was finally considered significant if the best of the 600 alignments had a score of at least 22 nats. The matching part of the PDB chain from a significant hit was used as the template structure for the corresponding region of the ORF.
Target–Template Alignment.
To obtain the target–template alignment for comparative modeling, the matching parts of the template structure and the ORF sequence were realigned by the use of the align2d command (R.S. and A.Š. unpublished work) of the modeller program (15–17). This command implements a global dynamic programming method for comparison of two sequences but also relies on the observation that evolution tends to place residue insertions and deletions in the regions that are solvent exposed, curved, outside secondary structure segments, and between two Cα positions close in space. Gaps in these structurally reasonable positions are favored by a variable gap penalty function that is calculated from the template structure alone. As a result, the alignment errors are reduced by approximately one-third relative to the standard sequence alignment techniques. Nevertheless, there is clearly a need for even more accurate sequence–structure alignments and for using multiple template structures, so that more accurate models are obtained (16).
Model Building.
The refined sequence–structure alignment was used by modeller to construct a 3D model of the ORF region (15–17). Model building began by extracting distance and dihedral angle restraints on the target sequence from its alignment with the template structure. These template-derived restraints were combined with most of the charmm energy terms (18) to obtain a full objective function. Finally, this function was optimized by conjugate gradients and molecular dynamics with simulated annealing to construct a model that satisfied all the spatial restraints as well as possible.
Assignment of a Model into the “Good” or “Bad” Class.
The overall accuracy of a model was measured by an overlap between the model and the actual structure. The overlap was defined as the fraction of residues whose Cα atoms are within 3.5 Å of each other in the globally superposed pair of structures. Models that overlap with the correct structures in >30% of their residues were defined here as “good” models. A method for predicting whether a given model is good was developed as follows. By using the PDB, 1,085 protein chains of known structure that had <30% sequence identity to each other were picked. Comparative models for these proteins were calculated by the standard procedure described above. In addition, many bad models were obtained by the same procedure, except that only target–template alignments with a relatively low alignment significance score from 15 to 20 nats were used. In the end, there were 3,993 and 6,270 good and bad models, respectively. There were more models than proteins because most proteins were modeled several times on a different template structure each time. The distribution of the target–template sequence identity for the good models was similar to that for the matching of the yeast ORFs with PDB chains (Fig. 2A). The quality score (Q_SCORE) of a model was defined as the ProsaII Z-Score (19) divided by the natural logarithm of sequence length, which made Q_SCORE almost independent of sequence length. The ProsaII Z-score approximates the difference in free energy of an evaluated model and the mean free energy of the same sequence threaded through unrelated folds, expressed in units of SD. The free energies were calculated with statistical potentials of mean force for single residues and pairs of residues (19). The distributions of Q_SCORE for good and bad models were obtained for different sequence length ranges. The posterior probability that a model was good, given that it had a certain Q_SCORE value, was obtained by using the Bayesian theorem (20) and assuming equal prior probabilities for good and bad models: p(GOOD/Q_SCORE) = p(Q_SCORE/GOOD)/[p(Q_SCORE/GOOD) + p(Q_SCORE/BAD)]. A model with p(GOOD/Q_SCORE) above 0.5 is predicted to be in the good class and thus have at least approximately correct fold. For proteins longer than 100 residues, it is possible to identify good models with <5% of false positives and <8% of false negatives (Fig. 1A).
Figure 2.
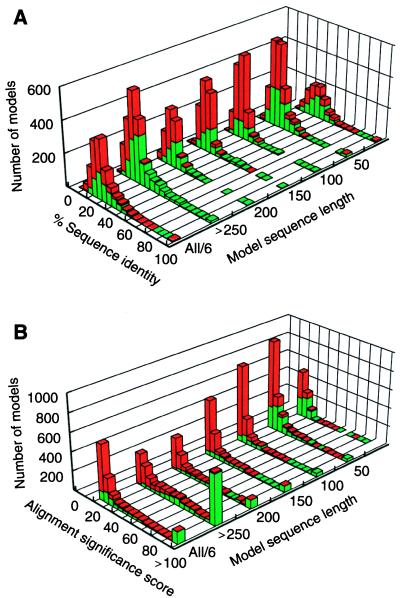
Protein structure models for yeast ORFs. (A) Distribution of the sequence identity between the models and the corresponding templates as a function of model sequence length. The 3,992 reliable models for substantial segments of 1,071 different ORFs that are predicted to be based on a correct template and approximately correct alignment are represented by the green bars, and the 4,588 unreliable models that are predicted to be based on a mostly incorrect alignment or an incorrect template are represented by the red bars. The last histogram at label “All/6” is the sum of the other six histograms divided by six. (B) The corresponding distribution of the alignment significance score calculated by the program align (13).
Figure 1.
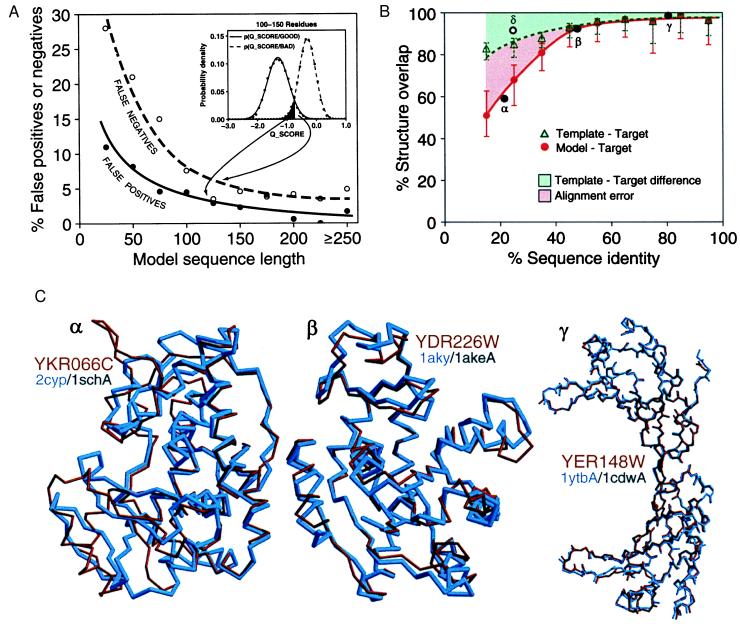
Predicting the overall accuracy of comparative models. The good and bad models for proteins of known structure are used to tune the prediction of reliability of a model when the actual structure is not known (Fig. 2). See Materials and Methods for details. (A) A rule for assigning a comparative model into either the good or bad class, based on its Q_SCORE. Inset shows the distributions of Q_SCORE for the good and bad models with 100 to 150 residues. Such distributions are used with the Bayes theorem to calculate the posterior probability that a model is good, given that it has a certain Q_SCORE value, p(GOOD/Q_SCORE). The main plot shows the percentages of false positives (bad models classified as good) and false negatives (good models classified as bad) as a function of sequence length. The curves were obtained by the jack-knife procedure. (B) A rule for estimating the accuracy of a reliable model (as predicted by its Q_SCORE), based on the percentage sequence identity to the template. The overlaps of an experimentally determined protein structure with its model (red continuous line) and with a template on which the model was based (green dashed line) are shown as a function of the target–template sequence identity. This identity was calculated from the modeling alignment. The structure overlap is defined as the fraction of the equivalent Cα atoms. For comparison of the model with the actual structure (filled circles), two Cα atoms were considered equivalent if they were within 3.5 Å of each other and belonged to the same residue. For comparison of the template structure with the actual target structure (open circles), two Cα atoms were considered equivalent if they were within 3.5 Å after alignment and rigid-body superposition by the align3d command in modeller (15). The points correspond to the median values, and the error bars in the positive and negative directions correspond to the average positive and negative differences from the median, respectively. Points labeled α, β, and γ correspond to the models in (C). The empty circle at 25% sequence identity corresponds to the unusually accurate model in Fig. 3B. (C) The range of accuracy for reliable comparative models is illustrated by a difficult, medium, and easy case. The Cα backbones of the models (red) for YKR066C and YDR226W and all mainchain atoms for YER148W are superposed with those of the actual structures (blue). The PDB codes of the target and template structures also are shown (target/template). The three target–template sequence identities are indicated in B (black filled circles). The number of yeast ORF models at each accuracy level can be determined from the red curve in B, or the sample comparisons in C, combined with Fig. 2A.
Prediction of the Overall Accuracy of a Model.
For the models predicted to be in the good class, the fraction of the Cα atoms modeled within 3.5 Å of the correct positions depends on the percentage sequence identity between the modeled sequence and the template. This dependence was determined by using the 3,993 good models for proteins of known structure described in the previous paragraph (Fig. 1B). Above 40% sequence identity, the median overlap between a model and the corresponding experimental structure is >90% (Fig. 1A). There are few errors in the alignment, and the model is as close to the correct structure as the template. Many models in this range have errors that are comparable to the differences between experimental structures of the same protein determined by different techniques or in different environments (12). For 30–40% sequence identity, the overlap between a model and the corresponding experimental structure is 75–90%. Because the alignment errors begin to appear, the models overlap with the correct structures less than the templates do. At very low sequence identity of <30%, the overlap drops to 50–75%. These model evaluation results can be understood in terms of the well known relationship between structural and sequence similarities of two proteins (7), the “geometrical” nature of modeling that forces the model to be as close to the template as possible (15), and the inability of any current modeling procedure to recover from an incorrect alignment (16).
RESULTS
Template Search.
The ORF–PDB matching procedure identified one or more possibly related structures for 2,256 or 36.3% of the ORFs (Fig. 2A). The average length of the local alignments was 174 residues, and the average pairwise sequence identity was 27%.
Evaluation of the Models.
Model evaluation indicates that 1,071 (17.2%) of the yeast ORFs have at least one segment of residues with a reliable model (Fig. 2). A small number of ORFs have a reliable model for more than one domain, resulting in the total of 1,168 nonoverlapping reliable models for all ORFs. In contrast, only 40 of the yeast proteins have had their structures determined experimentally (9). The average length of a reliable model is 176 residues, and 85% of the reliable models are longer than 50 residues. The average pairwise sequence identity on which the reliable models are based is 34%. Most of the models based on more than the average sequence identity are predicted to overlap with the correct structures in more than 80% of their residues (Fig. 1 B and C). In comparison, another study produced comparative models for 10–15% of the proteins in the E. coli genome (4,290 ORFs) (21). When our procedure was applied to the same genome, it resulted in reliable models for 18.1% of the proteins.
Fold Assignment Rate.
Fold recognition (22), sequence profile methods (23), and Hidden Markov Models (24) generally are considered to be more sensitive for detecting remote relationships than the local sequence alignment applied here. Thus, in the future, these methods will supplement the matching by pairwise sequence comparison in our pipeline for automated comparative modeling. However, it is not clear how many more accurate models can be calculated for the matches from the more sophisticated methods. The reason is that accurate 3D modeling requires both a correct fold assignment and an approximately correct target–template alignment. Unfortunately, it appears that when a correct target–template match is made in the absence of statistically significant sequence similarity already detectable by simple methods, it is rarely possible to produce an accurate alignment (25). Nevertheless, we now estimate what would have happened to the fold assignment rate alone if fold recognition and Hidden Markov Models were applied to the yeast genome. A recent automated fold recognition survey assigned folds to 103 (22.0%) of the 468 ORFs in the small M. genitalium genome (26). In comparison, when our procedure was applied to the same genome, it resulted in reliable models for 90 of the 468 ORFs (19.2%), 81 of which were shared with the fold recognition survey. For another benchmark, the pfam database obtained by Hidden Markov Models (27) related 315 yeast proteins to a protein of known structure, which is a relatively small fraction of the 1,071 matches obtained here. We identified 263 of the 315 pfam matches, and 248 of these corresponded to reliable models. Thus, fold recognition and Hidden Markov Models would provide a small but significant increase in the number of target–template matches for model evaluation by our combined alignment/modeling approach. However, even the existing procedure based on local sequence alignment appears to be able to identify some matches that were not identified by fold recognition (there are nine such cases for the M. genitalium genome). The reason is that, in the combined alignment/modeling procedure, the final decision about whether a given match is correct is made by evaluating the 3D model implied by the alignment rather than by scoring the alignment directly. Because model evaluation works well (Fig. 1A), the cutoff for accepting a match at the sequence matching stage can be lowered significantly, thus minimizing the loss of correct matches without adding many false positives. This results in a relatively large number of reliable models based on low sequence similarity (Fig. 2); for example, 261 yeast ORFs have at least one reliable model based on a match with a significance score worse than 24 nats (Fig. 2B), which is too low to establish a relationship on its own. The combined alignment/modeling approach to confirming a remote relationship already has been proven successful in several individual cases (16, 28, 29). Another example is the model of the component PRE4 of the yeast 20S proteasome complex (YFR050C). The model was based on the structure of subunit B of the Thermoplasma acidophilum proteasome (1pmaB); the target and the template have only 16% sequence identity, with the alignment significance score of 22 nats. However, the model of YFR050C was predicted to be good [p(GOOD/Q_SCORE) = 0.99]. The crystallographic structure of YFR050C, determined after the model was calculated (1rypN) (30), showed that the fold assignment was correct.
DISCUSSION
Usefulness of Models with Errors.
It is essential for assessing the value of 3D protein models to estimate their overall accuracy (19, 31). In general, mistakes in comparative modeling include sidechain packing errors, small distortions and rigid body shifts in correctly aligned regions, errors in inserted regions (loops), incorrect alignments, and incorrect templates (16). Fortunately, a 3D model does not have to be absolutely perfect to be helpful in biology (11). One reason is that knowing only the fold of a protein is frequently sufficient to predict its approximate biochemical function. For example, only 9 of 80 fold families known in 1994 contained proteins (domains) that were not in the same functional class, although 32% of all protein structures belonged to one of the nine superfolds (4). A model is likely to have the correct fold when the overlap with the actual structure is at least 30%. Such models are obtained when a correct template and an approximately correct alignment are used. This appears to be the case for 1,071 ORFs, as predicted by our model evaluation procedure (Fig. 2). Models for two yeast ORFs calculated before the actual structures were deposited to PDB are illustrated and discussed in Fig. 3. Almost half of the 1,071 reliably modeled ORFs share more than ∼35% sequence identity with their templates (Fig. 2A). In such cases, it is frequently possible to predict correctly important features of the target protein that do not occur in the template structure. For example, the location of a binding site can be predicted from clusters of charged residues (8), and the size of a ligand can be predicted from the volume of the binding site cleft (32).
Figure 3.
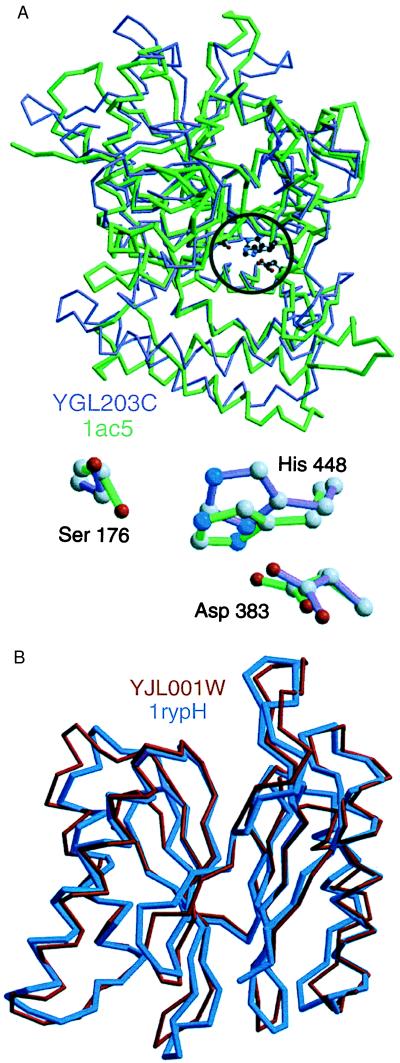
Sample models calculated before the crystallographic structures have been deposited to PDB. (A) A model for the yeast prohormone-processing carboxypeptidase (YGL203C, violet) is compared with its actual crystallographic structure (1ac5, green) (38). The model was constructed based on the crystal structure of the yeast serine carboxypeptidase (1cpy) with which it shares only 25% sequence identity. Although the overall structural overlap of the model and the actual structure is only 63%, the active site (Inset) and the neighboring residues have been modeled with useful accuracy; for example, it is possible to use the model to plan site-directed mutagenesis experiments for assessing residues critical for catalysis and binding specificity. The model also illustrates that the functionally important regions of the molecule tend to be modeled more accurately than the rest of the protein (Fig. 1B) because they are frequently more conserved in evolution than the rest of the fold. (B) A model for the yeast multi-catalytic protease (YJL001W, red) is compared with its actual crystallographic structure (1rypH) (30). Despite a low sequence identity of 24% to the template structure (1pmaB), the model overlaps with the actual x-ray structure in 92% of the residues (point δ in Fig. 1B). It was possible to predict that this particular model was unusually accurate given its sequence similarity to the template because it had a favorable Z-score of −8.3 and an energy profile with only one positive peak (19). The YJL001W subunit is part of the 20S proteasome, a highly ordered ring-shaped structure consisting of 14 similar subunits, all of which have been modeled in this study. The models are sufficiently accurate for use with protein–protein docking programs, which in turn are likely to predict correctly at least some of the interface residues between the subunits (17).
Usefulness of Comparative Models.
Comparative models are calculated from a sequence alignment between the protein to be modeled and a related protein of known structure. Thus, a question arises as to what additional insights that are not already possible from sequence matching alone can possibly be obtained by 3D modeling. The first advantage of 3D modeling is that it provides the best way of either confirming or rejecting a remote match (16), as discussed above. This is important because most of the related protein pairs share <30% sequence identity (Fig. 2A). For example, only 10.7% of the yeast ORFs have been matched reliably with known structures by Fasta (http://pedant.mips. biochem.mpg.de/frishman/pedant.html), as opposed to 17.2% in our study. Another case in point is that 236 of the 1,071 yeast ORFs with reliable models had no previously identified links to a protein of known structure in the major annotations of the yeast genome, including sacch3d, pedant, genequiz, and pfam (Table 1). Of these 236 proteins for which some structural information is now available, 41 also did not have a clear link to a protein sequence with known function. A subset of these 41 newly characterized proteins is listed in Table 1. Additional confidence in these matches is provided by the conservation of the known functionally important residues in the target models.
Table 1.
Examples of previously uncharacterized yeast proteins with reliable models
| ORF | residues | PDB code | residues | name | Percent sequence identity | Model accuracy | Conserved features |
|---|---|---|---|---|---|---|---|
| YDL117W | 13–64 | 1lckA | 65A–115A | P56-LCK SH3 domain | 30 (24.5) | 0.97 | W31 conserved; other binding residues conserved or similar. |
| YCR033W | 885–935 | 1idz | 140–190 | c-MYB DNA binding domain | 21 (22.3) | 0.99 | N interacting with DNA is conserved; K’s replaced by R’s. |
| YNL181W | 44–341 | 1fmcA | 2A–215A | 7-α-hydroxysteroid dehydrogenase | 14 (25.5) | 0.98 | K163 conserved; Y159F. |
| YOR221C | 124–368 | 1mla | 87–296 | malonyl-COA ACP transacylase | 17 (23.7) | 0.95 | Active site residues S92, R117, and H201 are conserved. |
| YPL217C | 63–182 | 1etu | 5–145 | elongation factor Tu (domain I) | 22 (22.7) | 0.86 | GTP binding loops are similar. Conserved GKTTL motif. |
These ORFs do not have clear similarity to any protein of known function according to the following sources (October 31, 1997): MIPS (http://www.mips.biochem.mpg.de/mips/yeast/index.html), Yeast Protein Database (http://quest7.proteome.com/YPDhome.html), genequiz (http://www.sander.ebi.ac.uk/genequiz), sacch3d (http://genome-www.stanford.edu/Sacch3D), pedant (http://pedant.mips.biochem.mpg.de/frishman/pedant.html), and pfam (27). The examples were selected partly by considering conservation of the functionally important residues (Conserved features). Thus, they have higher sequence similarity to known protein structures than most of the other previously uncharacterized yeast proteins. For each ORF and its corresponding template, the starting and ending residues of the matching regions are indicated. The number in parenthesis in the percent sequence identity column is the alignment significance score in nats (13). The overall model accuracy is given by p(GOOD/Q_SCORE). The complete list of 236 previously uncharacterized yeast proteins with reliable models is available at http://guitar.rockefeller.edu.
The second advantage of 3D modeling over sequence matching is that some binding and active sites cannot possibly be found by searching for local sequence patterns (33, 34) but frequently should be detectable by searching for small 3D motifs that are known to bind or act on specific ligands (35, 43). This is a consequence of the facts that (i) structure is more conserved than sequence (7), (ii) 3D motifs tend to consist of residues distant in sequence, and (iii) there are some 3D motifs whose residues do not follow the same order in sequence, even though they have the same arrangement in space. An example of this is the serine catalytic triad that almost certainly arose by convergent evolution in serine proteases of the trypsin and subtilisin type and also in some lipases (35). Enumeration of active and binding sites for many proteins in the genome, such as various metal and nucleotide binding sites, will facilitate experimental determination of protein function.
The third advantage of 3D modeling over sequence matching is that a 3D model frequently allows a refinement of the functional prediction based on sequence alone because the ligand binding is determined most directly by the structure of the binding site rather than its sequence. An example of this is provided by a predicted SH3 domain in the yeast ORF YDL117W (Table 1). Because there are known 3D structures of SH3 domains bound to proline-rich peptide ligands, it was possible to calculate a 3D model of such a complex for the putative yeast SH3 domain (Fig. 4). Based on the model, the SH3 residues that interact with the peptide were predicted. This can then be used to construct site-directed mutants with altered or destroyed binding capacity, which in turn could test hypotheses about the sequence–structure–function relationships for this SH3 domain.
Figure 4.
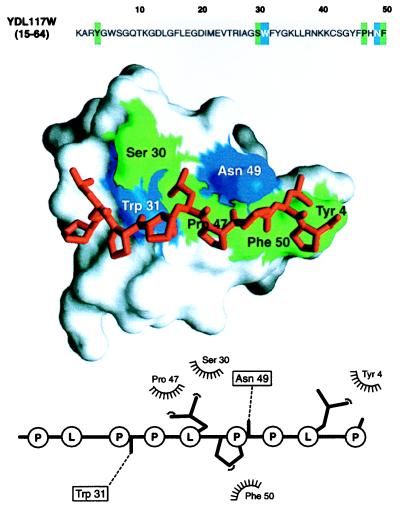
Modeling a putative interaction of a predicted YDL117W SH3 domain with a proline-rich peptide. A segment in the yeast ORF YDL117W sequence (Top) was predicted to be remotely related to the SH3 domains, many of which have known 3D structure (Table 1). The automated prediction was possible because of the sensitivity afforded by evaluating a 3D model implied by the match. The 3D model of the SH3 domain in turn allowed us to address the biochemical function of YDL117W by calculating a 3D model of a complex between the predicted SH3 domain and a putative ligand, a proline-rich peptide (Middle). Inspection of the YDL117W sequence revealed that there is a proline-rich segment downstream from the putative SH3 domain (PLPPLPPLP, positions 212–220). Because this peptide contains the signature PXXP sequence typical of the SH3 binding peptides (39), it was the ligand chosen for the modeling of the complex; both inter- and intramolecular interactions between SH3 domains and Pro-rich peptides already have been documented (39). A model of the complex was obtained by the same comparative method as the model of the SH3 domain (15), relying on the crystallographic structure of the complex between the FYN SH3 domain and its peptide ligand (PPAYPPPPVP) (40). The predicted SH3 domain is shown in the surface representation (41), with the ball-and-stick model of the peptide (red) lying in the binding site. The SH3 residues making hydrophobic contacts and hydrogen bonds to the ligand peptide are colored in green and blue, respectively. The bottom panel shows a schematic representation of the SH3-peptide interaction (42). The peptide atoms that interact with the SH3 residues are shown as filled spheres, hydrogen bonds are represented by dashed lines, and hydrophobic interactions are indicated by the spiked semicircles. This model facilitates designing experiments such as site-directed mutagenesis for maping of functionally important residues on the SH3 domain and its ligand. This should be compared to the starting point at which no functional information about this ORF or about the proteins related to it was known. More generally, the wealth of information in the bottom two panels relative to the top, sequence-only panel is a case in point for the utility of structural models in planning biological experiments (see also text). For the many proteins whose structures have not been determined by experiment, maximal structural information is obtained by both (i) establishing a match to a known protein structure and (ii) calculating an all-atom 3D model based on that match by using the methods described in this paper.
Conclusion.
Although an experimental structure or a comparative model are generally insufficient on their own to infer the biological function of a protein, they many times are complementary to sequence analysis and direct experiment. Our results show that comparative modeling efficiently increases the value of sequence information from the genome projects, although it is not yet possible to model all proteins with useful accuracy. The main bottlenecks are the absence of structurally defined members in many protein families and the difficulties in detection of weak similarities, both for fold recognition and sequence–structure alignment. However, although only 400 out of the total of a few thousand domain folds are known (36, 37), the structure of most globular folds likely is to be determined in <10 years (36). Thus, comparative modeling conceivably will be applicable to most of the globular protein domains close to the completion of the human genome project.
Acknowledgments
We are grateful to Drs. Azat Badretdinov, Stephen K. Burley, David Cowburn, John Kuriyan, and Richard L. Stevens for discussions about this project, to Dr. Stephen F. Altschul for the align program, to Dr. Rok Sosič of ActiveTools for the clustor program, and to Dr. Steve A. Chervitz for making links from the Saccharomyces Genome Database to our web site. R.S. is a Howard Hughes Medical Institute predoctoral fellow. A.Š. is a Sinsheimer Scholar and an Alfred P. Sloan Research Fellow. The investigation also has been aided by grants from National Institutes of Health (GM 54762) and National Science Foundation (BIR-9601845).
ABBREVIATIONS
- 3D
three-dimensional
- PDB
Protein Data Bank
Note Added in Proof
Two recent studies relied on sequence profile methods to assign folds to parts of 38% (44) and 37% (45) of the proteins in the M. genitalium genome.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Oliver S G. Nature (London) 1996;379:597–600. doi: 10.1038/379597a0. [DOI] [PubMed] [Google Scholar]
- 2.Koonin E V, Mushegian A R. Curr Opin Gen Dev. 1996;6:757–762. doi: 10.1016/s0959-437x(96)80032-3. [DOI] [PubMed] [Google Scholar]
- 3.Dujon B. Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- 4.Orengo C A, Jones D T, Thornton J M. Nature (London) 1994;372:631–634. doi: 10.1038/372631a0. [DOI] [PubMed] [Google Scholar]
- 5.Miklos G L G, Rubin G M. Cell. 1996;86:521–529. doi: 10.1016/s0092-8674(00)80126-9. [DOI] [PubMed] [Google Scholar]
- 6.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldman H, Galibert F, Hoheisel J D, Jacq C, Johnston M, et al. Science. 1996;274:563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 7.Chothia C, Lesk A M. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto R, Šali A, Ghildyal N, Karplus M, Stevens R L. J Biol Chem. 1995;270:19524–19531. doi: 10.1074/jbc.270.33.19524. [DOI] [PubMed] [Google Scholar]
- 9.Abola E E, Bernstein F C, Bryant S H, Koetzle T, Weng J. In: Crystallographic Databases: Information, Content, Software Systems, Scientific Applications. Allen F H, Bergerhoff G, Sievers R, editors. Bonn: Data Commission of the International Union of Crystallography; 1987. pp. 107–132. [Google Scholar]
- 10.Benson D A, Boguski M S, Lipman D J, Ostell J, Ouellette B F F. Nucleic Acids Res. 1997;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson M S, Srinivasan N, Sowdhamini R, Blundell T L. CRC Crit Rev Biochem Mol Biol. 1994;29:1–68. doi: 10.3109/10409239409086797. [DOI] [PubMed] [Google Scholar]
- 12.Sánchez R, Šali A. Curr Opin Struct Biol. 1997;7:206–214. doi: 10.1016/s0959-440x(97)80027-9. [DOI] [PubMed] [Google Scholar]
- 13.Altschul S F. Proteins. 1998;32:88–96. [PubMed] [Google Scholar]
- 14.Altschul S F, Gish W. Methods Enzymol. 1996;266:460–480. doi: 10.1016/s0076-6879(96)66029-7. [DOI] [PubMed] [Google Scholar]
- 15.Šali A, Blundell T L. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 16.Sánchez, R. & Šali, A. (1997) Proteins 1, Suppl., 50–58. [DOI] [PubMed]
- 17.Dunbrack R L, Jr, Gerloff D I, Bower M, Chen X, Lichtarge O, Cohen F E. Fold Des. 1997;2:R27–R42. doi: 10.1016/S1359-0278(97)00011-4. [DOI] [PubMed] [Google Scholar]
- 18.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 19.Sippl M J. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 20.Box G E P, Tiao G C. Bayesian Inference in Statistical Analysis. Wiley Interscience: New York; 1992. [Google Scholar]
- 21.Peitsch M C, Wilkins M R, Tonella L, Sánchez J C, Appel R D, Hochstrasser D F. Electrophoresis. 1997;18:498–501. doi: 10.1002/elps.1150180326. [DOI] [PubMed] [Google Scholar]
- 22.Bowie J U, Lüthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S F, Madden T L, Schaffer A A, Zhang J Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogh A, Brown M, Mian I S, Sjolander K, Haussler D. J Mol Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 25.Levitt, M. (1997) Proteins 1, Suppl., 92–104. [DOI] [PubMed]
- 26.Fischer D, Eisenberg D. Proc Natl Acad Sci USA. 1997;94:11929–11934. doi: 10.1073/pnas.94.22.11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonnhammer E L L, Eddy S R, Durbin R. Proteins. 1997;28:405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 28.Guenther B, Onrust R, Šali A, O’Donnell M, Kuriyan J. Cell. 1997;91:335–345. doi: 10.1016/s0092-8674(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 29.Wolf E, Vassilev A, Makino Y, Šali A, Nakatani Y, Burley S K. Cell. 1998;94:51–61. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 30.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 31.Lüthy R, Bowie J U, Eisenberg D. Nature (London) 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 32.Xu L Z, Sánchez R, Šali A, Heintz N. J Biol Chem. 1996;271:24711–24719. doi: 10.1074/jbc.271.40.24711. [DOI] [PubMed] [Google Scholar]
- 33.Bairoch A. Nucl Acids Res. 1992;20:2013–2018. doi: 10.1093/nar/20.suppl.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pawson T. Nature (London) 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 35.Wallace A, Borkakoti N, Thornton J M. Protein Sci. 1997;6:2308–2323. doi: 10.1002/pro.5560061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm L, Sander C. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 37.Hubbard T J P, Murzin A G, Brenner S E, Chothia C. Nucleic Acids Res. 1997;25:236–239. doi: 10.1093/nar/25.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shilton B H, Li Y, Tesier D, Thomas D Y, Cygler M. Protein Sci. 1996;5:395. doi: 10.1002/pro.5560050225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sicheri F, Kuriyan J. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 40.Musacchio A, Saraste M, Wilmanns M. Nat Struct Biol. 1994;1:546–551. doi: 10.1038/nsb0894-546. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 42.Wallace A C, Laskowski R A, Thornton J M. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 43.Fetrow J S, Skolnick J. J Mol Biol. 1998;281:949–968. doi: 10.1006/jmbi.1998.1993. [DOI] [PubMed] [Google Scholar]
- 44.Rychlewski L, Zhang B, Godzik A. Fold Des. 1998;3:229–238. doi: 10.1016/S1359-0278(98)00034-0. [DOI] [PubMed] [Google Scholar]
- 45.Huynen M, Doerks T, Eisenhaber F, Orengo C, Sunyaev S, Yuan Y, Bork P. J Mol Biol. 1998;280:323–326. doi: 10.1006/jmbi.1998.1884. [DOI] [PubMed] [Google Scholar]