

Abstract
Genetic events leading to the loss of heterozygosity (LOH) have been shown to play a crucial role in the development of cancer. However, LOH events do not occur only in genetically unstable cancer cells but also have been detected in normal somatic cells of mouse and man. Mice, in which one of the alleles for adenine phosphoribosyltransferase (Aprt) has been disrupted by gene targeting, were used to investigate the potency of carcinogens to induce LOH in vivo. After 7,12-dimethyl-1,2-benz[a]anthracene (DMBA) exposure, a 3-fold stronger mutagenic response was detected at the autosomal Aprt gene than at the X chromosomal hypoxantine-guanine phosphoribosyltransferase (Hprt) gene in splenic T-lymphocytes. Allele-specific PCR analysis showed that the normal, nontargeted Aprt allele was lost in 70% of the DMBA-induced Aprt mutants. Fluorescence in situ hybridization analysis demonstrated that the targeted allele had become duplicated in almost all DMBA-induced mutants that displayed LOH at Aprt. These results indicate that the main mechanisms by which DMBA caused LOH were mitotic recombination or chromosome loss and duplication but not deletion. However, after treatment with the alkylating agent N-ethyl-N-nitrosourea, Aprt had a similar mutagenic response to Hprt while the majority (90%) of N-ethyl-N-nitrosourea-induced Aprt mutants had retained both alleles. Unexpectedly, irradiation with x-rays, which induce primarily large deletions, resulted in a significant increase of the mutant frequency at Hprt but not at Aprt. This in vivo study clearly indicates that, in normal somatic cells, carcinogen exposure can result in the induction of LOH events that are compatible with cell survival and may represent an initiating event in tumorigenesis.
Tumorigenesis is an in vivo process that can be caused by exposure to various environmental factors, such as chemical or physical agents (1). Epidemiological studies have shown that exposure to tobacco smoke, aflatoxin B1, and UV light causes an increase in the rate of lung, liver, and skin cancers, respectively, in humans. Mutational spectra analysis in these cancers have revealed characteristic fingerprints associated with exposure to benzo[a]pyrene, aflatoxin B1, and UV light (2, 3). However, such strong relationships between genotoxic exposure, mutations, and cancer are not well defined for other human cancers. For example, the role of diet in the induction of colorectal cancer is still unclear because the most characteristic mutations found in p53 and APC tumor suppressor genes are C to T transitions, probably caused by spontaneous deamination of 5-methylcytosine residues (4). However, if predominantly genetic alterations are induced that result in loss of the wild-type allele, a carcinogen-specific fingerprint will not be detectable. This type of genetic events has been shown to be a major cause for loss of heterozygosity (LOH) in sporadic tumors as well as hereditary tumors with a germ-line mutation in one allele of a tumor suppressor gene such as Rb-1, WT-1, or p53 (5). Apart from intragenic mutations, the underlying mechanisms of LOH include chromosomal events such as mitotic recombination, mitotic nondisjunction, gene conversion, and deletion (6, 7).
Although it is not known whether genetic events causing LOH can initiate the neoplastic process, it is clear that LOH can take place in an early stage of tumorigenesis. For example, mutation of one allele of the APC tumor suppressor gene and consequent loss of the remaining allele are the first events in the formation of sporadic intestinal tumors (8). It already has been reported that LOH events can occur in normal somatic cells of mice and man that are, in contrast to tumor cells, genetically stable and have not undergone multiple genetic changes (9–12). However, little information is available on the relative contribution of the various mechanisms leading to LOH in normal diploid cells in vivo and what factors are involved. We and others have studied the mechanisms underlying LOH by using mice heterozygous for the autosomal adenine phosphoribosyltransferase (Aprt) gene on chromosome 8 (11, 13). These Aprt+/− mouse models have been generated by using a conventional gene targeting approach (13, 14) and allow the study of mutations occurring in vivo at both Aprt and Hprt. Recovery of mutants at the hemizygous X chromosomal Hprt locus is limited to intragenic mutations and deletions whereas at the autosomal Aprt locus both intragenic mutations and chromosomal changes can be monitored. In somatic cells from Aprt+/− mice or human individuals heterozygous for APRT, background mutant frequencies were found to be substantial higher at the Aprt locus than at the Hprt locus (9, 13). Molecular analysis of Aprt-deficient mutants showed that these differences were mainly caused by LOH events at the Aprt locus, i.e., mitotic recombination, which cannot be recovered at the Hprt locus (10–13).
The present study focuses on the role of carcinogens in inducing LOH in somatic cells in vivo. Aprt+/− mice were exposed to the model compounds 7,12-dimethyl-1,2-benz[a]anthracene (DMBA) and N-ethyl-N-nitrosourea (ENU) as well as to x-rays. The polycyclic aromatic hydrocarbon DMBA induces bulky DNA lesions, mainly at guanine and adenine residues, whereas the direct acting agent ENU ethylates O- and N-atoms of all four types of bases in the DNA, and x-rays induce mainly DNA strand breaks. All of these genotoxic agents induce high frequencies of Hprt mutants in mouse or rat somatic cells in vivo and are potent rodent carcinogens (refs. 15–17 and A. D. Tates, personal communication). In the present study, mutations were detected at the Aprt and Hprt loci in splenic T-lymphocytes by using a mouse splenocyte clonal assay (18). The Aprt mutant splenic T-lymphocyte clones induced by DMBA, ENU, or x-rays were analyzed at the molecular level by using an allele-specific PCR analysis. Dual colored fluorescence in situ hybridization (FISH) analysis was performed to investigate which mechanism is primarily responsible for LOH at the Aprt locus.
MATERIALS AND METHODS
Mice.
The generation of Aprt+/− mice in which one copy of the Aprt gene was inactivated by gene targeting with a neo construct in embryonic stem cells is described elsewhere (13). Aprt+/− mice used in these mutational studies were obtained by intercrossing either F1 (50% Ola129, 50% C57BL/6) heterozygotes or crossing F1 heterozygous mice with F1 homozygous deficient mice (Aprt+/− × Aprt+/− or Aprt+/− × Aprt−/−). Genotypes were determined by allele-specific multiplex PCR analysis of DNA isolated from tail tips. DNA isolation was carried out by the salting-out technique as described (19).
In the PCR, 3 primers were used:
aprt 531, 5′-CCCCAGGTCCAGAAGACTAG-3′;
aprt 831, 5′-CACGCTAAACTCACGTCAATC-3′ and;
Ums 1, 5′-GGGTTTGATATGCGTGCACAG-3′.
The PCR reaction was performed in a mix containing 6.7 mM MgCl2, 16.6 mM (NH4)2SO4, 5 mM 2-mercaptoethanol, 6.8 mM EDTA, 67 mM Tris⋅HCl (pH 8.8), 10% dimethyl sulfoxide, 0.2 mM of each of the four deoxyribonucleotide triphosphates, 20 pmol of each of the PCR primers, and 1.5 units of Amplitaq polymerase (Perkin–Elmer) in a total volume of 50 μl. After an initial denaturation step at 93°C for 5 min, 35 cycles of 1 min at 94°C, 1 min at 55°C, and 3 min at 72°C were performed in a Thermal Cycler (Perkin–Elmer). The wild-type allele (aprt 531–aprt 831) was identified as a 300-bp PCR product, and the targeted allele (aprt 531–Ums 1) was identified as a 170-bp PCR product.
Chemicals and Exposure.
Both male and female Aprt+/− mice were used that were 8–12 weeks of age at the time of treatment. ENU (Pfaltz and Bauer) was purified as described (20), and, shortly before use, it was dissolved in Sørensen’s phosphate buffer (pH 6.0). ENU was administered i.p. at single doses up to 100 mg/kg. DMBA (Sigma) was dissolved in tricaprilyn (Fluka) and was administered i.p. at single doses up to 60 mg/kg. For x-rays, Aprt+/− mice received a whole body exposure up to 3 Gy. The number of untreated control animals per experiment varied between two and nine mice. For the mutagen-treatments, 4–10 mice per dose group were used. Seven weeks after treatment, mice were killed, and spleens were isolated.
Isolation and Culturing of Splenic T-Lymphocytes.
Culture medium. Priming and cloning of T-lymphocytes was done in RPMI culture medium 1640 as described by Tates et al. (18) with some minor modifications. The serum-free medium DCCM-1 was replaced by AIM-V (Life Technologies, Grand Island, NY), and the antibiotics consisted of 100 units/ml penicillin and 100 μg/ml streptomycin sulfate.
Isolation and priming of T-lymphocytes.
Mouse T-lymphocytes were isolated from the spleen by rubbing the spleen through a sterile 70-μm nylon mesh (Falcon Cell Strainer, 2350) and subsequently were frozen in RPMI medium 1640 supplemented with 10% dimethyl sulfoxide and 40% fetal bovine serum by using a Cryomed freezing apparatus (Forma Scientific, Marietta, OH). When required, frozen cells were thawed at 37°C and immediately were stored on ice. Subsequently, 10 ml of RPMI medium 1640 supplemented with 40% fetal bovine serum was added slowly to the cells. Priming of the cells was performed in 15 ml of culture medium supplemented with 4 μg/ml Con A (Pharmacia) for 44 hr at 37°C, 5% CO2.
Selection of 6-thioguanine- and 8-azaadenine-resistant mutants.
Stimulated T-lymphocytes were cultured and selected for Hprt deficiency in the presence of lethally irradiated feeder cells as described by Tates et al. (18). Similar conditions were used to select Aprt-deficient mutants except that 50 μg/ml 8-azaadenine (8-AA; Sigma) was used as a selecting agent. The Sp2/0 feeder cells were mouse lymphoblastoid cells irradiated with 30 Gy of x-rays. After incubation for 6–8 days at 37°C, 5% CO2, plates were scored for colony growth by using an inverted microscope. Cloning efficiencies and mutant frequencies were calculated as described (18).
Molecular Analysis of Aprt-Deficient Mutants.
8-AA-resistant (8-AAr) clones were selected and diluted 1:3 in culture medium containing 8-AA (50 μg/ml). After 3 days of culturing, crude cell lysates were made (21). Cells were incubated for 1 hr at 55°C in 100 μl of Nonidet-lysis buffer (50 mM KCl/10 mM Tris⋅HCl, pH 8.3/2.5 mM MgCl2/0.1 mg/ml gelatin/0.45% Nonidet P40/0.45% Tween 20) with 60 μg/ml proteinase K. Proteinase K was heat inactivated for 10 min at 95°C. A portion (10 μl) of this crude cell lysate was used in an allele-specific multiplex PCR in a total volume of 50 μl, as described for the tail tips.
FISH.
Dilution and culturing of clones was performed as described for molecular analysis of Aprt mutants, followed by a second 1:2 dilution. After another 3 days of culturing, colcemid (0.1 μg/ml) was added to the cells 2 hr before harvesting. The cells were fixed by the conventional methanol-acetic acid procedure after a hypotonic shock with 0.075 M KCl at 4°C. Cell suspensions were dropped on clean slides at room temperature.
Probes used for FISH analysis were (i) a 20-kilobase genomic SalI fragment containing the mouse Aprt gene in pGEMEX-2 (APRT) and (ii) a 2-kilobase PGKneopA fragment in a pSPORT-1 vector (NEO).
The probes were labeled with either biotin-16-dUTP or digoxigenin-11-dUTP (Boehringer Mannheim) in a standard nick translation procedure. Dual-colored FISH was carried out by using avidin fluorescein isothiocyanate and Texas Red as described (22) with minor modifications. In the case of APRTbio/NEOdig hybridization, 250 ng of APRTbio was preannealed with 10 μg of mouse Cot-1 DNA for 20–30 min, after which 250 ng of NEOdig was added. The APRTdig/NEObio hybridization was performed without preannealing, and the hybridization mix contained 250 ng of each of the probes and 10 μg of mouse-Cot-1 DNA. Pretreatment of the slides, hybridization, washing, and immunochemical detection was performed as described (23). All metaphases on a slide were evaluated. The dual-colored spots (yellow) at chromosome 8 were checked with separate microscope filters for the individual colors (red and green) to ascertain colocalization of the two probes.
RESULTS
Carcinogen-Induced Aprt and Hprt Mutant Frequencies.
Young adult Aprt+/− mice were exposed to different exposure levels of DMBA, ENU, or x-rays. Seven weeks after treatment, both Aprt and Hprt mutant frequencies were determined in the same splenic T-lymphocyte cell population of each animal. The mean cloning efficiency (cloning efficiency ± SEM) per dose group varied between 9.5 (± 0.4%) and 20.5% (± 6.6%). After DMBA-treatment, a dose-dependent, significant increase in mutant frequency was seen at both the Aprt and Hprt genes (Fig. 1A). At equal dose levels, however, DMBA was 3- to 4-fold more mutagenic at the Aprt locus than at the Hprt locus. ENU also induced 8-AAr and 6-thioguanine-resistant mutants in a dose-dependent manner, the induction curves for mutant frequencies having the same kinetics (Fig. 1B). Exposure to different doses of x-rays resulted in a clear increase in the Hprt mutant frequency, which is linear with dose (Fig. 1C). Surprisingly, mutant frequencies at Aprt were not significantly different between x-irradiated and control animals, although there appeared to be a slight increase in Aprt mutant frequency at the highest dose. However, the high background mutant frequencies at Aprt versus Hprt (7.4 ± 1.2 × 10−6 vs. 0.6 ± 0.4 × 10−6 in this experiment) made it more difficult to detect small increases in mutant frequency at Aprt than at Hprt. No differences in mutant frequencies were found between males and females for all three treatments.
Figure 1.

Mutant frequencies (± SEM) in splenic T-lymphocytes of carcinogen-exposed Aprt+/− mice. Mutant frequencies were determined at two endogenous loci, i.e., Aprt (closed triangles) and Hprt (closed circles) after exposure to either DMBA (A), ENU (B), or x-rays (C).
These results indicate (i) that both the Aprt and Hprt loci appear to be similarly sensitive targets to the types of mutations induced by ENU, (ii) that DMBA induces a class of mutations at the Aprt locus that is not detectable at the Hprt locus, and (iii) that the hemizygous Hprt locus is a better target for the recovery of x-ray-induced genetic alterations than is the heterozygous Aprt locus.
Molecular Analysis of x-ray-, DMBA-, and ENU-Induced Mutants at the Aprt Locus.
An allele-specific PCR-analysis was performed to detect the extent of loss of the normal, nontargeted Aprt allele in isolated mutants obtained from mice exposed to DMBA (40 and 60 mg/kg), ENU (60 and 100 mg/kg), or x-rays (2 and 3 Gy) (Fig. 2). After DMBA treatment, 70% of the mutant clones had lost the normal Aprt allele (64 of 92 mutants; Table 1). However, 70% of the DMBA-induced Hprt mutant clones still produced Hprt mRNA as determined by reverse transcription–PCR (data not shown). This finding suggests that deletions play at most a minor role in DMBA mutagenesis at Hprt. Among the ENU-induced mutants, only 13 of 125 Aprt mutants had lost the normal Aprt allele whereas x-rays induced loss of the normal Aprt locus in 30 of 43 mutants (70%) analyzed (Table 1). It should be noted that spontaneous mutants may contribute significantly to this latter set of mutants because x-rays caused only a small increase of the Aprt mutant frequency above the background level and 69% of the spontaneous Aprt mutants lack the normal Aprt allele (Table 1).
Figure 2.

Allele-specific PCR analysis for the detection of loss of the wild-type Aprt allele in Aprt mutant clones. Loss of heterozygosity in A, DMBA-induced 8-AAr clones; in B, ENU-induced 8-AAr clones; in C, x-ray-induced 8-AAr clones. All lanes represent different mutant clones. The normal allele is shown as a 300-bp fragment; the targeted allele is shown as a 170-bp fragment.
Table 1.
Allele-specific PCR analysis of Aprt mutant clones
| Total no. of clones | No. of clones lacking the nontargeted Aprt allele | LOH*, % | |
|---|---|---|---|
| Background† | 140 | 97 | 69 |
| DMBA | 92 | 64 | 70 |
| ENU | 125 | 13 | 10 |
| x-rays | 43 | 30 | 70 |
Loss of heterozygosity.
Ref. 13.
FISH Analysis of Mutant Clones.
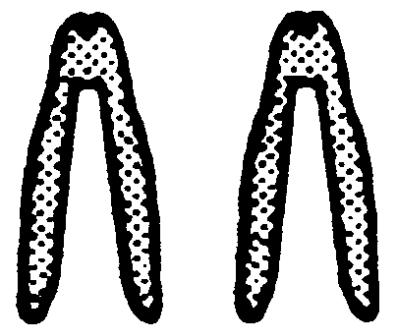
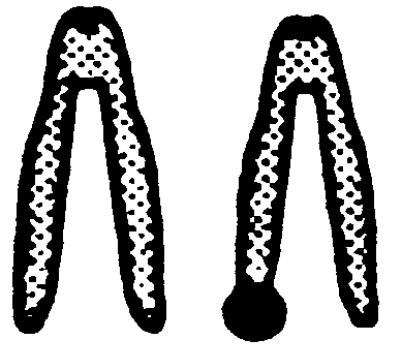
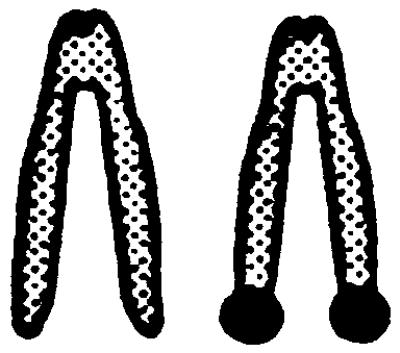
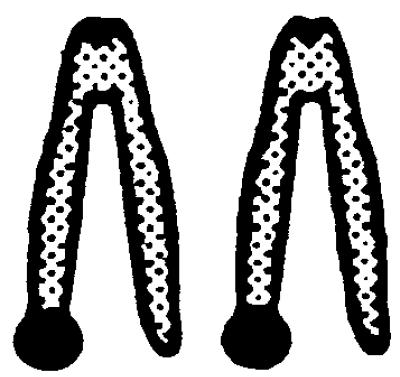
The underlying mechanisms by which LOH events arise include deletion, nondisjunction with or without chromosome duplication, mitotic recombination, and gene conversion. To investigate which mechanism is responsible for LOH at the Aprt locus in mutagen-induced 8-AAr clones, a dual-colored FISH analysis was performed by using an APRT- and a NEO-specific probe. In Aprt+/− cells, the APRT-specific probe will hybridize to both chromosomes 8 because it recognizes the normal and targeted Aprt allele whereas the NEO-probe will hybridize solely to chromosome 8 harboring the targeted Aprt allele. Therefore, in metaphase spreads of nonmutated cells, the chromosome 8 homologue containing the targeted Aprt gene will have a dual-colored signal at both chromatids whereas the other homologue of chromosome 8 will contain a single-colored signal at both chromatids. The efficiency of the FISH analysis was determined by using Aprt+/− cells. In 10–22% of the metaphases, both chromatids of the targeted chromosome 8 contained a dual-colored spot (Table 2), which is the expected efficiency of FISH with small probes (24) such as the 2-kilobase NEO-probe used in these experiments.
Table 2.
Dual-colored FISH analysis of 2 Aprt+/− clones and 20 Aprt mutant clones
| Clone | Total no. of metaphases | Hybridization patterns on chromosomes 8
|
LOH by PCR§ | ||||||
|---|---|---|---|---|---|---|---|---|---|
 |
 |
 |
 |
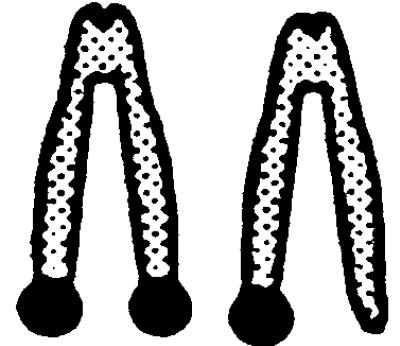 |
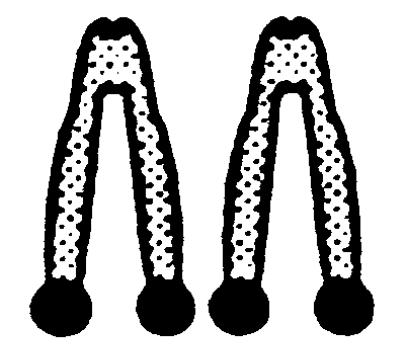 |
||||
| Aprt+/−* | 1 | 30 | 17 | 10 | 3 | 0 | 0 | 0 | |
| 2 | 100 | 46 | 32 | 22 | 0 | 0 | 0 | ||
| DMBA† | I.1 | 51 | 19 | 16 | 8 | 3 | 5 | 0 | Y* |
| I.2 | 15 | 12 | 1 | 2 | 0 | 0 | 0 | N | |
| I.3 | 51 | 7 | 4 | 25 | 3 | 1 | 11 | Y* | |
| I.4 | 96 | 36 | 34 | 11 | 13 | 1 | 1 | Y* | |
| I.5 | 24 | 9 | 9 | 5 | 1 | 1 | 0 | Y | |
| II.1 | 176 | 74 | 47 | 31 | 19 | 2 | 3 | Y* | |
| II.2 | 59 | 23 | 19 | 16 | 1 | 0 | 0 | N | |
| II.3 | 46 | 36 | 6 | 4 | 0 | 0 | 0 | N | |
| III.1 | 84 | 24 | 14 | 25 | 4 | 13 | 4 | Y* | |
| III.2 | 101 | 3 | 13 | 37 | 14 | 18 | 16 | Y* | |
| III.3 | 28 | 17 | 6 | 4 | 0 | 1 | 0 | N | |
| ENU‡ | IV.1 | 71 | 35 | 21 | 14 | 0 | 0 | 1 | N |
| IV.2 | 61 | 34 | 12 | 12 | 0 | 3 | 0 | N | |
| IV.3 | 19 | 15 | 4 | 0 | 0 | 0 | 0 | N | |
| IV.4 | 37 | 21 | 10 | 5 | 0 | 1 | 0 | N | |
| IV.5 | 53 | 5 | 22 | 23 | 2 | 1 | 0 | N | |
| V.1 | 61 | 28 | 7 | 22 | 3 | 2 | 1 | N | |
| V.2 | 105 | 61 | 36 | 8 | 0 | 0 | 0 | N | |
| V.3 | 166 | 106 | 49 | 11 | 0 | 0 | 0 | N | |
| V.4 | 74 | 21 | 28 | 21 | 3 | 1 | 0 | N | |
Nonselected Aprt+/− clones* were isolated from cloning efficiency plates. 8-AAr mutant clones were derived from five different animals (I, II, III, IV, V) treated with either 40 mg/kg DMBA
or 100 mg/kg ENU
.  represents a metaphase chromosome 8; • represents simultaneous staining of the APRT- and NEO-probes. All clones also were analyzed by allele-specific PCR and were checked for loss of heterozygosity
represents a metaphase chromosome 8; • represents simultaneous staining of the APRT- and NEO-probes. All clones also were analyzed by allele-specific PCR and were checked for loss of heterozygosity
. The ratio of the number of dual-colored spots at both chromosomes 8 (2, 3, or 4 spots) divided by the number of dual-colored spots at one chromosome 8 (1 or 2 spots) was used to determine whether duplication of the targeted Aprt allele had occurred in the mutant clones showing LOH by PCR. In the clones marked with an asterisk, a statistically significant difference (χ2 test, P < 0.0001) was found in this ratio compared to the ratio for the combined group of DMBA- and ENU-induced mutants without LOH by PCR, indicating that, in the marked clones, the targeted Aprt allele had become duplicated.
In all clones tested, the majority of the metaphases (85–90%) were diploid. To determine whether the targeted Aprt allele had become duplicated in mutant clones, the ratio of the number of dual-colored spots at both chromosomes 8 (two, three, or four) divided by the number of dual-colored spots at one chromosome 8 (one or two) of the individual mutants was calculated. In six of seven DMBA-induced 8-AAr clones that showed loss of the nontargeted Aprt allele by PCR (I.1, I.3, I.4, II.1, III.1, and III.2; Table 2), this ratio was significantly different (P < 0.0001) from the ratio for the combined group of DMBA- and ENU-induced mutants that had retained the nontargeted allele (Table 2). This result indicates that, in these six mutants, both chromosomes 8 contain the neo-insert (Fig. 3). Apparently, DMBA-induced LOH at the Aprt locus in Aprt+/− splenic T-lymphocytes frequently is accompanied by duplication of the targeted allele.
Figure 3.
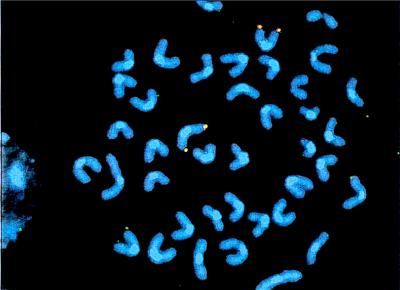
Dual-colored FISH-analysis of a DMBA-induced Aprt mutant clone. Duplication of the targeted Aprt allele can be recognized because both chromosomes 8 are hybridized with the APRT- (red) and NEO-specific (green) probe. Colocalization of these two probes gives rise to a red/yellow spot at the telomeric end of chromosome 8.
In all nine ENU-induced 8-AAr clones, no LOH of the Aprt locus was detected by using allele-specific PCR. These mutants contained almost exclusively dual-colored signals at the chromosome 8 harboring the targeted allele and single-colored signals at the other chromosome 8 (Table 2). However, occasionally, because of relatively high background signals of the NEO-probe, some metaphases were detected that contained dual-colored spots at both chromosomes 8 (Table 2).
DISCUSSION
To gain information on the nature of adducts that are capable of inducing LOH in vivo, Aprt+/− mice were exposed to three mutagens causing different types of DNA-alterations, i.e., bulky lesions, alkylation damage, and DNA breaks. Exposure of Aprt+/− mice to the potent rodent carcinogen DMBA (17), a model-agent for polycyclic aromatic hydrocarbons, resulted in a 3- to 4-fold increase in the induction of Aprt mutant clones compared with the induction of Hprt mutant clones, at the same exposure level. Dual-colored FISH analysis revealed that six of seven DMBA-induced Aprt mutants showing LOH had lost the normal, nontargeted Aprt allele whereas the targeted Aprt allele had become duplicated. DMBA-induced mutations at the Hprt locus consisted presumably of intragenic mutations, as was found in rats in which the majority of DMBA-induced Hprt mutants carried bp substitutions (16). ENU treatment of Aprt+/− mice gave rise to comparable mutant frequencies at the Aprt gene and the Hprt gene by using the same dose. The fraction of Aprt mutants that showed LOH by allele-specific PCR was small (10%). Mutational spectra analysis at the Hprt gene in skin fibroblasts or T-lymphocytes of rats, mice, and monkeys exposed in vivo to ENU have shown that the predominant types of mutation were transversions at A:T base pairs probably caused by O-ethylated thymines mispairing with thymine or cytosine during DNA replication (15, 25, 26).
The relative potency of DMBA and ENU to induce chromosomal type of LOH events as shown here is in good agreement with literature data. DMBA-induced mouse skin carcinomas had a wide spectrum of alterations at chromosome 7, including trisomy, mitotic recombination, deletion, and gene duplication whereas the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine-induced tumors only rarely exhibited chromosomal alterations (27). Also, DMBA-induced mouse mammary tumors showed LOH on chromosomes 4 (in 25% of the analyzed tumors), 8 (in 20%), and 11 (in 30%) (28). Moreover, ENU treatment has been shown to cause more frequently intragenic mutations than recombinational events at the Chinchilla locus by using the spot test in mice (29).
Whole body exposure of Aprt+/− mice to a dose of 1, 2, or 3 Gy of x-rays resulted in a significant mutation induction at the Hprt locus but surprisingly not at the Aprt locus. The predominant type of mutations induced by x-rays have been shown to be large multilocus deletions (30, 31). The recovery of x-ray-induced mutants, therefore, is expected to be better for a heterozygous locus than for a hemizygous locus because concomitant deletion of a nearby essential gene will lead to cell death in the latter case. Indeed, in various mammalian cell lines, a large difference in mutant frequency after x-irradiation was reported between autosomal loci in a heterozygous versus hemizygous configuration (32, 33). In cultured mouse embryonal carcinoma cells, x-ray-induced Aprt mutants contained large chromosomal events at chromosome 8 that were predominantly interstitial deletions (34, 35). However, in this in vivo study, large deletions appear to be tolerated better at the hemizygous Hprt gene than at the heterozygous Aprt gene. The reasons for this discrepancy in sensitivity of the mouse Aprt locus to x-rays are unclear. Genetic events might be induced that are viable in transformed embryonal carcinoma cells but are lethal to normal somatic cells in vivo. Possibly, because of the relatively telomeric location of the mouse Aprt gene, its loss is accompanied by loss of the telomere, resulting in a chromosome instability and reduced cell viability. Alternatively, an essential gene may be located close to the mouse Aprt gene of which a double gene dosage is necessary for cell survival, so that deletion of this gene together with Aprt will result in a cell lethal phenotype. Recently, such a candidate gene, thought to be essential for cell survival, was identified just downstream of the Aprt gene (36). However, hamster and human cells hemizygous for this gene seem to have normal growth characteristics. The weak mutagenic response to x-rays does not only indicate that large deletions are recovered poorly at the Aprt locus but also that LOH events of the type induced by DMBA do not occur after ionizing radiation.
Several cellular mechanisms can result in loss of the normal Aprt allele. The finding that, in most of the DMBA-induced Aprt mutants, the targeted Aprt allele had become duplicated indicates that the main mechanisms causing LOH at the Aprt gene were mitotic recombination or chromosome loss and duplication but not deletion. In contrast, alkylation damage induced by ENU gave rise predominantly to intragenic mutations at Aprt whereas exposure to x-rays resulted in a marginal induction of Aprt mutants. Among spontaneous human and mouse Aprt mutants in vivo, mitotic recombination seems to be the major pathway leading to LOH (10–13). In vitro studies in mammalian cell lines also showed, for both the autosomal APRT and TK genes, that mitotic recombination is the main mechanism leading to LOH in spontaneous and induced mutants (37, 39). Molecular analysis of small-colony mouse lymphoma Tk mutants revealed that, because of somatic recombination, the majority of the mutants had lost the Tk+ allele whereas the Tk− allele had become duplicated (40). Additionally, chromosome loss and duplication appeared to be a major mechanism for LOH in vitro, as determined at the human leukocyte antigen gene in 4 different human lymphoblastoid cell lines (41).
Which aspect of DNA lesions trigger the induction of recombinational events? Studies using duplicated mutant reporter genes have shown that DNA damage-induced intrachromosomal recombination is increased in the absence of nucleotide excision repair, suggesting that single-stranded nicks induced by this repair pathway are not a major cause for induction of recombinational events (42). More likely, blocked DNA replication forks might be the initiating events leading to recombination, as has been suggested for the induction of sister chromatid exchanges (43). Bulky DNA adducts such as those caused by DMBA probably represent a much stronger block for DNA replication than ethylated bases, which are bypassed more readily during DNA replication. If so, the relative potency of a mutagen to induce intragenic versus chromosomal mutations will depend on the ability of DNA replication to bypass the induced mutagenic adducts.
From epidemiological studies, it is known that human cancer rates are influenced by environmental factors, suggesting that environmental mutagens contribute to the induction of somatic mutations during lifetime (1). Carcinogenic polycylic aromatic hydrocarbons have been identified in many foods, including broiled, barbecued, and smoked meat or fish (44). However, there is often no clear relationship between specific carcinogenic components in the diet and cancer (4), and no characteristic mutational fingerprint can be found in colorectal tumors. The results obtained with DMBA-exposed Aprt+/− mice indicate that environmental carcinogens such as polycyclic aromatic hydrocarbons can induce chromosomal types of changes that lead to LOH in normal somatic cells without interfering with cell viability and that may contribute to the initiation of cancer. No mutational fingerprint will appear if a carcinogen predominantly induces chromosomal type of LOH events, providing an explanation for the absence of a carcinogen-specific fingerprint in colorectal cancer.
In the present study, we show that some carcinogens can cause LOH of a housekeeping gene in normal somatic cells in vivo, similar to the alterations causing allelic loss of tumor suppressor genes during cancer development. The mechanisms involved in the generation of LOH now can be analyzed carefully by using Aprt+/− mice crossed with transgenic mouse models defective in pathways that guard genome integrity. These studies should provide more insight into the role of LOH events in early stages of tumorigenesis.
Acknowledgments
We thank Bep Smit for technical assistance in the FISH experiments and Jaap Jansen for critical reading of the manuscript. This work was supported financially by the Dutch Cancer Society (Project 96–1321) and the Division of Medical Sciences of the Netherlands Organization for Scientific Research (Contract 900–568-116).
ABBREVIATIONS
- DMBA
7,12-dimethyl-1,2-benz[a]anthracene
- ENU
N-ethyl-N-nitrosourea
- LOH
loss of heterozygosity
- FISH
fluorescence in situ hybridization
- 8-AA
8-azaadenine
- 8-AAr
8-AA-resistant
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Doll R, Peto R. J Nat Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 2.Harris C C, Hollstein M. New Engl J Med. 1993;329:1318–1319. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 3.Denissenko M F, Pao A, Tang M S, Pfeifer G P. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler K W, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 5.Knudson A G. Proc Natl Acad Sci USA. 1993;90:10914–10921. doi: 10.1073/pnas.90.23.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavenee W K, Dryja T P, Phillips R A, Benedict W F, Godbout R, Gallie B L, Murphree A L, Strong L C, White R L. Nature (London) 1983;305:779–784. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- 7.Smith L E, Grosovsky A J. Mutat Res. 1993;289:245–254. doi: 10.1016/0027-5107(93)90075-q. [DOI] [PubMed] [Google Scholar]
- 8.Fearon E R, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 9.Hakoda M, Nishioka K, Kamatani N. Cancer Res. 1990;50:1738–1741. [PubMed] [Google Scholar]
- 10.Hakoda M, Yamanaka H, Kamatani N. Am J Hum Genet. 1991;48:552–562. [PMC free article] [PubMed] [Google Scholar]
- 11.Stambrook P J, Shao C, Stockelman M, Boivin G, Engle S J, Tischfield J A. Environ Mol Mutagen. 1996;28:471–482. doi: 10.1002/(SICI)1098-2280(1996)28:4<471::AID-EM25>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 12.Gupta P K, Sahota A, Boyadjiev S A, Bye S, Shao C, O’Neill J P, Hunter T C, Albertini R J, Stambrook P J, Tischfield J A. Cancer Res. 1997;57:1188–1193. [PubMed] [Google Scholar]
- 13.Van Sloun, P. P. H., Wȳnhoven, S. W. P., Kool, J. M., Weeda, G., Slater, R., Lohman, P. H. M., van Zeeland, A. A. & Vrieling, H. (1998) Nucleic Acids Res., in press. [DOI] [PMC free article] [PubMed]
- 14.Engle S J, Stockelman M G, Chen J, Boivin G, Yum M N, Davies P M, Ying M Y, Sahota A, Simmonds H A, Stambrook P J, et al. Proc Natl Acad Sci USA. 1996;93:5307–5312. doi: 10.1073/pnas.93.11.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skopek T R, Walker V E, Cochrane J E, Craft T R, Cariello N F. Proc Natl Acad Sci USA. 1992;89:7866–7870. doi: 10.1073/pnas.89.17.7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heflich R H, Mittelstaedt R A, Manjanatha M G, Lyncook L E, Aidoo A. Environ Mol Mutagen. 1996;28:5–12. doi: 10.1002/(SICI)1098-2280(1996)28:1<5::AID-EM3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 17.Gold L S, Zeiger E. Handbook of Carcinogenic Potency and Genotoxicity Databases. Boca Raton, FL: CRC; 1997. [Google Scholar]
- 18.Tates A D, van Dam F J, de Zwart F A, van Teijlingen C C M, Natarajan A T. Mutat Res. 1994;309:299–306. doi: 10.1016/0027-5107(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 19.Miller S A, Dykes D D, Polesky H F. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansen J G, de Groot A J, van Teijlingen C C M, Lohman P H, Mohn G R, Vrieling H, van Zeeland A A. Mutat Res. 1994;307:95–105. doi: 10.1016/0027-5107(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 21.Higuchi R. Amplifications. 1989;2:1–3. [Google Scholar]
- 22.Hagemeijer A, Buijs A, Smit E, Janssen B, Creemers G J, Van der Plas D, Grosveld G. Genes Chromosomes Cancer. 1993;8:237–245. doi: 10.1002/gcc.2870080406. [DOI] [PubMed] [Google Scholar]
- 23.Arnoldus E P, Wiegant J, Noordermeer I A, Wessels J W, Beverstock G C, Grosveld G C, van der Ploeg M, Raap A K. Cytogenet Cell Genet. 1990;54:108–111. doi: 10.1159/000132972. [DOI] [PubMed] [Google Scholar]
- 24.Wiegant J, Galjart N J, Raap A K, d’Azzo A. Genomics. 1991;10:345–349. doi: 10.1016/0888-7543(91)90318-9. [DOI] [PubMed] [Google Scholar]
- 25.Jansen J G, Mohn G R, Vrieling H, van Teijlingen C M M, Lohman P H M, van Zeeland A A. Cancer Res. 1994;54:2478–2485. [PubMed] [Google Scholar]
- 26.Walker V E, Gorelick N J, Andrews J L, Craft T R, Deboer J G, Glickman B W, Skopek T R. Cancer Res. 1996;56:4654–4661. [PubMed] [Google Scholar]
- 27.Bremner R, Kemp C J, Balmain A. Mol Carcinogen. 1994;11:90–97. doi: 10.1002/mc.2940110206. [DOI] [PubMed] [Google Scholar]
- 28.Aldaz C M, Liao Q Y, Paladugu A, Rehm S, Wang H. Mol Carcinogen. 1996;17:126–133. doi: 10.1002/(SICI)1098-2744(199611)17:3<126::AID-MC4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 29.Fahrig R, Steinkamp-Zucht A. Mutat Res. 1996;354:59–67. doi: 10.1016/0027-5107(96)00036-x. [DOI] [PubMed] [Google Scholar]
- 30.Vrieling H, Simons J W, Arwert F, Natarajan A T, van Zeeland A A. Mutat Res. 1985;144:281–286. doi: 10.1016/0165-7992(85)90065-x. [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson F. J Mol Biol. 1995;254:372–380. doi: 10.1006/jmbi.1995.0624. [DOI] [PubMed] [Google Scholar]
- 32.Bradley W E, Belouchi A, Messing K. Mutat Res. 1988;199:131–138. doi: 10.1016/0027-5107(88)90238-2. [DOI] [PubMed] [Google Scholar]
- 33.Evans H H. Radiat Res. 1994;137:131–144. [PubMed] [Google Scholar]
- 34.Turker M, Walker K A, Jennings C D, Mellon I, Yusufji A, Urano M. Mutat Res. 1995;329:97–105. doi: 10.1016/0027-5107(95)00046-l. [DOI] [PubMed] [Google Scholar]
- 35.Turker M S, Pieretti M, Kumar S. Mutat Res. 1997;374:201–208. doi: 10.1016/s0027-5107(96)00230-8. [DOI] [PubMed] [Google Scholar]
- 36.Harwood J, Meuth M. Somatic Cell Mol Genet. 1995;21:151–160. doi: 10.1007/BF02254767. [DOI] [PubMed] [Google Scholar]
- 37.Yandell D W, Dryja T P, Little J B. Mutat Res. 1990;229:89–102. doi: 10.1016/0027-5107(90)90011-r. [DOI] [PubMed] [Google Scholar]
- 38.Li C Y, Yandell D W, Little J B. Somatic Cell Mol Genet. 1992;18:77–87. doi: 10.1007/BF01233450. [DOI] [PubMed] [Google Scholar]
- 39.Fujimori A, Tachibana A, Tatsumi K. Mutat Res. 1992;269:55–62. doi: 10.1016/0027-5107(92)90160-4. [DOI] [PubMed] [Google Scholar]
- 40.Applegate M L, Moore M M, Broder C B, Burrell A, Juhn G, Kasweck K L, Lin P F, Wadhams A, Hozier J C. Proc Natl Acad Sci USA. 1990;87:51–55. doi: 10.1073/pnas.87.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Nooij-van Dalen A G, van Buuren-van Seggelen V H, Mulder A, Gelsthorpe K, Cole J, Lohman P H, Giphart- Gassler M. Mutat Res. 1997;374:51–62. doi: 10.1016/s0027-5107(96)00218-7. [DOI] [PubMed] [Google Scholar]
- 42.Tsujimura T, Maher V M, Godwin A R, Liskay R M, McCormick J J. Proc Natl Acad Sci USA. 1990;87:1566–1570. doi: 10.1073/pnas.87.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rainaldi G, Sessa M R, Mariani T. Chromosoma. 1984;90:46–49. doi: 10.1007/BF00352277. [DOI] [PubMed] [Google Scholar]
- 44.Grimmer G. Environmental Carcinogens: Polycyclic Aromatic Hydrocarbons. Boca Raton, FL: CRC; 1983. [Google Scholar]