

Abstract
An important signaling pathway for the differentiation of T helper type 2 (TH2) cells from uncommitted CD4 T cell precursors is activation of the STAT6 transcription factor by interleukin 4 (IL-4). The protooncogene BCL-6 is also involved in TH2 differentiation, as BCL-6 −/− mice develop an inflammation of the heart and lungs associated with an overproduction of TH2 cells. Surprisingly, IL-4 −/− BCL-6 −/− and STAT6 −/− BCL-6 −/− double-mutant mice developed the same TH2-type inflammation of the heart and lungs as is characteristic of BCL-6 −/− mice. Furthermore, a TH2 cytokine response developed in STAT6 −/− BCL-6 −/− and IL-4 −/− BCL-6 −/− mice after immunization with a conventional antigen in adjuvant. In contrast to these in vivo findings, STAT6 was required for the in vitro differentiation of BCL-6 −/− T cells into TH2 cells. BCL-6, a transcriptional repressor that can bind to the same DNA binding motifs as STAT transcription factors, seems to regulate TH2 responses in vivo by a pathway independent of IL-4 and STAT6.
The differentiation of CD4 T helper cells into T helper type 1 or 2 (TH1 or TH2) subsets plays a critical role in the outcome of an immune response. Depending on the nature of the immune challenge, TH1 or TH2 cells can play either a beneficial or detrimental role in the progression of the immune response (reviewed in refs. 1–4). TH1 cells specifically produce interferon-γ (IFN-γ) and tumor necrosis factor β; these cytokines are effective in activating macrophages, mediating delayed-type hypersensitivity, and promoting the elimination of intracellular parasites. TH2 cells specifically produce interleukin 4 (IL-4), IL-5, and IL-13, which down-regulate macrophage functions, recruit eosinophils, and promote the production of IgG1 and IgE antibodies. TH2 cells are suited particularly to the elimination of large extracellular parasites such as helminths and nematodes. An inappropriate TH2 response can result in an ineffective immune response and even a progressive and fatal disease, as in the well characterized system of BALB/c mice infected with the parasite Leishmania major (5). Other manifestations of an abnormal production of TH2 cells are immune disorders such as allergic diseases, hypereosinophilia, TH2-type granuloma formation, chronic graft-vs.-host disease, and progressive systemic sclerosis (2, 4). Elucidating the signaling pathways that control TH2 differentiation is therefore critical if we are to manipulate the immune system effectively.
Signaling by IL-4, which activates the STAT6 transcription factor, is the best characterized pathway by which naive CD4 T cells differentiate into TH2 cells (6). The initial source of IL-4 that promotes TH2 differentiation is under debate (7), but some evidence suggests that on activation, naive CD4 T cells themselves can make a small amount of IL-4 that can promote TH2 responses (8–10). Another source for IL-4 is NK1.1+ CD4 T cells (11, 12), although in some circumstances these cells clearly are not required for TH2 differentiation (13–15). The fundamental importance of both IL-4 and STAT6 in TH2 differentiation has been shown by IL-4-deficient and STAT6-deficient mice (16–19). Both IL-4 −/− and STAT6 −/− mice have profound defects in IgG1 and IgE antibody responses, which are normally promoted by TH2 cells. T cells from IL-4 −/− and STAT6 −/− mice are almost completely defective in differentiating into TH2 cells in vitro, and T cells from these same mice produce very decreased levels of TH2 cytokines in response to infection with the nematode Nippostrongylus braziliensis (17, 19). A more recent study found STAT6 to be important for TH2 cytokine production and granuloma formation in response to the parasitic worm Schistosoma mansoni (20). Two recent studies that used a mouse model of allergy found that loss of STAT6 resulted in an almost complete abrogation of antigen-induced airway hyperresponsiveness (21, 22). Thus, IL-4 and STAT6 play important roles in the regulation of TH2 responses in vivo and in vitro.
Recently, several other transcription factors besides STAT6 have been implicated in TH2 differentiation, including GATA-3, c-maf, NFAT, and BCL-6 (23–30). The mechanism by which these other factors are activated in TH2 cells and how they relate to STAT6 is unclear. Evidence that the BCL-6 protooncogene also regulates TH2 differentiation comes from BCL-6 mutant mice that accumulate TH2 cells in peripheral lymphoid organs and die at an early age from a severe TH2-type inflammation of the heart and lungs (23, 28). Although BCL-6 and STAT6 bind to similar DNA elements, BCL-6 is a transcriptional repressor, whereas STAT6 is a transcriptional activator (26). One theory, therefore, is that during T cell differentiation, BCL-6 may repress STAT6 gene targets and inhibit TH2 differentiation. In the absence of BCL-6, T cell activation might be skewed toward TH2 differentiation because of an absence of repression of STAT6 target genes. Alternatively, BCL-6 may regulate TH2 differentiation by controlling a different set of genes than STAT6. To understand the relationship of BCL-6, IL-4, and STAT6 in TH2 development, we took a genetic approach and created mice deficient in both BCL-6 and either STAT6 or IL-4. Unexpectedly, BCL-6 −/− STAT6 −/− mice also developed a lethal TH2-type inflammation; also, immunization could induce them to produce TH2 cytokines. Similar results were seen with BCL-6 −/− IL-4 −/− mice. BCL-6 may therefore control an alternative pathway of TH2 development that does not require IL-4 and STAT6.
MATERIALS AND METHODS
Mice.
Mice were housed under sterile conditions in an animal facility certified by the American Association of Laboratory Animal Care. STAT6 −/− mice (18) on a mixed B6–129 strain background were mated to BCL-6 +/− mice on a mixed B6–129 background. Mice were genotyped for BCL-6 by a three-oligonucleotide PCR assay on genomic DNA with the following oligonucleotides: (i) 5′-CCA-GCC-AAC-CTG-AAG-ACC-CAC-AC-3′, (ii) 5′-TGT-GGA-TGC-GCA-GAT-GGC-TCT-TCA-GAG-3′, and (iii) 5′-AAA-TGT-GTC-AGT-TTC-ATA-GCC-TGA-AGA-ACG-3′. Mice were genotyped for STAT6 by a three-oligonucleotide PCR assay on genomic DNA with the following oligonucleotides: (i) 5′-ATG-TCT-CTG-TGG-GGC-CTA-ATT-TCC-AAG-3′, (ii) 5′-ACA-GAA-AGC-ATC-TGA-ACC-GAC-CAG-GAA-3′, and (iii) 5′-GCC-TTC-TAT-CGC-CTT-CTT-GAC-GAG-TTC-3′. IL-4 −/− mice (17), bred for 10 generations to B10.A mice, were mated to BCL-6 +/− mice on a mixed B6–129 background. Mice were genotyped for IL-4 by a three-oligonucleotide PCR assay on genomic DNA with the following oligonucleotides: (i) 5′-GCA-CAG-AGC-TAT-TGA-TGG-GTC-3′, (ii) 5′-GCT-GTG-AGG-ACG-TTT-GGC-3′, and (iii) 5′-TCA-GGA-CAT-AGC-GTT-GGC-3′.
Heart- and Lung-Cell Preparation and Stimulation.
Heart and lung cells were prepared as described (23). Cells were stimulated for 48 h at 106 cells per ml with plate-bound anti-CD3 (145–2C11 at 2 μg/ml; PharMingen) and anti-CD28 (37.51 at 5 μg/ml; PharMingen).
Cytokine Measurements.
Cytokines were measured by ELISA: IFN-γ, IL-4, and IL-5 with PharMingen antibody sets, and IL-13 with reagents from R & D Systems.
Immunizations and Lymph-Node Cell Stimulations.
Mice were immunized i.p. with 100 μg of trinitrophenyl-coupled keyhole limpet hemocyanin suspended in a precipitate of aluminum sulfate. The draining mesenteric lymph nodes were taken for cell preparation 14 days later. CD4+ lymph-node cells were prepared by adherence to plastic for 1 h at 37°C to remove macrophages and by treatment with anti-CD8 and anti-B220 magnetic beads (Dynal, Great Neck, NY). Cells prepared in this manner are typically 90–95% CD4+.
In Vitro Differentiation Assays.
Lymph-node cells were stained with fluorescein isothiocyanate-coupled anti-CD62L (PharMingen) and phycoerythrin-coupled anti-CD4 (PharMingen), and the CD4+CD62Lhigh population was purified by flow cytometry. Cells obtained in this manner were routinely 98–99% pure. TH1 differentiation conditions involved stimulation on plates coated with anti-CD3 (2 μg/ml) and anti-CD28 (5 μg/ml) at 106 cells per ml in the presence of recombinant human IL-2 (10 units/ml, a gift from Cetus), anti-IL-4 antibody (11B11, 10 μg/ml), and recombinant mouse IL-12 (10 ng/ml, R & D Systems). TH2 differentiation conditions involved stimulation on plates coated with anti-CD3 and anti-CD28 at 106 cells per ml in the presence of recombinant human IL-2 (10 units/ml), anti-IFN-γ antibody (XMG1.2, 10 μg/ml), and recombinant mouse IL-4 (1,000 units/ml; ref. 31). Cells were stimulated and grown for 7 days, then washed extensively, and restimulated at 106 cells per ml on plates coated with anti-CD3 (2 μg/ml) and anti-CD28 (5 μg/ml). Supernatants were harvested after 24 h and tested for cytokine levels by ELISA.
RESULTS
Because BCL-6 −/− mice are generally infertile, BCL-6 +/− mice were mated to IL-4-deficient (IL-4 −/−; ref. 17) and STAT6-deficient (STAT6 −/−; ref. 18) mice. These sets of double-heterozygote mice were mated together to obtain BCL-6 −/− IL-4 −/− and BCL-6 −/− STAT6 −/− mice. Double-mutant mice of both genotypes were born at the expected frequency of 1 in 16. BCL-6 −/− IL-4 −/− and BCL-6 −/− STAT6 −/− mice were phenotypically indistinguishable from single BCL-6 −/− mice; all three types of mice were growth-retarded and frequently died at an early age (28 of 82 BCL-6 −/−, 9 of 23 BCL-6 −/− STAT6 −/− mice, and 5 of 13 BCL-6 −/− IL-4 −/− mice died between 4 and 8 weeks of age). The average age of death for both types of double-mutant mice was 5.5 weeks, compared with 6.5 weeks for BCL-6 −/− mice. This early death severely limited the number of double-mutant mice that could be studied experimentally. Pathological examination of BCL-6 −/− IL-4 −/− and BCL-6 −/− STAT6 −/− mice that died prematurely showed that these mice typically had a severe inflammation of the heart and lungs (Fig. 1) that was identical to the inflammation that strikes BCL-6 −/− mice (23). Overall, 8 of 9 BCL-6 −/− STAT6 −/− mice and 9 of 10 BCL-6 −/− IL-4 −/− mice examined had heart and lung inflammation. The TH2 nature of the inflammation was indicated first by the presence of large numbers of eosinophils, a feature of TH2 responses resulting from IL-5 secretion (32).
Figure 1.
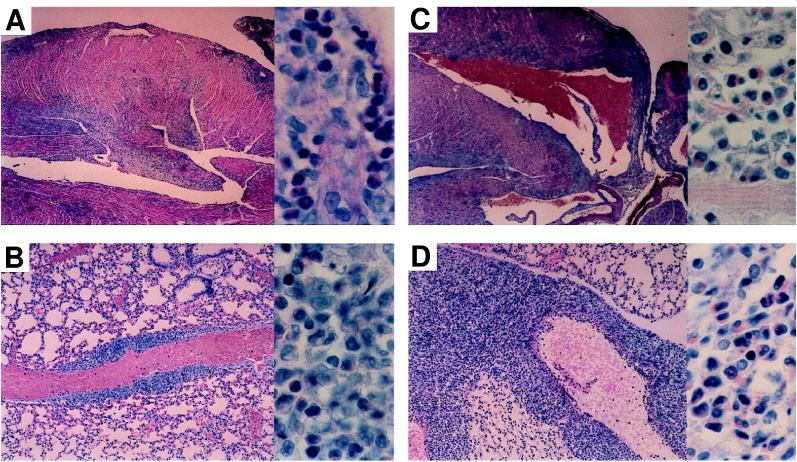
Histopathology of hearts (A and C) and lungs (B and D) from BCL-6 −/− STAT6 −/− mice (A and B) and BCL-6 −/− IL-4 −/− mice (C and D). All sections are stained with Giemsa stain and show extensive myocarditis and pulmonary vasculitis. High power views of each tissue are shown in the panels to the right; they show the presence of large numbers of eosinophils. Eosinophils were confirmed by Giemsa stain in four of four BCL-6 −/−STAT6 −/− mice and four of four BCL-6 −/− IL-4 −/− mice.
To confirm the presence of TH2 cells in the inflamed sites, infiltrating cells in the hearts and lungs of BCL-6 −/−, BCL-6 −/− IL-4 −/−, and BCL-6 −/− STAT6 −/− mice were extracted and stimulated in vitro with antibodies to CD3 and CD28. T cells from the hearts and lungs of BCL-6 −/− STAT6 −/− mice produced levels of the TH2 cytokines IL-4, IL-5, and IL-13 that were 7- to 1,000-fold greater than the levels produced by heart and lung T cells of STAT6 −/− mice (Fig. 2). T cells taken from the hearts and lungs of BCL-6 −/− IL-4 −/− mice also produced levels of IL-5 and IL-13 that were 3- to 50-fold higher than the levels produced by heart and lung T cells from IL-4 −/− mice (Fig. 2). The levels of TH2 cytokines produced by the BCL-6 −/− IL-4 −/− and BCL-6 −/− STAT6 −/− inflammatory cells were comparable to those produced by heart and lung T cells from BCL-6 −/− mice (ref. 23; data not shown). In contrast, generally, levels of the signature TH1 cytokine IFN-γ were not elevated in any of the inflammatory cell stimulations (Fig. 2). There is variation in the relative levels of TH2 cytokines produced by the BCL-6 −/− IL-4 −/− and BCL-6 −/− STAT6 −/− inflammatory cells. Most likely, this variation is caused by the nature of the heart and lung inflammation; i.e., the onset and course of the disease differs from mouse to mouse, as was observed in the BCL-6 −/− mice as well (ref. 23; data not shown). Nevertheless, T cells from each BCL-6 −/− STAT6 −/− and BCL-6 −/− IL-4 −/− mouse tested produced significantly elevated levels of TH2 cytokines compared with STAT6 −/− and IL-4 −/− controls, respectively.
Figure 2.

Cytokine expression of cells extracted from the hearts and lungs of STAT6 −/− BCL-6 −/−, STAT6 −/−, IL-4 −/− BCL-6 −/−, and IL-4 −/− mice. Gray bars represent either STAT6 −/− or IL-4 −/− littermate controls; black bars represent either STAT6 −/− BCL-6 −/− or IL-4 −/− BCL-6 −/− double-mutant mice. Cytokine units are ng/ml. N.D., not done.
We next investigated whether a TH2 inflammatory reaction could be induced by immunizing the mice. Previously, we observed that a TH2-type inflammation could be induced in the spleen of BCL-6 −/− mice by i.p. injection of antigen plus adjuvant (23). BCL-6 −/−, BCL-6 −/− STAT6 −/−, STAT6 −/−, and wild-type mice were immunized i.p. with trinitrophenyl-coupled keyhole limpet hemocyanin, a typical hapten-coupled protein antigen, suspended in the adjuvant aluminum sulfate. After a 2-week period, spleens from immunized BCL-6 −/−, BCL-6 −/− STAT6 −/−, and BCL-6 −/− IL-4 −/− mice contained a large percentage of granulocytes and were enlarged relative to spleens from immunized control mice, consistent with the induction of an inflammatory response (data not shown). Draining mesenteric lymph nodes were taken from the immunized mice and restimulated in vitro to measure cytokine responses. Strikingly, polyclonal stimulation of lymph-node T cells from immunized BCL-6 −/− STAT6 −/− mice led to secretion of TH2 cytokines at levels at least 10-fold higher than those observed from lymph nodes from wild-type and STAT6 −/− immunized mice (Fig. 3). The TH2 cytokines in these cultures seemed to be derived from CD4+ T cells, because cultures of purified CD4+ T cells yielded comparable results (Fig. 3). A large increase in TH2 cytokine production relative to control mice was also obtained by using lymph-node cells from immunized BCL-6 −/− IL-4 −/− mice (Fig. 3). High levels of TH2 cytokines were also generated when the lymph-node T cells from the immunized BCL-6 −/− and BCL-6 −/− STAT6 −/− mice were rechallenged in vitro with the immunizing antigen, but the antigen-specific production of TH2 cytokines was lower than the levels produced from polyclonal stimulation (data not shown). After immunization, both BCL-6 −/− and BCL-6 −/− STAT6 −/− lymph-node cells produced elevated levels of IFN-γ compared with wild-type and STAT6 −/− lymph-node cells, suggesting that a TH1 response was also increased in the absence of BCL-6. However, there was a greater increase in TH2 cytokines (8- to 310-fold) than the increase in IFN-γ (3- to 4-fold). These data show that the in vivo T cell response of the BCL-6 −/− mice is skewed toward the production of TH2 cytokines, and surprisingly, this effect is independent of STAT6.
Figure 3.

TH2 cytokine expression from mesenteric lymph-node cells from immunized BCL-6 −/−, wild-type, BCL-6 −/− STAT6 −/−, and STAT6 −/− mice. Gray bars represent either wild-type, STAT6 −/−, or IL-4 −/− littermate controls; black bars represent either BCL-6 −/− STAT6 −/− or BCL-6 −/− IL-4 −/− double-mutant mice. Numbers at the top of the graph bars indicate n-fold increase of BCL-6 −/− or BCL-6 −/− STAT6 −/− cytokine levels over the highest levels obtained from control lymph-node cell stimulations. Cytokine units are ng/ml. N.D., not done.
We next addressed whether the overproduction of TH2 cells by the BCL-6 −/− mice could be caused by an intrinsic bias toward TH2 differentiation or whether BCL-6 −/− CD4 T cells have a defect in differentiation into TH1 cells. We used an in vitro culture system in which CD4+CD62+ naive T cells can be induced to differentiate into either TH1 cells in response to IL-12 or TH2 cells in response to IL-4 (6, 31). As shown in Fig. 4, IL-12 induced the differentiation of BCL-6 −/− naive CD4 T cells into phenotypic TH1 cells that produced high levels of IFN-γ and very little IL-4. Conversely, addition of IL-4 to the cultures induced both wild-type and BCL-6 −/− naive T cells to differentiate into IL-4-producing TH2 cells. Highly purified BCL-6 −/− STAT6 −/− naive CD4 T cells could be induced to differentiate in vitro into TH1 cells in response to IL-12 (Fig. 4). These same cells were defective in developing into IL-4-producing cells in vitro in response to IL-4, showing that STAT6 is required for in vitro TH2 differentiation even in the absence of BCL-6 (Fig. 4). These results indicate that the bias toward TH2 development in vivo in the BCL-6 −/− and BCL-6 −/− STAT6 −/− mice could not be reproduced by in vitro activation in the presence of IL-4.
Figure 4.

In vitro T cell differentiation cultures with naive CD4+CD62+ sorted T cells from BCL-6 −/−, wild-type (WT), BCL-6 −/− STAT6 −/−, and STAT6 mice. Supernatants were taken after 24 h. The data shown for WT and BCL-6 −/− naive CD4 cells are representative of four experiments with sorted cells. The results shown for STAT6 −/− and BCL-6 −/− STAT6 −/− naive CD4 cells were repeated four times with similar results with unsorted lymph-node cells. Cytokine units are ng/ml.
DISCUSSION
In this study, we have investigated how two transcription factors, BCL-6 and STAT6, impinge on TH2 cell differentiation. We have found that BCL-6-deficient mice develop TH2 inflammation and produce TH2 cytokines independent of a functional STAT6 gene. Our results from in vitro cultures are consistent with previous studies showing an absolute requirement for STAT6 in TH2 differentiation in vitro (16, 18, 19). Our in vivo findings, however, challenge the conclusion that STAT6 is required invariably for TH2 generation and point to an in vivo pathway of TH2 differentiation normally inhibited by BCL-6.
As assayed by immunohistochemistry, BCL-6 protein expression is high in germinal-center B cells, a subset of germinal-center T cells, and a small number of CD4 T cells scattered throughout peripheral lymphoid tissues (33–36). BCL-6 protein is not expressed at high levels in most other cell types of the body. As yet, very little is known about the signals that regulate BCL-6 expression in T cells, and attempts to up-regulate BCL-6 protein expression in T cells in vitro have been unsuccessful (A.L.D., unpublished results). It is not yet clear whether BCL-6 expression in CD4 T cells represents a transient state of activation through which all CD4 T cells pass or whether BCL-6 expression represents a unique form of T cell activation that occurs under certain conditions. In this regard, one study found an inverse correlation between expression of BCL-6 and expression of CD40-ligand in T cells, suggesting that BCL-6 protein expression does not reflect the same stage or type of T cell activation that results in CD40-ligand expression (37). In either case, T cells that receive signals that would normally induce BCL-6 expression would presumably be the cells that differentiate abnormally into TH2 cells in the absence of a functional BCL-6 gene. Because most T cells in vivo are negative for BCL-6 expression, but a subset of T cells express BCL-6, it is clear that BCL-6 expression in T cells is a regulated process. An interesting hypothesis is that in a subset of T cells, BCL-6 blocks an alternative pathway of TH2 differentiation that does not require STAT6. This alternative pathway for TH2 differentiation may be amplified specifically by the absence of BCL-6 in mice.
We also observed a discrepancy between the production of TH2 cytokines by cells stimulated ex vivo from BCL-6 −/− STAT6 −/− mice and the ability to drive TH2 differentiation in vitro with exogenous IL-4. This finding confirms previous findings that there is a unique role for STAT6 in TH2 differentiation driven by exogenous IL-4 (16, 18, 19) and suggests that the TH2 differentiation in BCL-6 −/− STAT6 −/− mice is mechanistically different from in vitro TH2 differentiation. An important cytokine that promotes the differentiation of naive T cells into TH1 effector cells is IL-12 (38), and it is possible that the increased TH2 differentiation in the BCL-6 −/− mice is caused by a defect in IL-12 production. This explanation seems relatively unlikely, because both freshly isolated macrophages and macrophage colony-stimulating factor-treated macrophages from BCL-6 −/− and BCL-6 −/− STAT6 −/− mice could be induced to make normal levels of IL-12 after lipopolysaccharide stimulation (A.L.D., unpublished results).
BCL-6 is a transcriptional repressor that can bind to a consensus DNA motif that is strikingly similar to the “GAS” motif recognized by the cytokine-induced STAT transcription factors (39, 40). Indeed, BCL-6 can bind to a STAT6 binding site and repress IL-4-activated transcription (23). Thus, a mechanistic explanation for the effect of BCL-6 on TH2 differentiation is that BCL-6 may repress the transcription of genes that control TH2 differentiation by binding to DNA elements that are positively regulated by STAT6. An alternative possibility is that BCL-6 negatively regulates a separate set of target genes that control TH2 differentiation. Because the critical target genes for TH2 differentiation are unknown, we cannot distinguish between these two possibilities at present. If BCL-6 and STAT6 regulate the same target genes involved in TH2 differentiation, our data suggest that BCL-6 represses the “basal” transcriptional activity of these genes and that, in the absence of BCL-6 and STAT6, these TH2 differentiation genes can be expressed at levels sufficient to drive TH2 differentiation. Our results show clearly that in the absence of BCL-6, transcriptional activity by STAT6 is not required for TH2 differentiation absolutely. An understanding of the mechanism by which BCL-6 affects TH2 cell differentiation will require both the elucidation of the in vivo signals that control BCL-6 expression in T cells and the identification of the target genes regulated by BCL-6 to control TH2 differentiation.
Acknowledgments
We thank Cyndy Watson for help in establishing the double-knockout mouse matings.
ABBREVIATIONS
- TH1 and TH2
T helper type 1 and 2
- IL
interleukin
- IFN-γ
interferon-γ
References
- 1.Romagnani S. Annu Rev Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 2.Romagnani S. Immunol Today. 1997;18:263–266. doi: 10.1016/s0167-5699(97)80019-9. [DOI] [PubMed] [Google Scholar]
- 3.Mosmann T R, Coffman R L. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 4.Abbas A K, Murphy K M, Sher A. Nature (London) 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 5.Reiner S L, Locksley R M. Annu Rev Immunol. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- 6.Seder R A, Paul W E. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 7.Coffman R L, von der Weid T. J Exp Med. 1997;185:373–375. doi: 10.1084/jem.185.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamogawa Y, Minasi L A, Carding S R, Bottomly K, Flavell R A. Cell. 1993;75:985–995. doi: 10.1016/0092-8674(93)90542-x. [DOI] [PubMed] [Google Scholar]
- 9.Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell R A. J Exp Med. 1997;185:461–469. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rocken M, Saurat J H, Hauser C. J Immunol. 1992;148:1031–1036. [PubMed] [Google Scholar]
- 11.Bendelac A. Curr Opin Immunol. 1995;7:367–374. doi: 10.1016/0952-7915(95)80112-x. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimoto T, Paul W E. J Exp Med. 1994;179:1285–1295. doi: 10.1084/jem.179.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von der Weid T, Beebe A M, Roopenian D C, Coffman R L. J Immunol. 1996;157:4421–4427. [PubMed] [Google Scholar]
- 14.Smiley S T, Kaplan M H, Grusby M J. Science. 1997;275:977–979. doi: 10.1126/science.275.5302.977. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y H, Chiu N M, Mandal M, Wang N, Wang C R. Immunity. 1997;6:459–467. doi: 10.1016/s1074-7613(00)80289-7. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan M H, Schindler U, Smiley S T, Grusby M J. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 17.Kopf M, Le Gros G, Bachmann M, Lamers M C, Bluethmann H, Kohler G. Nature (London) 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 18.Shimoda K, van Deursen J, Sangster M Y, Sarawar S R, Carson R T, Tripp R A, Chu C, Quelle F W, Nosaka T, Vignali D A, et al. Nature (London) 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Nature (London) 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan M H, Whitfield J R, Boros D L, Grusby M J. J Immunol. 1998;160:1850–1856. [PubMed] [Google Scholar]
- 21.Kuperman D, Schofield B, Wills-Karp M, Grusby M J. J Exp Med. 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, Takeda K, Akira S. J Exp Med. 1998;187:1537–1542. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dent A L, Shaffer A L, Yu X, Allman D, Staudt L M. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 24.Ho I C, Hodge M R, Rooney J W, Glimcher L H. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 25.Hodge M R, Ranger A M, de la Brousse F C, Hoey T, Grusby M J, Glimcher L H. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- 26.Kiani A, Viola J P, Lichtman A H, Rao A. Immunity. 1997;7:849–860. doi: 10.1016/s1074-7613(00)80403-3. [DOI] [PubMed] [Google Scholar]
- 27.Ranger A M, Hodge M R, Gravallese E M, Oukka M, Davidson L, Alt F W, de la Brousse F C, Hoey T, Grusby M, Glimcher L H. Immunity. 1998;8:125–134. doi: 10.1016/s1074-7613(00)80465-3. [DOI] [PubMed] [Google Scholar]
- 28.Ye B H, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti R S, et al. Nat Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida H, Nishina H, Takimoto H, Marengere L E, Wakeham A C, Bouchard D, Kong Y Y, Ohteki T, Shahinian A, Bachmann M, et al. Immunity. 1998;8:115–124. doi: 10.1016/s1074-7613(00)80464-1. [DOI] [PubMed] [Google Scholar]
- 30.Zheng W, Flavell R A. Cell. 1997;89:587–596. [Google Scholar]
- 31.Hu-Li J, Huang H, Ryan J, Paul W E. Proc Natl Acad Sci USA. 1997;94:3189–3194. doi: 10.1073/pnas.94.7.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wardlaw A J, Moqbel R, Kay A B. Adv Immunol. 1995;60:151–266. doi: 10.1016/s0065-2776(08)60586-6. [DOI] [PubMed] [Google Scholar]
- 33.Carbone A, Gloghini A, Gaidano G, Dalla-Favera R, Falini B. Blood. 1997;90:2445–2450. [PubMed] [Google Scholar]
- 34.Cattoretti G, Chang C C, Cechova K, Zhang J, Ye B H, Falini B, Louie D C, Offit K, Chaganti R S, Dalla-Favera R. Blood. 1995;86:45–53. [PubMed] [Google Scholar]
- 35.Flenghi L, Bigerna B, Fizzotti M, Venturi S, Pasqualucci L, Pileri S, Ye B H, Gambacorta M, Pacini R, Baroni C D, et al. Am J Pathol. 1996;148:1543–1555. [PMC free article] [PubMed] [Google Scholar]
- 36.Onizuka T, Moriyama M, Yamochi T, Kuroda T, Kazama A, Kanazawa N, Sato K, Kato T, Ota H, Mori S. Blood. 1995;86:28–37. [PubMed] [Google Scholar]
- 37.Falini B, Bigerna B, Pasqualucci L, Fizzotti M, Martelli M F, Pileri S, Pinto A, Carbone A, Venturi S, Pacini R, et al. Blood. 1996;87:465–471. [PubMed] [Google Scholar]
- 38.Trinchieri G. Annu Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 39.Ihle J N. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 40.Darnell J E, Jr, Kerr I M, Stark G R. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]