

Abstract
The blood-brain barrier (BBB) is dramatically but transiently compromised in the cerebella of myelin basic protein immunized mice at least one week prior to the development of the paralytic phase of experimental allergic encephalomyelitis (EAE). Treatment of mice with the peroxynitrite-dependent radical scavenger uric acid (UA) during the first week after immunization blocks the early increase in cerebellar BBB permeability and the subsequent development of clinical signs of EAE. These results indicate that the early loss of BBB integrity in the cerebellum is likely to be a necessary step in the development of paralytic EAE.
Keywords: experimental allergic encephalomyelitis (EAE), multiple sclerosis (MS), blood-brain barrier (BBB), uric acid (UA), peroxynitrite (ONOO−), cerebellum
1. Introduction
Despite the identification of a variety of factors that contribute to the pathogenesis of multiple sclerosis (MS) and its animal model Experimental Allergic Encephalomyelitis (EAE), the precise mechanisms responsible for the development of the CNS inflammatory lesions that are characteristic of these neurodegenerative diseases remain poorly understood. Lesion formation is clearly associated with enhanced expression of a number of cytokines, chemokines, adhesion molecules and matrix metalloproteinases (MMPs) as well as the accumulation of immune/inflammatory cells in CNS tissues (reviewed Brown, 2001; Fazekas and Tabira, 2000; Hemmer et al., 2002; Imitola et al., 2005). Free radicals including nitric oxide (NO.) and peroxynitrite (ONOO−), the product of NO. and superoxide, have also been implicated in lesion formation both through neurotoxicity and the induction of the changes in blood-brain barrier (BBB) integrity that are associated with immune cell invasion into CNS tissues (Cross et al., 1997; Cross et al., 1998; Hooper et al., 1997; Hooper et al., 2000; Liu et al, 2001; Scott et al., 2001; van der Veen et al., 1997). Studies in EAE have provided considerable insight into the development of immune processes that lead to CNS pathology but information about contributory events in the CNS tissues that precede lesion formation is limited. Just as it is impossible to predict the onset of MS, or the appearance of new plaques in an existing case, it is difficult to predict whether an individual animal will develop EAE following appropriate immunization. For example, while immunized PLSJL mice all develop similar immune responses to myelin basic protein (MBP) by day 10–14 after immunization (Kean et al., 2000), only 30–70% of the mice generally progress to exhibit clinical signs of EAE in a particular experiment (Hooper et al., 1998). Moreover, some animals may become sick as early as day 12 after immunization while others take more than 30 days for symptoms to appear (Fabis et al., 2007). This is likely a reflection of the fact that the development of CNS pathology in EAE is dependent upon contributions from a number of processes including the induction of an appropriate myelin-antigen specific immune response as well as pro-inflammatory changes in the neurovasculature and CNS tissue. The latter are required to promote the invasion of the circulating immune/inflammatory cells responsible for lesion formation across the BBB and into CNS tissues.
The development of a strong pro-inflammatory response in the CNS tissues of mice immunized with myelin antigens, identified by the enhanced expression of TNF-α, is associated with the loss of BBB integrity and the development of clinical signs of EAE (Scott et al., 2004). However, there is evidence of functional changes occurring in the neurovasculature prior to the onset of disease. This includes elevated adhesion molecule expression (Archelos and Hartung, 1999; Kieseir et al., 1999; Scott et al., 2004) and transiently enhanced BBB permeability which has been detected in the cerebella of SJL/J mice 6 days following immunization with proteolipid protein (PLP) peptide aa139-151 (Tonra et al., 2001). While it is clear that the elevation of adhesion molecules on the neurovasculature would facilitate immune/inflammatory cell invasion into CNS tissues, the contribution of transient BBB permeability in the cerebellum to the development of clinical signs of EAE several days later is unknown. We have previously demonstrated that ONOO− makes an important contribution toward the induction of enhanced BBB permeability in EAE. For example, the administration of uric acid (UA), a natural scavenger of peroxynitrite-dependent radicals, prevents the loss of BBB integrity in mice immunized with myelin antigens as well as CNS inflammation and the development of EAE (Hooper et al., 1997, 2000). Our findings indicate that UA treatment suppresses the BBB permeability changes associated with the symptomatic phase of EAE without interfering with the induction of myelin-specific immunity (Kean et al., 2000). With a view toward establishing whether or not this early loss of BBB integrity is an essential step in lesion formation and the development of clinical signs of EAE, in this investigation we have assessed the effects of UA treatment on the transient neurovascular permeability seen in the cerebella of mice immunized 6 days previously with myelin basic protein (MBP) and the subsequent development of disease.
2. Materials and Methods
2.1. Induction of EAE and treatment of mice
Female, 8–10 week old, PLSJL mice (Jackson Laboratory, Bar Harbor, ME) were immunized subcutaneously at 3 sites with 200 µl of an emulsion of 100 µg MBP in complete Freund’s adjuvant containing 0.05% M. Butyricum plus an additional 4mg/ml M. tuberculosis H37 RA (Difco). Pertussis toxin, 400 ng, was given intra-peritoneally twice, on days 0 and 2. Mice were scored twice daily for clinical signs of EAE on the basis of the presence of the following symptoms: 0, normal mouse; 1, piloerection, tail weakness; 2, tail paralysis; 3, tail paralysis plus hindlimb weakness; 4, tail paralysis plus partial hindlimb paralysis; 5, total hindlimb paralysis; 6, hind and forelimb paralysis; 7, moribund/dead. (Hooper et al.,1998). UA (Sigma Chemical Co., St. Louis, MO) was administered i.p. 4 times per day, 10 mg in 100 µl saline. Control groups of mice received saline alone.
2.2. Assessment of BBB permeability
BBB permeability was assessed using sodium-fluorescein (MW 376) as a tracer molecule. Each animal received 100 µl of 10% sodium fluorescein (Sigma) in PBS i.p. After 10 min, mice were anesthetized by i.p. administration of sodium pentobarbital (20 mg/kg body weight), cardiac blood was collected, and the animals were transcardially perfused with PBS-heparin and PBS. Sodium-fluorescein uptake into the spinal cord was determined as detailed previously (Hooper et al., 2000). Briefly, spinal cord tissue was homogenized in 7.5% trichloroacetic acid and centrifuged for 10 min at 10,000 rpm to remove insoluble precipitates. Following the addition of 5 N NaOH, the fluorescence of the sample was determined at excitation 485 nm and emission 530 nm using a Cytofluor II fluorimeter (PerSeptive Biosystems, Cambridge, MA). Serum levels of sodium fluorescein were assessed as described (Hooper et al., 2000). Levels of sodium-fluorescein in spinal cord tissue were normalized against serum levels using the following formula: (g fluorescence spinal cord/mg protein)/ (g fluorescence sera/l blood). To visualize sodium-fluorescein accumulation, the excised brain was exposed to a UV light and photographed using a Nikon digital camera on a Leitz Microlab microscope.
2.3. Quantitative RT-PCR
Peripheral blood was collected into heparinized tubes and white blood cells were isolated by density centrifugation on Lymphoprep (Nycomed, Oslo, Norway) as previously detailed (Plebanski et al., 1992). Mice were anesthetized and transcardially perfused with PBS-heparin (1000 U/l) prior to the collection of organs. RNA was isolated from peripheral blood cells, cerebellum and other organs using TRIzol Reagent (GibcoBRL, Grand Island, NY). DNA contamination was removed by treatment with DNA-free Reagent (Ambion, Austin, TX) according to the manufacturer’s recommendations. cDNA was synthesized from 5µg of total RNA using M-MLV reverse transcriptase (Promega, Madison, WI) and dT15 primer. An equivalent of 50ng of total RNA was used for the PCR reaction with Taqman PCR Core Reagent Kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using a Bio-Rad iCycler iQ Real Time Detection System (Hercules, CA) as described (Hooper et al., 2001; Scott et al., 2004). Primers and probes have been designed using the Web Primer 3 program (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi). Double labeled probes were purchased from Integrated DNA Technologies (Coralville, IA). 5’ ends of probes are labeled with reporter dyes Hex or 6-Fam and 3’ ends with the quencher BHQ-1. Primers were synthesized by the Nucleic Acid Facility, Kimmel Cancer Institute, Thomas Jefferson University (Philadelphia, PA). Data are expressed as the number of copies or fold increase in mRNA expression based on the copy number of a specific mRNA per copy of housekeeping gene. Copy numbers of mRNA are calculated based on internal standards for each gene as detailed (Phares et al., 2006). Two housekeeping genes were tested: GAPDH and L13. Since results using both housekeeping genes were similar (correlation >90%) only GAPDH data are presented. Probes and primers for the following gene-specific mRNAs have been previously described: L-13, ICAM-1, Mac-1 (CD11b), CD4 and CD8 (Phares et al., 2006). Probes and primers for mRNAs specific for GAPDH, inducible nitric oxide synthase (iNOS) and CD68 are presented in Table 1 and primers for the corresponding standards in Table 2. All primers/probe sets had efficiencies in real-time RT-PCR >85%.
Table 1.
Primer and probe sequences used for real time quantitative RT-PCR
| Gene | 5’ Primer | 3’ Primer | |
|---|---|---|---|
| Probe | |||
| GAPDH | GGCAAATTCAACGGCACAG | AGATGGTGATGGGCTTCCC | AGGCCGAGAATGGGAAGCTTGTCATC |
| iNOS | TGGCTACCACATTGAAGAAGCTG | TCTGGCTCTTGAGCTGGAAGAAA | TGGCCACCAAGCTGAACTTGAGCGA |
| CD68 | GTGCTCATCGCCTTCTGCATCA | GGCGCTCCTTGGTGGCTTAC | CCAGCCCCTCTGAGCATCTGCCCC |
Table 2.
Primer sequences used to synthesize cDNA standards for real time quantitative RT-PCR
| Gene | 5’ Primer | 3’ Primer |
|---|---|---|
| GAPDH | GAACGGATTTGGCCGTATTG | GGATGCAGGGATGATGTTCT |
| iNOS | TCCCAGCCTTGCATCCTCATT | TACTCAGTGCCAGAAGCTGGA |
| CD68 | ATACCCAATTCAGGGTGGAAG | GTTGAGTCAGTGGCATGGTG |
2.4. Statistical analyses
Results are expressed as the mean ±S.D. or S.E. for replicate observations as indicated in the figure legends. Evaluation of the significance of differences between parameters was performed using Student’s t- test or Mann-Whitney test as indicated. In all cases, p<0.05 was considered significant.
3. Results
3.1. BBB permeability changes occur in the cerebellum prior to the onset of EAE
It is well established that the severity of the clinical disease seen in most EAE models is correlated with increased BBB permeability in the spinal cord (Fabis et al., 2007). However, a transient reduction in BBB integrity specific to the cerebellum has been reported in PLP139–151 immunized SJL/J mice several days before the onset of symptomatic EAE (Tonra et al., 2001). This observation led us to re-examine the development of elevated BBB permeability in PLSJL mice immunized with MBP. We first compared the amount of leakage of a fluid phase marker, Na-fluorescein, from the circulation into spinal cord versus cerebellar tissue in mice during the clinical phase of EAE. Mice with a mean clinical score of 3.7 exhibited extensive permeability changes in both the cerebellum and spinal cord neurovasculature with the latter being somewhat greater (Figure 1). As previously reported for SJL/J mice (Tonra et al., 2001), PLSJL mice immunized with myelin antigens in adjuvant develop enhanced BBB permeability 6 days later which is approximately 8–14 days prior to the onset of clinical signs of EAE in these animals Figure 2. The administration of complete Freund’s adjuvant or pertussis toxin either separately or in combination has lesser effects on BBB integrity (Figure 2). A small but statistically insignificant increase in Na-fluorescein accumulation was detected in spinal cord tissues but not in the cerebral cortex, kidney, liver, thymus, spleen, or lymph nodes (Table 3).
Figure 1. BBB permeability in the spinal cords and cerebellum of PLSJL mice with EAE.
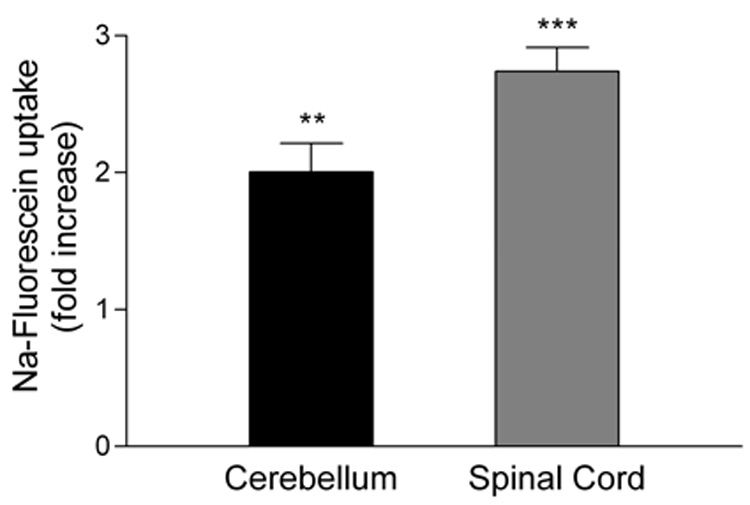
BBB permeability to Na-fluorescein was assessed in non-immune and MBP-immunized mice with clinical EAE (mean score 3.7; range 1–6) collected 18 to 24 days after immunization as described in Materials and Methods. The results are presented as a fold increase in Na-fluorescein content in the MBP immunized group (24 mice) by comparison with that of the non-immune group (10 mice). Statistically significant differences determined by Student’s t test are denoted by ** (p<0.005) and *** (p<0.0005).
Figure 2. The BBB in the cerebellum of MBP-immunized PLSJL mice becomes permeable to Na-fluorescein 6 days after immunization.
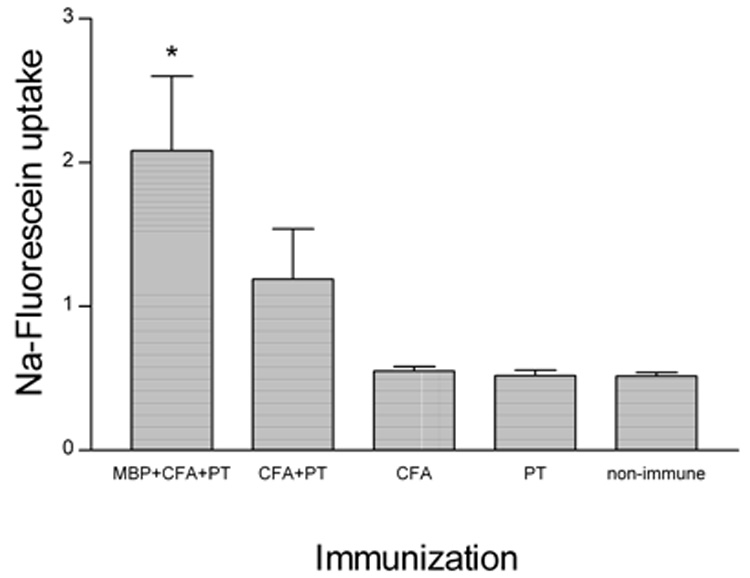
Groups of 6 PLSJL mice were either immunized with MBP in CFA with PT or given the adjuvants separately or in combination without antigen and BBB permeability in the cerebellum was assessed by Na-fluorescein uptake 6 days later as described in Materials and Methods. As determined by Student’s t test, Na-fluorescein uptake was only significantly elevated over non-immune mice in those receiving MBP in CFA with PT (*; p<0.05).
Table 3.
Na-fluorescein uptake by various organs
| Tissue source | Na-fluorescein uptake | Significance (p-value) | |
|---|---|---|---|
| Immunized | Non-immune | ||
| Spinal cord | 1.27±0.38 | 0.9±0.45 | 0.097 |
| Cortex | 0.8±0.17 | 0.71±0.19 | 0.374 |
| Kidney | 2.25±0.58 | 2.15±0.67 | 0.73 |
| Liver | 4.59±0.74 | 4.3±0.43 | 0.464 |
| Thymus | 3.03±0.44 | 2.92±0.52 | 0.651 |
| Spleen | 2.59±0.38 | 2.59±0.12 | 0.997 |
| Lymph nodes | 3.9±0.74 | 4.21±1.51 | 0.684 |
Five non-immune, and 15 PLSJL mice immunized with MBP six days previously were given Na-fluorescein, transcardially-perfused and various tissues assessed for Na-fluorescein uptake as described in Materials and Methods. Results, presented as mean Na-fluorescein levels ± S.D. for each group, were assessed for significant differences between the tissues of non-immune and immunized mice using Student’s t test.
As a first step towards understanding whether or not this preclinical, transient increase in BBB permeability may have any impact on the later development of clinical signs of EAE, we determined the proportions of mice that show such changes with the incidence of EAE. A group of 40 PLSJL mice were immunized with MBP, 15 were used to assess BBB permeability changes 6 days later and the remainder were monitored for the development of clinical signs of EAE. Na-fluorescein levels greater than the mean plus 4x S.D. of those seen in non-immune controls were detected in 6 of the 15 mice assessed at day 6 p.i. (Figure 3A; denoted by (#). The accumulation of Na-fluorescein in the cerebella of these mice can be readily visualized under UV illumination (Figure 3B 1, 2). In 3 of the animals the fluorescein uptake was only moderately higher than in non-immune control mice (Figure 3A; denoted by (+), at least 2 S.D. above non-immune). The visual areas of Na-fluorescein accumulation in these animals were considerably smaller (Figure 3B 3, 4). The remaining 6 of the 15 mice assessed for day 6 BBB permeability failed to develop Na-fluorescein uptake that was elevated over that of non-immune control mice nor did their brains exhibit more than a superficial fluorescence (Figure 3B 5). Overall, 9 of the 15 (60%) mice analyzed showed fluorescein uptake into the cerebellum which is greater than the mean + 2x S.D. found in controls (significantly higher incidence by Fisher’s exact test, P < 0.04). This is comparable with the 14 out of 25 (56%) mice that developed EAE symptoms, ranging in severity from 1 to 6, during a 25 day monitoring period following immunization (Figure 3C).
Figure 3. Equivalent proportions of MBP-immunized PLSJL mice exhibit Increased cerebellar BBB permeability to Na-fluorescein at 6 days following immunization and develop clinical EAE.
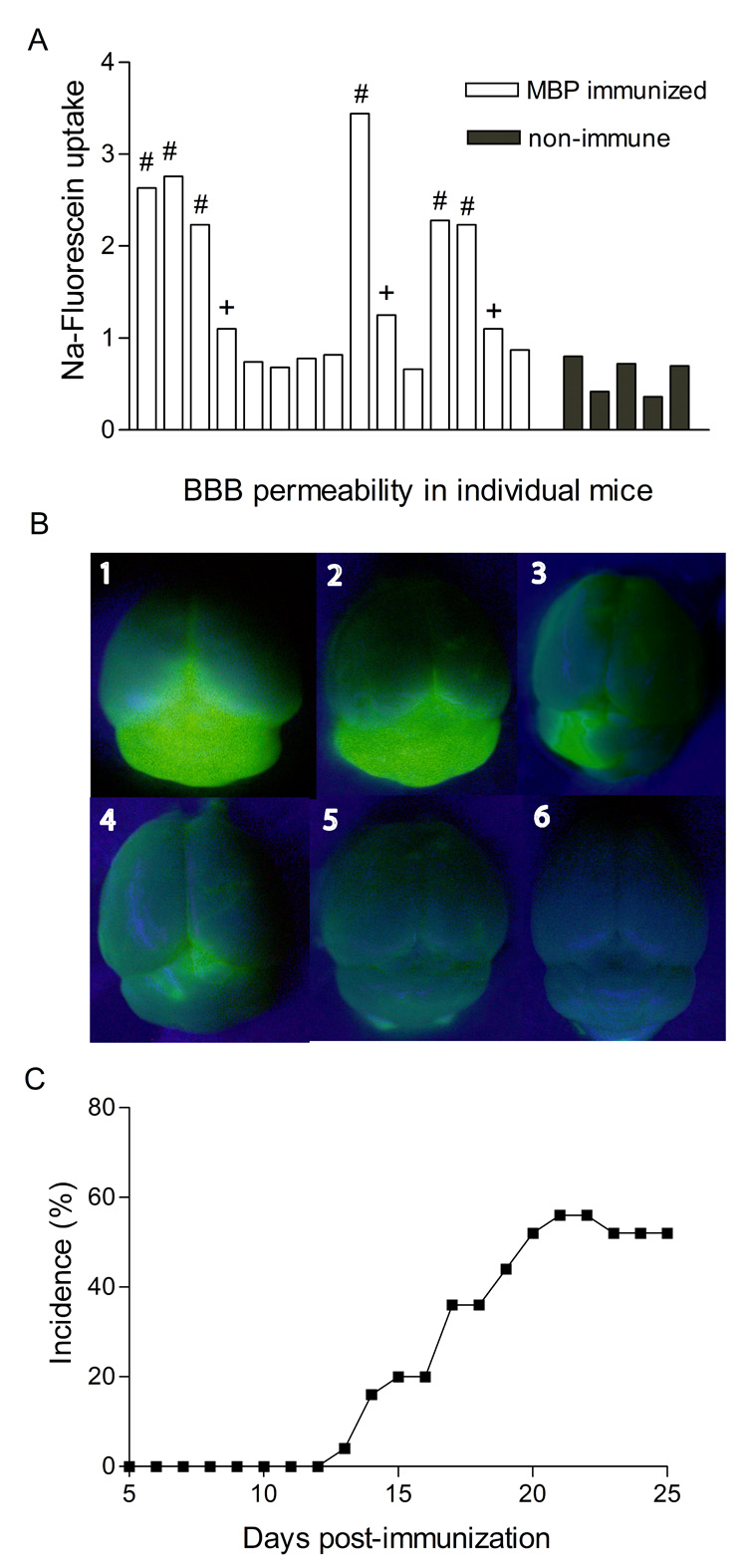
40 PLSJL mice were immunized with MBP and the uptake of Na-fluorescein into CNS tissues was assessed in 15 of the immunized mice on day 6 after immunization as well as in 5 non-immune controls, as detailed in Materials and Methods. The results are presented as the Na-fluorescein content of cerebella from individual animals (panel A). Na-fluorescein levels greater than the mean plus 4x S.D. of those seen in non-immune controls are denoted by (#) while (+) identifies cerebellar tissues with Na-fluorescein levels at least 2 S.D. but less than 4x S.D. above the mean of non-immune tissues. Photographs of representative brains (1 and 2 from the group denoted by #; 3 and 4 from the + group; 5, no substantial increase; and 6 control) were taken under UV illumination prior to isolation of the cerebella and Na-fluorescein extraction (panel B). The remaining twenty five MBP-immunized mice were observed for the development of clinical signs of EAE over a 25 day period and the disease incidence is shown (C).
3.2. Inflammatory markers are expressed in the peripheral blood and cerebellum in association with BBB permeability changes
We have previously demonstrated that the loss of BBB integrity in the spinal cord of mice developing clinical signs of EAE is associated with an increase in the levels of circulating iNOS-positive cells (Kean et al., 2000) as well as proinflammatory changes and cell infiltration in the spinal cord tissues (Hooper et al., 2000; Scott et al., 2004). Somewhat similar patterns are seen for peripheral blood lymphocytes (PBLs) and cerebellar tissues 6 days following MBP-immunization. There is a strong elevation in iNOS expression by PBLs (Figure 4) and elevated expression of the proinflammatory molecules (iNOS, ICAM-1) and the cell markers (CD4, CD8, Mac-1, CD68) in tissues from permeable areas of the cerebellum (Figure 5). For the latter experiments, brains were collected from Na-fluorescein-treated, perfused mice immunized with MBP 6 days previously and cerebellar tissues exhibiting different levels of fluorescence as detected by UV light (see Fig. 3B) were collected for mRNA content assessment by real-time quantitative RT-PCR. Cerebellar tissues from non-immune mice were used as controls. While the levels of mRNAs specific for all of the markers assessed were elevated in cerebella exhibiting extensive BBB permeability, the patterns of changes seen differed with varying extents of permeability (Figure 5). Certain markers, such as mRNAs specific for CD8 and for the adhesion molecule ICAM-1, were elevated in all MBP-immunized mice regardless of the extent of BBB permeability. On the other hand, mRNA specific for the macrophage markers Mac-1 (CD11b) and CD68 were elevated only in cerebella which exhibited increased BBB permeability but the extent of these changes appear to have no correlation. Finally, iNOS and CD4 markers, which showed the highest increase in mRNA levels, were observed in mice with the greatest BBB disruption.
Figure 4. iNOS mRNA expression is elevated in peripheral blood cells after immunization with MBP.
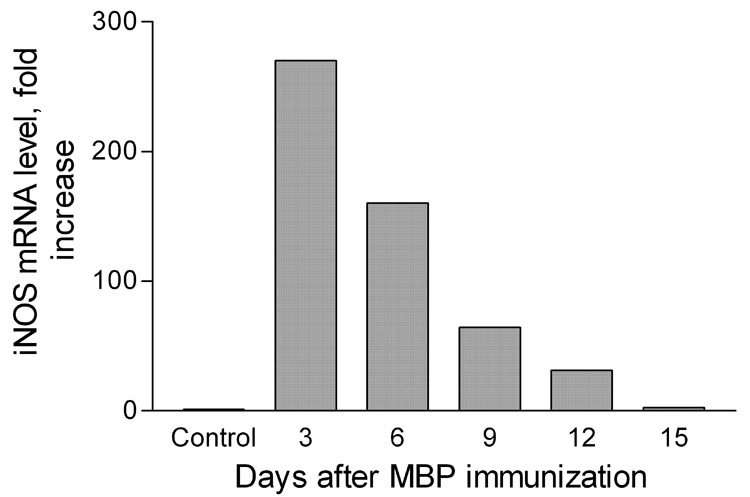
Real-time RT-PCR was used to assess iNOS-specific mRNA levels in PBLs isolated from 6 non-immune PLSJL mice as well as from groups of 6 PLSJL mice at 3, 6, 9, 12, and 15 days after immunization with MBP in adjuvant as described in Materials and Methods. Results presented as fold increase with the mean level detected in PBLs from the non-immune mice taken as 1.0
Figure 5. Elevated BBB permeability in the cerebellum of MBP immunized PLSJL mice is accompanied by the increased expression of mRNAs for pro-inflammatory factors.
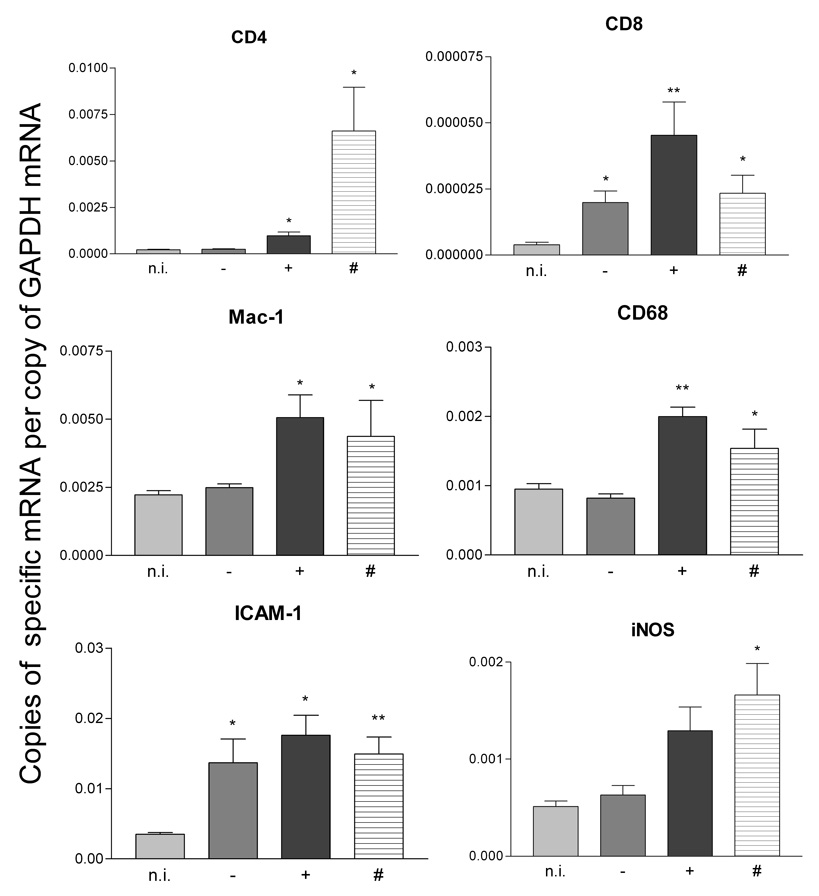
Six days after MBP-immunization of 21 PLSJL mice, Na-fluorescein was administered, the mice were transcardially perfused, and cerebella isolated as detailed in Materials and Methods. Cerebella were observed under UV light and sorted into three groups: (−) no visible Na-fluorescein intake (5 mice); (+) intermediate uptake (fluorescence in less than 50% of the visible cerebellum) (11 mice); and (#) extensive fluorescein uptake (fluorescence in more than 50% of the visible cerebellum) (5 mice). As described in Materials and Methods, total RNA was isolated from the cerebellum and real time quantitative RT PCR was used to assess the expression levels of the indicated mRNAs. Cerebella from non-immune mice (5) were similarly analyzed. Results are expressed as the mean gene-specific mRNA copy number/copy of GAPDH mRNA. Statistically significant differences in copy number between immunized and non-immune samples determined by the Mann-Whitney test are denoted by * (p<0.05) and ** (p<0.005).
3.3. UA treatment inhibits early onset BBB permeability changes in the cerebellum as well as the subsequent development of EAE
The increased BBB permeability in the spinal cord that is associated with symptomatic EAE can be blocked by treatment with uric acid, a natural scavenger of peroxynitrite-dependent radicals (Hooper et al., 2000; Kean et al., 2000). The presence of iNOS-positive cells in the circulation at the time of early onset cerebellar BBB permeability changes (Figure 4) suggested to us that peroxynitrite-dependent radicals might also be involved in this process and UA may have similar effects. As shown in Figure 6A, UA treatment throughout the first week after immunization inhibits the early onset BBB permeability changes in the cerebellum. Inhibition of this transient permeability has a profound impact on the clinical course of EAE. The administration of UA during the first 7 days after MBP immunization delayed the onset of EAE (Figure 6B) as well as reduced its incidence, severity, and mortality (Table 4) despite the fact that treatment was terminated at least a week prior to the appearance of clinical disease signs.
Figure 6. UA treatment during the first week after immunization prevents BBB permeability changes in the cerebellum at day 6 post MBP-immunization and inhibits the subsequent development of clinical EAE.
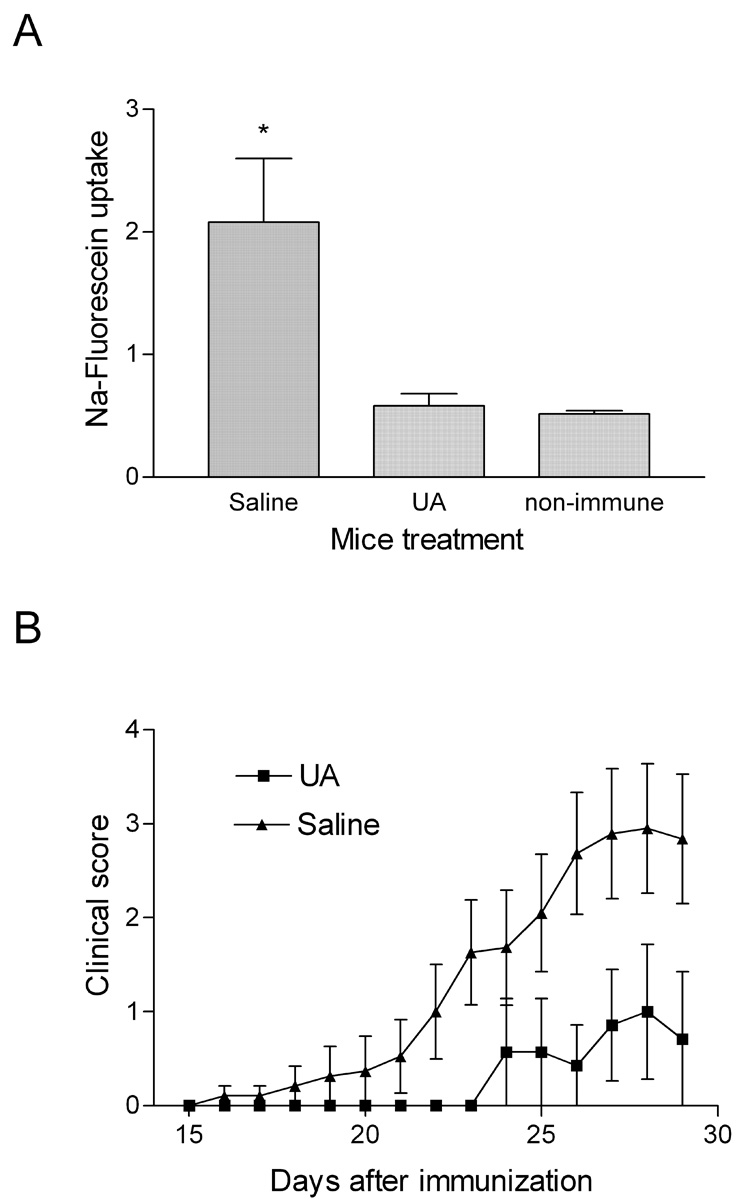
PLSJL mice were immunized with MBP and, beginning on the day of immunization, were treated with 4 daily i.p doses of 10mg UA or saline as described in Materials and Methods. At 6 days after immunization groups of 10 each, as well as non-immune controls (10) were assessed for cerebellar BBB permeability to Na-fluorescein. The data is expressed as Na-fluorescein levels with significant increases over control mice determined by Student’s t test denoted by * (p<0.05) (A). Treatment of the remaining saline (19) and UA (14) –treated mice was terminated at day 7 post-immunization and disease development was monitored for 30 days. The results are presented in (B) as the mean clinical score ± SD.
Table 4.
UA treatment of MBP-immunized PLSJL mice during the first seven days after immunization decrease incidence of paralytic EAE and suppress mortality.
| Incidence of paralytic EAE (%) (score > 2) | % mortality | |
|---|---|---|
| UA treated mice | 28.6 | 0 |
| Saline treated mice | 52.6 | 21.1 |
PLSJL mice were immunized and treated for 7 days with UA (14 mice) or saline (19 mice) as described in a legend for figure 6B. Disease development was monitored for 30 days. Data are represented as percent of mice showing clinical signs of EAE or as a percent mortality.
4. Discussion
Pathological changes occur in the spinal cord and cerebellum during the onset of the symptomatic phase of EAE (Archambault et al., 2006). The current study extends prior observations that associate the pathogenesis of EAE with the loss of neurovascular integrity in the spinal cord (Cross et al., 1993; Pan et al., 1996; Simmons et al., 1982; Fabis et al., 2007) by confirming that elevated BBB permeability also occurs in the cerebellum as clinical signs of EAE develop. However, a transient increase in neurovascular permeability that is limited to the cerebellum occurs considerably before the development of clinical disease in both SJL/J mice immunized with PLP139–151 (Tonra et al., 2001) and, as we show here, PLSJL mice immunized with MBP. This permeability change develops approximately 6 days after immunization, is not seen at other sites and requires both MBP and adjuvant for optimal effect. Thus, both antigen-specific and non-specific factors are likely to contribute and the neurovasculature of the cerebellum is particularly susceptible. Following the restoration of this early elevation in cerebellar BBB permeability there is an interval of at least 1 week before BBB integrity is lost in both the cerebellum and spinal cord and clinical signs of EAE appear.
The clinical signs of EAE are generally manifested as an ascending paralysis and the extent of BBB permeability in the spinal cord correlates with the severity of EAE (Fabis et al., 2007). Moreover, there are differences in the pathological changes between the spinal cord and cerebellum that raise questions about the contribution of the latter to the clinical signs of EAE (Archambault et al., 2006). Thus the possibility that a transient increase in BBB permeability in the cerebellum may contribute to the subsequent development of clinical disease may seem remote despite the fact that the proportions of PLSJL mice that show disruptions of the cerebellar BBB at day 6 post-immunization with MBP and develop clinical signs of EAE several weeks afterwards are similar. To develop a means to more directly examine the possibility of a relationship between early changes in the integrity of the cerebellar BBB and the subsequent development of clinical EAE we first assessed whether or not we could inhibit the early onset BBB permeability changes using an approach that does not interfere with the induction of other parameters of MBP-specific immunity. We have previously shown that inhibiting ONOO−-dependent reactions in MBP immunized mice by raising their normally low serum UA levels prevents the disease as well as facilitates recovery from pre-existing EAE (Hooper et al., 1997; 2000). At least one aspect of the protective effects of UA in these models is evidently manifested at the BBB. Treatment with UA prior to the onset of EAE prevents the enhanced BBB permeability associated with EAE (Kean et al., 2000) while treatment of mice with preexisting EAE returns BBB integrity to normal (Hooper et al., 2000). Extensive studies of the effects of UA on immunity in general have failed to demonstrate that UA has any immunosuppressive properties outside of its effects on BBB permeability and cell infiltration into CNS tissues (Hooper et al., 2001; Kean et al., 2000). For example, UA treatment has no effect on the T-cell proliferative response to MBP, antigen presenting cell function, or on the elevation of iNOS expression in vivo or in vitro (Kean et al., 2000). Our detection of increased iNOS mRNA levels at day 6 in both circulating PBLs as well as cerebellar tissues suggested that ONOO− is being made by circulating cells as they infiltrate the tissues and that urate treatment may have some value in preventing the associated BBB permeability changes. This proved to be the case as treatment with the ONOO− -dependent radical scavenger UA during the first week after MBP immunization blocked not only the early loss of BBB integrity but also the subsequent development of EAE.
The fact that preventing the early, transient elevation of BBB permeability in the cerebellum with UA indicates that a ONOO− -dependent radical mediated process, although asymptomatic, is critical for the subsequent development of EAE. In our prior EAE experiments, BBB permeability changes in the spinal cord and CNS inflammation were prevented by urate treatment begun at 10–15 days post-immunization with MBP. Thus the changes caused by the early response are not sufficient to overcome the effects of UA treatment at later stages. At present we can only speculate as to which features of the early response may promote the development of clinical EAE. From our studies of neurotrophic virus clearance we have concluded that the cerebellum is the principle site where immune effectors gain access to CNS tissues during a therapeutic neuroimmune response (Phares et al., 2006). Other studies have demonstrated that cerebellar tissue is particularly susceptible to BBB disruption under inflammatory conditions (Muller et al., 2005; Silwedel and Forster 2006). Although antigen specificity is not necessary to cause enhanced BBB permeability in our studies or in an adoptive transfer EAE model (Smorodchenko et al., 2007), an antigen-specific reaction is evidently required to induce clinical signs of EAE in both cases. Therefore, we speculate that either cells involved in the induction of an MBP specific response may infiltrate into the CNS tissues approximately 6 days after MBP immunization and that this is critical for the development of EAE weeks later. Small increases in mRNAs specific for CD4, Mac-1, CD68, and iNOS are detectable in the cerebellum of MBP-immunized mice that develop early onset permeability. On the other hand, CD8 mRNA levels in the cerebellum are increased by immunization regardless of whether or not elevated permeability is detected, as is the case for ICAM-1 mRNA levels. This suggests that the invasion of CD4 cells and iNOS-positive monocytes may be more important for the later development of EAE but further experiments are necessary to fully understand the mechanisms involved.
Acknowledgments
We thank T. Mikheeva and A. D. Pekala for technical help. This work was supported by a grant from Commonwealth of Pennsylvania Department of Health to Biotechnology Foundation Laboratories (to H.K.) and grant from NIH/NCAM (AT 0013025) to D.C.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sergei Spitsin, Thomas Jefferson University, 1020 Locust St., JAH room 470C, Philadelphia, PA, 19107.
Carla Portocarrero, Thomas Jefferson University , 1020 Locust St., JAH room 470C, Philadelphia, PA, 19107.
Timothy W. Phares, Lerner Research Institute, 9500 Euclid Avenue, Cleveland, OH 44195
Rhonda B. Kean, Thomas Jefferson University, 1020 Locust St., JAH room 454, Philadelphia, PA, 19107
Christine M. Brimer, Thomas Jefferson University, 1020 Locust St., JAH room 454, Philadelphia, PA, 19107
Hilary Koprowski, 1020 Locust St., JAH room M-85, Philadelphia, PA, 19107.
D.Craig Hooper, 1020 Locust St., JAH room 452, Philadelphia, PA, 19107.
References
- 1.Archambault AS, Sim J, McCandless EE, Klein RS, Russell JH. Region-specific regulation of inflammation and pathogenesis in experimental autoimmune encephalomyelitis. J. Neuroimmunology. 2006;181(1–2):122–132. doi: 10.1016/j.jneuroim.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Archelos JJ, Hartung HP. Frontiers in Multiple Sclerosis II. London: Martin Dunitz Ltd; 1999. Adhesion molecules in multiple sclerosis: a review; pp. 85–116. [Google Scholar]
- 3.Brown KA. Factors modifying the migration of lymphocytes across blood-brain barrier. International Immunopharmacology. 2001;1:2043–2062. doi: 10.1016/s1567-5769(01)00129-1. [DOI] [PubMed] [Google Scholar]
- 4.Cross AH, O'Mara T, Raine CS. Chronologic localization of myelin-reactive cells in the lesions of relapsing EAE: implications for the study of multiple sclerosis. Neurology. 1993;43:1028–1033. doi: 10.1212/wnl.43.5.1028. [DOI] [PubMed] [Google Scholar]
- 5.Cross AH, Manning PT, Stern MK, Misko TP. Evidence for the production of ONOO-in inflammatory CNS demyelination. J. Neuroimmunol. 1997;80:121–130. doi: 10.1016/s0165-5728(97)00145-8. [DOI] [PubMed] [Google Scholar]
- 6.Cross AH, Maning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J. Neuroimmunol. 1998;88:45–56. doi: 10.1016/s0165-5728(98)00078-2. [DOI] [PubMed] [Google Scholar]
- 7.Fabis MJ, Scott GS, Kean RB, Koprowski H, Hooper DC. Loss of blood-brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc Natl Acad Sci U S A. 2007;104(13):5656–5661. doi: 10.1073/pnas.0701252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fazekas G, Tabira T. What transgenic and knockout mouse models teach us about experimental autoimmune encephalomyelitis. Reviews in Immunogenetics. 2000;2:115–132. [PubMed] [Google Scholar]
- 9.Hemmer B, Cepok S, Nessler S, Sommer N. Pathogenesis of multiple sclerosis: an update on immunology. Current Opinion in Neurol. 2002;15:227–231. doi: 10.1097/00019052-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Hooper DC, Bagasra O, Marini JC, Zborek A, Ohnishi ST, Kean R, Champion JM, Sarker AB, Bobroski L, Farber JL, Akaike T, Maeda H, Koprowski H. Prevention of experimental allergic encephalomyelitis by targeting nitric oxide and ONOO-: Implications for the treatment of multiple sclerosis. P. N. A. S. 1997;94:2528–2533. doi: 10.1073/pnas.94.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, Koprowski H. Uric acid, a natural scavenger of ONOO-, in experimental allergic encephalomyelitis and multiple sclerosis. P. N. A. S. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 13.Hooper DC, Kean RB, Scott GS, Spitsin SV, Morimoto K, Bette M, Röhrenbeck AM, Dietzschold B, Weihe E. The CNS inflammatory response to neurotropic virus infection is peroxynitrite-dependent. J. Immunol. 2001;167:3470–3477. doi: 10.4049/jimmunol.167.6.3470. [DOI] [PubMed] [Google Scholar]
- 14.Imitola J, Chitnis T, Khoury SJ. Cytokines in multiple sclerosis: from bench to bedside. Pharmacol Ther. 2005;106:163–177. doi: 10.1016/j.pharmthera.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Kean RB, Spitsin SV, Mikheeva T, Scott GS, Hooper DC. The peroxynitrite scavenger uric acid prevents inflammatory cell invasion into the CNS in experimental allergic encephalomyelitis through maintenance of blood-CNS barrier integrity. J. Immunol. 2000;165:6511–6518. doi: 10.4049/jimmunol.165.11.6511. [DOI] [PubMed] [Google Scholar]
- 16.Kieseier BC, Storch MK, Archelos JJ, Martino G, Hartung HP. Effector pathways in immune mediated central nervous system demyelination. Current Opinion in Neurology. 1999;12:323–336. doi: 10.1097/00019052-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Liu JS-H, Zhao M-L, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am. J. Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller DM, Pendera MP, Greer JM. Blood—brain barrier disruption and lesion localisation in experimental autoimmune encephalomyelitis with predominant cerebellar and brainstem involvement. J. Neuroimmunology. 2005;160:162–169. doi: 10.1016/j.jneuroim.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Pan W, Banks WA, Kennedy MK, Gutierrez EG, Kastin AJ. Differential permeability of the BBB in acute EAE: enhanced transport of TNT-alpha. Am J Physiol. 1996;271:E636–E642. doi: 10.1152/ajpendo.1996.271.4.E636. [DOI] [PubMed] [Google Scholar]
- 20.Phares TW, Kean RB, Mikheeva T, Hooper DC. Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J Immunol. 2006;176(12):7666–7675. doi: 10.4049/jimmunol.176.12.7666. [DOI] [PubMed] [Google Scholar]
- 21.Plebanski M, Saunders M, Burtles SS, Crowe S, Hooper DC. Primary and secondary human in vitro T cell responses to soluble antigens are mediated by subsets bearing different CD45 isoforms. Immunology. 1992;75:86–91. [PMC free article] [PubMed] [Google Scholar]
- 22.Scott GS, Hake P, Kean RB, Virág L, Szabó C, Hooper DC. Role of poly (ADP-ribose) synthetase activation in the development of experimental allergic encephalomyelitis. J. Neuroimmunol. 2001;117:78–86. doi: 10.1016/s0165-5728(01)00329-0. [DOI] [PubMed] [Google Scholar]
- 23.Scott GS, Kean RB, Fabis MJ, Mikheeva T, Brimer CM, Phares TW, Spitsin S, Hooper DC. ICAM-1 upregulation in the spinal cords of PLSJL mice with experimental allergic encephalomyelitis is dependent upon TNF-a production triggered by the loss of blood-brain barrier integrity. Journal of Neuroimmunology. 2004;155:32–42. doi: 10.1016/j.jneuroim.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Silwedel C, Förster CJ. Differential susceptibility of cerebral and cerebellar murine brain microvascular endothelial cells to loss of barrier properties in response to inflammatory stimuli. J. Neuroimmunol. 2006;179:37–45. doi: 10.1016/j.jneuroim.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Simmons RD, Bernard CC, Singer G, Carnegie PR. Experimental autoimmune encephalomyelitis. An anatomically-based explanation of clinical progression in rodents. J Neuroimmunol. 1982;3:307–318. doi: 10.1016/0165-5728(82)90034-0. [DOI] [PubMed] [Google Scholar]
- 26.Smorodchenko A, Wuerfel J, Pohl EE, Vogt J, Tysiak E, Glumm R, Hendrix S, Nitsch R, Zipp F, Carmen Infante-Duarte C. CNS-irrelevant T-cells enter the brain, cause blood-brain barrier disruption but no glial pathology. European Journal of Neuroscience. 2007;26:1387–1398. doi: 10.1111/j.1460-9568.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 27.Tonra JR, Reiseter BS, Kolbeck R, Nagashima K, Robertson R, Keyt B, Lindsay RM. Comparison of the timing of acute blood-brain barrier breakdown to rabbit immunoglobulin G in the cerebellum and spinal cord of mice with experimental autoimmune encephalomyelitis. Journal of Comparative Neurology. 2001;430:131–144. [PubMed] [Google Scholar]
- 28.Tonra JR. Cerebellar susceptibility to experimental autoimmune encephalomyelitis in SJL/J mice: potential interaction of immunology with vascular anatomy. Cerebellum. 2002;1:57–68. doi: 10.1080/147342202753203096. [DOI] [PubMed] [Google Scholar]
- 29.van der Veen RC, Hinton DR, Incardonna F, Hofman FM. Extensive ONOO- activity during progressive stages of central nervous system inflammation. J. Neuroimmunol. 1997;77:1–7. doi: 10.1016/s0165-5728(97)00013-1. [DOI] [PubMed] [Google Scholar]