

Abstract
Practical methodology is reported for the synthesis of a homologous series of side chain extended amino acids containing aminooxy functionality bearing orthogonal protection suitable for Fmoc peptide synthesis. These reagents may be useful for the preparation of libraries containing fragments joined by peptide linkers.
Keywords: oxime, peptidomimetic, aldehydes, combinatorial chemistry
Fragment-based approaches to drug discovery have gained increasing importance in the pharmaceutical industry.1 Tethered libraries represent a subclass of fragment-based methodologies. However, to date, tethered libraries have typically employed structurally simple linker elements, consisting mainly of polymethylene segments bearing terminal aminoxy groups that serve as anchoring points for fragment attachment.2 However, increased library complexity, leading potentially to enhanced bioactivites, may be possible by combining the structural diversity of peptide scaffolds together with linker-based functionalized oxime ethers. Amino acids 1a - 1f and 2a were designed to serve as key components of linker-based peptide libraries by providing protected aminooxy groups appended onto the peptide backbone via methylene chains of increasing lengths (Figure 1). Final tethered products would be obtained by acid-catalyzed oxime ether deprotection and conjugation with fragment libraries. However, only the core structures of 1a,3 1c4 and 1d5 are known in the literature and efficient syntheses of the series 1a - f and 2a have not yet been reported. Practical methodology for the preparation of 1a - f and 2a bearing orthogonal protection designed for Fmoc-based peptide synthesis was the focus of the current study.
Figure 1.
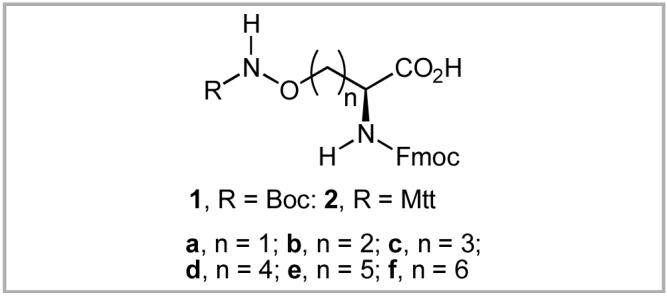
Structures of protected aminooxy amino acid analogues.
Cross metathesis (CM) has found wide application for coupling through highly efficient carbon - carbon bond formation.6 A CM approach for the synthesis of target compounds 1d - 1f required the N-Boc-protected aminooxy building blocks 4a - 4c,7 which could be obtained by reaction of the corresponding bromides 3a - 3c with N-Boc hydroxylamine and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in CH2Cl2(Scheme 1).8 The CM coupling of N-Fmoc-l-allylglycine O-benzyl ester (5)6c with five equivalents of the respective substrates 4a - 4c in refluxing CH2Cl2 using 5% Grubbs 2nd generation catalyst, [((PCy3)(Im(Mes)2)Ru=CHPh)]9 provided the reaction products 6a - 6c (Scheme 2), which were contaminated with small quantities of unwanted material resulting from homodimerization of reagent 4. It was advantageous to subject the crude mixtures directly to hydrogenation to provide the easily purified final products 1d - 1f. Based on combined yields over two steps, it was found that CM efficiency improved with increasing substrate chain length (1d, 40% yield; 1e, 53% yield and 1f, 72% yield).
Scheme 1. Reagents and conditions.
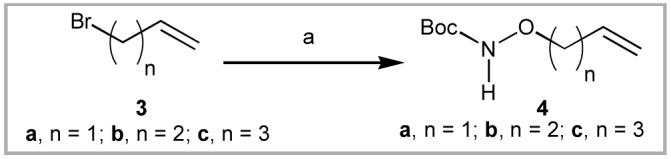
(a) Boc-NH-OH, DBU, CH2Cl2.
Scheme 2. Reagents and conditions.
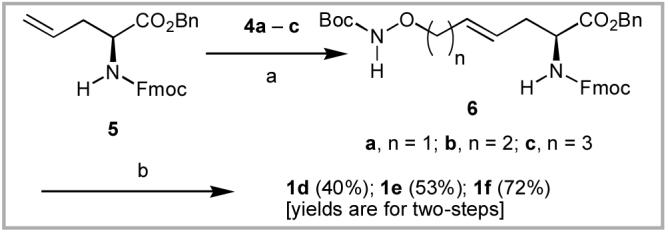
(a) Grubbs 2nd generation catalyst, CH2Cl2, Δ; (b) H2, Pd·C, EtOH.
The shorter homologues 1a - 1c were prepared from l-serine (Ser), l-homoserine (HSer) and l-glutamic acid (Glu), respectively. Near quantitative conversion of Ser to N-Trt-L-serine O-methyl ester (N-Trt-Ser-OMe, 710) and reaction with N-hydroxyphthalimide under Mistunobu conditions, provided the globally-protected product 8 (79% yield, Scheme 3). Use of the N-Trt group was to minimize unwanted beta-elimination during the Mitsunobu reaction.11 Having served its purpose, the N-Trt group was replaced by Cbz protection to provide intermediate 9 (84% yield).
Scheme 3. Reagents and conditions.
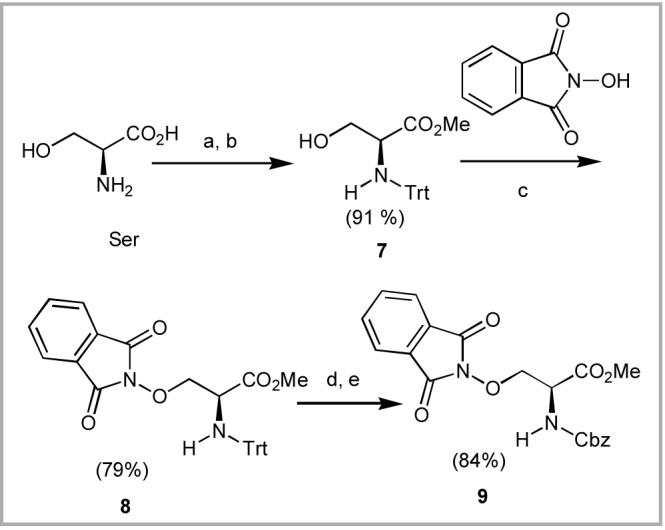
(a) HCl, MeOH; (b) TrtCl, NEt3; (c) Ph3P, DEAD; (d) HCl (37%), CH2Cl2; CbzCl, THF, H2O.
Conversion of HSer to N-Cbz-l-homoserine O-methyl ester (N-Cbz-HSer-OMe, 10) was by literature procedures,12 while the side chain-elongated analogue 12 was obtained from commercially available N-Cbz-Glu-OBn (11)13 by reduction of the free carboxylic acid group (Scheme 4).14 When direct transformation of 10 to the phthalimidooxy product 15 under Mitsunobu conditions was accompanied by poor yields and the formation of cyclized by-products, the alcohols 10 and 12 were converted in quantitative yield to the corresponding mesyl esters (13 and 1415). Subsequent nucleophilic displacement using N-hydroxyphthalimide provided 15 and 16, respectively in approximately 60 - 70% yields.12
Scheme 4. Reagents and conditions.
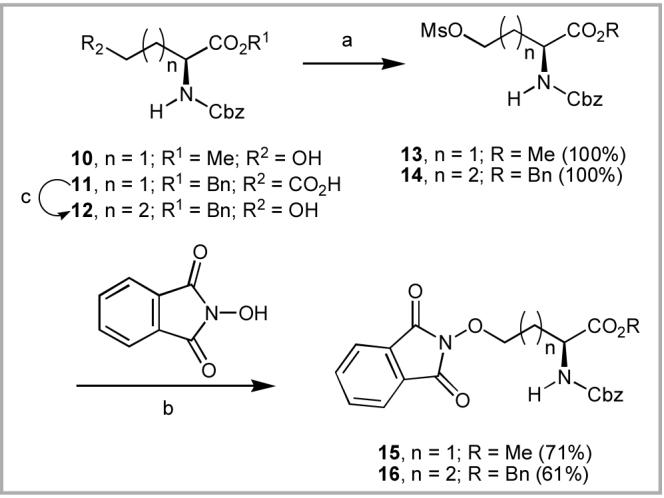
(a) MsCl, NEt3, CH2Cl2; (b) DBU, DMF; (c) ClCO2Et, 4-Methyl morpholine, THF followed by NaBH4, MeOH.
To complete the synthesis of the target compounds the phthalimidooxy intermediates 9, 15 and 16 were cleaved by treatment with methylhydrazine in CH2Cl2 at 0° C, then protected as their N-Boc derivatives (17 - 19) by reaction with Boc anydride (near quantitative yields for two steps, Scheme 5). Cleavage of the amino acid methyl esters of 17 and 18 (LiOH in aqueous THF) followed by Cbz hydrogenolysis and reprotection as the N-Fmoc derivatives provided final products 1a and 1b in 58% and 81% yields over three steps. Hydrogenation of 19 and N-Fmoc reprotection gave 1c directly (47% yield). To determine whether the DBU/DMF conditions resulted in racemization at the α-amino center, 1c was evaluated for enantiomeric purity and found to exhibit an ee value greater than 98%.
Scheme 5. Reagents and conditions.
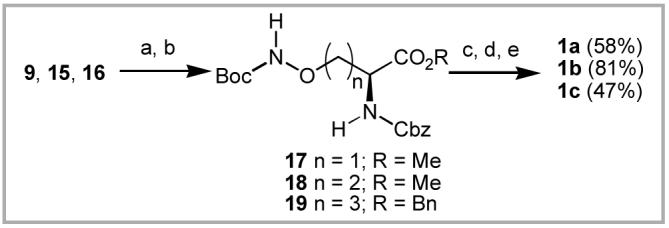
(a) MeNHNH2, CH2Cl2; (b) Boc2O, NEt3, THF; (c) LiOH, THF, H2O; (d) H2, Pd·C, MeOH; (e) FmocOSu, dioxane.
Use of the Boc-protected aminooxy-containing analogue 1a in solid-phase peptide synthesis is potentially limited by β-elimination during piperidine-mediated Fmoc deprotection. Indeed, attempted incorporation on NovaSyn® TGR resin of 1a in place of threonine in the Tsg101-binding peptide, FITC-Ava-PEPTAPPEE-amide16 gave only product resulting from β-elimination.17 Therefore, the aminooxy Boc protecting group in 1a was replaced with the more electron donating 4-methyl-triphenylmethyl (Mtt) (final product 2a). This was achieved by deprotection of phthalamide 9 using methyl hydrazine in CH2Cl2 followed by reaction with Mtt-Cl [N(i-Pr)2Et, CH2Cl2] to give the corresponding Mtt-protected aminooxy analogue 20 in quantitative yield (Scheme 6). Conversion to the N-Fmoc final product 2a was as described above for the preparation of target compounds 1a - 1c. Repeating the synthesis of the Tsg101-binding peptide described above using 2a rather than 1a gave the desired aminooxy-containing peptide as the major product with no β-elimination side product as detected by HPLC.18 This homologous series of aminooxy-containing amino acids analogues are intended to serve as valuable building blocks for the post solid-phase construction of tethered oxime-based peptide libraries.
Scheme 6. Reagents and conditions.
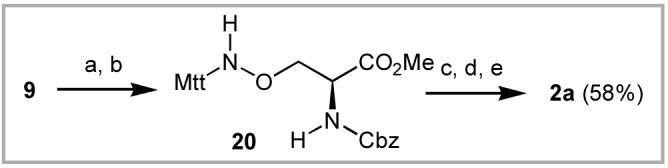
a) MeNHNH2, CH2Cl2; (b) MttCl, N(i-Pr)2Et, CH2Cl2; (c) LiOH, THF, H2O; (d) H2, Pd·C, MeOH; (e) FmocOSu, dioxane.
General Procedure for the Preparation of 4a - 4c
To a mixture of alkenyl bromide (3a - 3c) (10 mmol) and N-Boc-hydroxylamine (25 mmol) in CH2Cl2 (1.0 mL) at 0 °C, was added DBU (7.5 mL) carefully. The reaction mixture was warmed to room temperature and stirred (overnight). The mixture was diluted with EtOAc (150 mL) and washed with H2O (50 mL) then brine (50 mL) and purified by silica gel column chromatography (hexanes:EtOAc) to yield 4a - 4c as colorless oils.
N-(2-Propen-1-yloxy)-carbamic Acid 1,1-Dimethylethyl Ester (4a)7
Prepared from 3a (1.18 g, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.28 (brs, 1 H), 5.88 (m, 1 H), 5.28 - 5.18 (m, 2 H), 4.27 (dd, J = 6.2, 1.0 Hz, 2 H), 1.41 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 156.74, 132.52, 119.54, 81.37, 77.24, 28.08. IR (KBr) νmax: 3290.9, 2979.5, 2361.4, 1717.3, 1456.0, 1368.3, 1249.7, 1165.8, 1107.9, 928.6. ESI-MS (+VE) m/z: 196.1 (M + Na)+.
N-(3-Buten-1-yloxy)-carbamic Acid 1,1-Dimethylethyl Ester (4b)7
Prepared from 3b (1.10 g, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1 H), 5.74 (m, 1 H), 5.03 (m, 1 H), 4.96 (m, 1 H), 3.81 (t, J = 6.8 Hz, 2 H), 2.30 (m, 2 H), 1.39 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 156.90, 134.31, 116.64, 81.36, 75.45, 32.40, 28.09. IR (KBr) νmax: 3297.7, 2979.5, 1721.2, 1478.2, 1368.3, 1245.8, 1165.8, 1108.9, 773.3. ESI-MS (+VE) m/z: 210.1 (M + Na)+.
N-(4-Penten-1-yloxy)-carbamic Acid 1,1-Dimethylethyl Ester (4c)7
Prepared from 3c (1.20 g, 60% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 (s, 1 H), 5.71 (m, 1 H), 4.92 (m, 1 H), 4.86 (m, 1 H), 3.75 (t, J = 6.6 Hz, 2 H), 2.04 (m, 2 H), 1.62 (m, 2 H), 1.38 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 156.90, 137.80, 114.90, 81.41, 75.96, 29.92, 28.13, 27.12. IR (KBr) νmax: 3300.6, 2978.5, 1721.2, 1368.3, 1246.8, 1166.7, 1108.9, 912.2, 775.2. ESI-MS (+VE) m/z: 224.1 (M + Na)+.
General Procedure for the Preparation of Final Products 1d - 1f
A solution of alkene 4a - 4c (5.0 eq.) and protected allylglycine 56c (1.0 eq) in anhydrous CH2Cl2 (20 mL) was degassed under argon (5 min), then Grubbs 2nd generation catalyst [((PCy3)(Im(Mes)2)Ru=CHPh)]9 (0.05 eq.) was added and the mixture was refluxed (8 h). The solvent was evaporated by rotary evaporatoration and the residue was purified by silica gel column chromatography (hexanes:EtOAc) to yield crude 6a - 6c as colorless oils. Without further purification, a solution of 6 in EtOH (10 mL) was hydrogenated at room temperature under 1 atm H2 over 10% Pd·C (10% by weight) (1 h). Catalyst was removed by filtration and the filtrate was concentrated and purified by silica gel column chromatography (CH2Cl2:MeOH) to yield 1d - 1f as white waxes.
(3S)-3-Carboxy-12,12-dimethyl-10-oxo-8,11-dioxa-2,9-diazadecanoic Acid 1-(9H-Fluoren-9-ylmethyl) Ester (1d)
Prepared from 4a in 40% yield over 2 steps. [α]20D + 9.60 (c 0.70, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.55 Hz, 2 H), 7.59 (dd, J = 7.07, 4.13 Hz, 2 H), 7.38 (t, J = 7.46 Hz, 2 H), 7.29 (t, J = 7.45 Hz, 2 H), 7.18 (brs, 1 H), 5.67 (d, J = 7.95 Hz, 1 H), 4.40 - 4.38 (m, 3 H), 4.20 (m, 1 H), 3.84 (t, J = 5.97 Hz, 2 H), 1.91 (m, 1 H), 1.76 (m, 1 H), 1.68 - 1.59 (m, 3 H), 1.54 - 1.46 (m, 10 H). 13C NMR (100 MHz, CDCl3) δ 176.16, 157.53, 156.35, 143.75, 143.61, 141.24, 127.67, 127.04, 125.05, 119.93, 82.30, 76.09, 67.17, 53.60, 47.05, 31.71, 28.16, 27.24, 21.58. IR (KBr) νmax: 3296.7, 2923.6, 1701.9, 1522.5, 1449.2, 1160.9, 909.3, 733.8. ESI-MS (+VE) m/z: 507.2 (M + Na)+. HR-ESI/APCI MS calcd for C26H32N2NaO7 (M +Na) +: 507.2107, Found: 507.2104.
(3S)-3-Carboxy-13,13-dimethyl-11-oxo-9,12-dioxa-2,10-diazadecanoic Acid 1-(9H-Fluoren-9-ylmethyl) Ester, (1e)
Prepared from 4b in 53% yield over 2 steps. [α]20D + 7.04 (c 0.48, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 7.55 Hz, 2 H), 7.66 - 7.50 (m, 3 H), 7.37 (m, 2 H), 7.29 (t, J = 7.41 Hz, 2 H), 5.54 (d, J = 8.13 Hz, 1 H), 4.55 - 4.30 (m, 3 H), 4.19 (m, 1 H), 3.82 (m, 2 H), 1.88 (m, 1 H), 1.72 (m, 1 H), 1.64 - 1.55 (m, 2 H), 1.46 (s, 9 H), 1.44 - 1.35 (m, 3 H), 1.30 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 176.23, 157.51, 156.22, 143.76, 143.61, 141.24, 127.69, 127.04, 125.04, 119.93, 82.16, 76.44, 67.11, 53.66, 47.06, 31.96, 28.17, 27.50, 25.30, 24.77. IR (KBr) νmax: 3285.1, 2933.2, 2364.3, 1700.9, 1521.6, 1449.2, 1162.8, 909.3, 732.8. ESI-MS (+VE) m/z: 521.2 (M + Na)+. HR-ESI/APCI MS calcd for C27H34N2NaO7 (M +Na) +: 521.2264, Found: 521.2261.
(3S)-3-Carboxy-14,14-dimethyl-12-oxo-10,13-dioxa-2,11-diazadecanoic Acid 1-(9H-Fluoren-9-ylmethyl) Ester (1f)
Prepared from 4c in 72% yield over 2 steps. [α]20D + 7.67 (c 1.11, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.47 Hz, 2 H), 7.64 - 7.51 (m, 3 H), 7.37 (t, J = 7.32 Hz, 2 H), 7.29 (d, J = 2.29 Hz, 2 H), 5.55 (brs, 1 H), 4.57 - 4.27 (m, 3 H), 4.19 (t, J = 6.84 Hz, 1 H), 3.80 (t, J = 6.44 Hz, 2 H), 1.86 (m, 1 H), 1.70 (m, 1 H), 1.64 - 1.52 (m, 2 H), 1.52 - 1.41 (m, 10 H), 1.41 - 1.19 (m, 5 H). 13C NMR (100 MHz, CDCl3) δ 157.31, 156.15, 143.70, 141.24, 127.67, 127.04, 125.10, 119.93, 113.43, 81.82, 76.60, 67.04, 47.10, 32.13, 28.76, 28.20, 27.74, 25.49, 24.94. IR (KBr) νmax: 3292.9, 2928.4, 1702.8, 1450.2, 1248.7, 1162.9, 1107.9, 909.3, 731.9. ESI-MS (+VE) m/z: (M + Na)+ 535.2. HR-ESI/APCI MS calcd for C28H36N2NaO7 (M +Na) +: 535.2420, Found: 535.2414.
N-(Triphenylmethyl)-l-serine Methyl Ester (7)10
To cooled MeOH (100 mL) at 0 °C was added acetyl chloride (10.0 mL) dropwise. The resulting solution was stirred (15 min) then L-Serine (5.0 g, 47.6 mmol) was added and the solution was stirred at reflux (2 h) then cooled to room temperature. The solvent was evaporated to provide H-Ser-OMe·HCl as a white solid (7.40 g, quantitative). To a suspension of this material (1.50 g, 9.74 mmol) in CH2Cl2 (50 mL) at 0 °C was added triethylamine (3.0 mL, 21.4 mmol) and trityl chloride (2.90 g, 10.22 mmol) and the mixture was stirred at room temperature (overnight). The reaction mixture was diluted with EtOAc (200 mL), washed with H2O (50 mL) and brine (50 mL) and purified by silica gel column chromatography (hexanes:EtOAc) to yield 7 as a white solid (3.20 g, 91% yield). [α]20D + 3.02 (c 1.80, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.48 - 7.44 (m, 6 H), 7.27 - 7.17 (m, 9 H), 3.69 (m, 1 H), 3.55 (m, 2 H), 3.27 (s, 3 H), 2.96 (brs, 1 H), 2.29 (brs, 1 H). 13C NMR (100 MHz, CDCl3) δ 173.90, 145.57, 128.72, 127.91, 126.59, 70.94, 64.92, 57.78, 51.96. ESI-MS (+VE) m/z: 384.1(M + Na)+.
O-(1,3-Dihydro-1,3-dioxo-2H-isoindol-2-yl)-N-(triphenylmethyl)-L-serine Methyl Ester (8)
To a solution of alcohol 7 (2.80 g, 7.76 mmol), N-hydroxyphthalimide (2.53 g, 15.51 mmol) and triphenyl-phosphine (4.50 g, 17.06 mmol) in THF (100 mL) at 0 °C under argon was slowly added diethyl azodicarboxylate (DEAD) (40% in toluene, 7.80 mL, 17.06 mmol). The mixture was warmed to room temperature and stirred (overnight). The reaction mixture was diluted with EtOAc (200 mL), washed with H2O (2 × 50 mL) and brine (50 mL) and dried (Na2SO4). Purification by silica gel column chromatography (hexanes:EtOAc) provided 8 as a white wax (3.10 g, 79% yield). [α]20D + 39.6 (c 1.34, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.83 - 7.81 (m, 2 H), 7.74 - 7.72 (m, 2 H), 7.55 - 7.52 (m, 6 H), 7.27 - 7.23 (m, 6 H), 7.19 - 7.15 (m, 3 H), 4.48 (dd, J = 9.2, 4.0 Hz, 1 H), 4.13 (dd, J = 9.2, 6.0 Hz, 1 H), 3.70 (m, 1 H), 3.42 (s, 3 H), 3.10 (d, J = 10.0 Hz, 1 H). 13C NMR (100 MHz, CDCl3) δ 172.51, 162.94, 145.58, 134.46, 128.82, 128.72, 127.90, 126.49, 123.48, 80.18, 71.20, 55.74, 52.15. IR (KBr) νmax: 2360.4, 1735.6, 1026.9, 701.0. ESI (+VE) m/z: 529.1 (M + Na)+. HR-ESI/APCI MS calcd for C31H26N2NaO5 (M +Na) +: 529.1739, Found: 529.1760.
O-(1,3-Dihydro-1,3-dioxo-2H-isoindol-2-yl)-N-[(phenylmethoxy)carbonyl]-l-serine Methyl Ester (9)
To a solution of 8 (2.00 g, 3.95 mmol) in CH2Cl2 (50 mL) at room temperature was added 37% HCl (0.50 mL) and the suspension was stirred (1 h) then quenched by addition of NaHCO3. The reaction mixture was extracted with CH2Cl2 and the combined organic layer was washed with brine and dried (Na2SO4). The solvent was removed by rotary evaporation and the residue was dissolved in THF (30 mL) with H2O (10 mL). To this was added NaHCO3 (498 mg, 5.92 mmol) and benzyl chloroformate (0.59 mL, 4.15 mmol) and the mixture was stirred at room temperature (overnight). THF was removed by rotary evaporation and the resulting aqueous phase was extracted with EtOAc and the combined organic layer was washed with H2O (2 × 50 mL) and brine (50 mL) and dried (Na2SO4), then purified by silica gel column chromatography (hexanes:EtOAc) to yield 9 as a viscous colorless oil (1.32 g, 84% yield). [α]20D + 31.8 (c 0.75, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.75 - 7.73 (m, 2 H), 7.67 - 7.65 (m, 2 H), 7.30 - 7.20 (m, 5 H), 6.26 (d, J = 8.8 Hz, 1 H), 5.07 (s, 2 H), 4.73 (dd, J = 10.6, 3.0 Hz, 1 H), 4.57 (m, 1 H), 4.33 (dd, J = 10.6, 3.4 Hz, 1 H), 3.63 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 169.56, 163.19, 156.11, 136.21, 134.76, 128.56, 128.45, 128.06, 127.97, 123.71, 77.61, 67.11, 53.45, 52.80. IR (KBr) νmax: 3366.1, 2954.4, 2363.3, 1727.9, 1516.7, 1211.1, 1054.9, 876.5, 698.1. HR-ESI/APCI MS cacld for C20H18N2NaO7 (M +Na) +: 421.1011, Found: 421.1007.
General Procedure for the Preparation of 13 and 14
To a solution of N-Cbz-L-homoserine O-methyl ester (10)12 or 5-hydroxy-N-[(phenylmethoxy)carbonyl]-l-norvaline (12)14 (1.0 eq.) in CH2Cl2 (30 mL) at 0 °C, was added triethylamine (1.5 equiv.) and methylsulfonyl chloride (1.2 equiv.) and the mixture was stirred (1 h). The mixture was diluted with CH2Cl2 (100 mL) then washed with H2O (50 mL) and brine (50 mL) and dried (Na2SO4). Purificaiton by silica gel column chromatography (hexanes: EtOAc) provided 13 (from 10) and 14 (from 12) as viscous colorless oils.
N-Benzyloxycarbonyl-O-mesyl-L-serine Methyl Ester (13)
Prepared from 10 (2.70 g, quantitative yield). [α]20D + 5.0 (c 0.30, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.36 - 7.30 (m, 5 H), 5.61 (d, J = 6.0 Hz, 1 H), 5.11 (s, 2 H), 4.50 (m, 1 H), 4.28 (m, 2 H), 3.76 (s, 3 H), 2.96 (s, 3 H), 2.33 (m, 1 H), 2.14 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 173.73, 171.86, 155.93, 135.97, 128.55, 128.29, 128.12, 67.21, 65.69, 52.76, 50.76, 37.14, 31.79. IR (KBr) νmax: 3327.6, 2940.0, 1696.1, 1538.0, 1341.3, 1172.5, 1005.7, 740.5. ESI-MS (+VE) m/z: 368.0 (M + Na)+.
5-[(Methylsulfonyl)oxy]-N-[(phenylmethoxy)carbonyl]-L-norvaline Phenylmethyl Ester (14)
Prepared from 12 (3.95 g, quantitative yield). [Note: This material was used in the next step without further purification.] 1H NMR (400 MHz, CDCl3) δ 7.40-7.30 (10H, m), 5.36 (1H, d, J = 8.0 Hz) 5.18 (2H, m), 5.11 (2H, s), 4.45 (1H, q, J = 8.0 Hz), 4.20 (2H, t, J = 8.0 Hz), 2.95 (3H, s), 2.01 (1H, m), 1.84-1.70 (3H, m). ESI-MS m/z 525.1 (M + Na+).
General Procedure for the Preparation of 15 and 16
To a solution of N-hydroxyphthalimide (2.0 equiv.) in DMF (10 mL) at room temperature was added a solution of DBU (2.0 equiv.) in DMF (10 mL). The mixture was cooled to 0 °C and stirred (30 min), then a solution of either 13 (to prepare 15) or 14 (to prepare 16) (1.0 eq.) in DMF (10 mL) was added dropwise. The mixture was warmed to room temperature and stirred (2 days). The solution was diluted with EtOAc (200 mL) and washed with H2O (50 mL) and brine (50 mL) and dried (Na2SO4). Purification by silica gel column chromatography (hexanes:EtOAc) provided products 15 and 16 as white solids.
O-(1,3-Dihydro-1,3-dioxo-2H-isoindol-2-yl)-N-[(phenylmethoxy)carbonyl]-l-homoserine Methyl Ester (15)12
Prepared from 13 (2.20 g, 71% yield). [α]20D + 8.6 (c 1.95, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.77 - 7.74 (m, 2 H), 7.71 - 7.67 (m, 2 H), 7.31 - 7.22 (m, 5 H), 6.16 (d, J = 8.4 Hz, 1 H), 5.07 (s, 2 H), 4.58 (q, J = 6.8 Hz, 1 H), 4.26 (m, 2 H), 3.70 (s, 3 H), 2.27 (m, 2 H). 13C NMR (100 MHz, CDCl3) δ 172.02, 163.47, 156.14, 136.39, 134.60, 128.74, 128.40, 127.97, 127.91, 123.60, 74.98, 66.85, 52.55, 51.44, 30.19. IR (KBr) νmax: 3331.4, 2364.3, 2343.1, 1725.0, 1682.6, 1529.3, 1216.9, 698.1. ESI (+VE) m/z: 435.1 (M + Na)+. HR-ESI/APCI MS calcd for C21H20N2NaO7 (M +Na)+: 435.1168, Found: 435.1176.
5-[(1,3-Dihydro-1,3-dioxo-2H-isoindol-2-yl)oxy]-N-[(phenylmethoxy)carbonyl]-l-norvaline Phenyl-methyl Ester (16)
Prepared from 14 (2.70 g, 61% yield). [α]20D-16.34 (c 0.35, CHCl3). mp: 101 - 105 °C. 1H NMR (400 MHz, CDCl3) δ 7.83-7.80 (2H, m), 7.76-7.73 (2H, m), 7.40-7.29 (10H, m), 5.47 (1H, d, J = 8.0 Hz), 5.20 (2H, s), 5.10 (2H, s), 4.49 (1H, m), 4.19 (2H, m), 2.16 (1H, m), 2.02 (1H, m), 1.81 (2H, m). 13C NMR (100 MHz, CDCl3) δ; 171.95, 163.57, 155.94, 136.23, 135.23, 134.45, 128.86, 128.56, 128.44, 128.38, 128.23, 128.06, 127.98, 123.50, 67.20, 66.91, 53.63, 28.83, 24.28. IR (neat) νmax = 3324, 3066, 3033, 2965, 2936, 2361, 2336, 1725, 1698, 1539, 1276, 1186, 1127, 1018, 876, 736, 696. HR-ESI/APCI MS calcd for C28H26N2NaO7 (M + Na) +: 525.1638, Found: 525.1641.
General Procedure for the Preparation of 17 - 20
To a solution of intermediates 9, 15 or 16 (1.0 eq.) in CH2Cl2 (20 mL) at 0 °C was added methylhydrazine (1.5 equiv.) and the reaction mixture was stirred at 0 °C (1 h). The mixture was passed through celite and the filtrate was concentrated and dried under high vacuum (30 min). For product 20 see below.* For products 17 - 19, the residue was re-dissolved in THF (50 mL) and to this was added triethylamine (2.0 equiv.) and Boc2O (2.0 equiv.) and the solution was stirred at room temperature (overnight). The reaction mixture was diluted with EtOAc (200 mL) and washed with H2O (2 × 50 mL) and brine (50 mL) and dried (Na2SO4). Purification by silica gel column chromatography (hexanes:EtOAc) provided 17 - 19 as viscous colorless oils. For product 20, the residue above* prepared by treatment of 9 with methylhydrazine was dissolved in CH2Cl2 (10 mL) and to this was added diisopropylethylamine (DIPEA) (2.0 equiv.) and 4-methyltrityl chloride (1.2 equiv.) and the mixture was stirred room temperature (1 h). The mixture was diluted with EtOAc (100 mL) and washed with H2O (50 mL) and brine (50 mL) and dried (Na2SO4). Purification by column chromatography (hexanes:EtOAc) using triethylamine-pretreated silica gel provided 20 as a white wax.
3-[[(1,1-Dimethylethoxy)carbonyl]aminooxy]-N-[(phenylmethoxy)carbonyl]-l-serine Methyl Ester (17)
Prepared from 9 (1.05 g, 95% yield). [α]20D - 11.7 (c 1.33, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1 H), 7.31 - 7.23 (m, 5 H), 6.18 (d, J = 8.4 Hz, 1 H), 5.09 (s, 2 H), 4.54 (m, 1 H), 4.19 (dd, J = 7.2 Hz, 4.4 Hz, 1 H), 4.03 (dd, J = 11.0, 3.4 Hz, 1 H), 3.70 (s, 3 H), 1.42 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 170.54, 156.87, 156.32, 136.24, 128.44, 128.06, 127.97, 82.30, 75.89, 67.00, 53.18, 52.60, 28.09. IR (KBr) νmax: 3297.7, 2978.5, 2361.4, 1716.3, 1519.6, 1210.1, 1055.8, 775.2. ESI-MS (+VE) m/z: 391.1 (M + Na)+. HR-ESI/APCI MS calcd for C17H24N2NaO7 (M +Na) +: 391.1481, Found: 391.1482.
4-[[(1,1-Dimethylethoxy)carbonyl]aminooxy]-N-[(phenylmethoxy)carbonyl]-l-homeserine Methyl Ester (18)
Prepared from 15 (0.90 g, 97% yield). [α]20D + 5.8 (c 0.65, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1 H), 7.37 - 7.26 (m, 5 H), 6.10 (d, J = 7.6 Hz, 1 H), 5.12 (s, 2 H), 4.52 (dd, J = 13.6, 6.4 Hz, 1 H), 4.00 - 3.87 (m, 2 H), 3.73 (s, 3 H), 2.12 (m, 2 H), 1.47 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 172.56, 156.94, 156.13, 136.34, 128.41, 128.00, 127.91, 81.88, 72.87, 66.81, 52.44, 51.62, 30.28, 28.12. IR (KBr) νmax: 3308.3, 2977.6, 2361.4, 1703.8, 1527.4, 1246.7, 1162.9, 739.6. ESI-MS (+VE) m/z: 405.1 (M + Na)+. HR-ESI/APCI MS calcd for C18H26N2NaO7 (M +Na) +: 405.1638, Found: 405.1647.
5-[[(1,1-Dimethylethoxy)carbonyl]aminooxy]-N-[(phenylmethoxy)carbonyl]-l-norvaline Phenylmethyl Ester (19)
Prepared from 16 (1.85 g, 80% yield). [α]20D -5.92 (c 0.58, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.38-7.28 (10H, m), 7.15 (1H, s), 5.56 (1H, d, J = 8.0 Hz), 5.22-5.13 (2H, m), 5.10 (2H, s), 4.42 (1H, q, J = 8.0 Hz), 3.81 (2H, t, J = 6.0 Hz), 2.23-1.92 (1H, m), 1.90-1.80 (1H, m), 1.72-1.60 (2H, m), 1.45 (9H, s). 13C NMR (100 MHz, CDCl3) 172.15, 157.00, 156.05, 136.22, 135.27, 128.56, 128.44, 128.39, 128.23, 128.07, 81.70, 75.74, 67.09, 66.93, 53.77, 29.00, 28.14, 27.57, 23.93. IR (neat) νmax = 3310, 3065, 3034, 2976, 2940, 2881, 1711, 1523, 1454, 1367, 1247, 1165, 1110, 1044, 911, 734, 697. ESI-MS m/z 495.1 (M + Na+). HR-ESI/APCI MS calcd for C25H32N2NaO7 (M + Na) +: 495.2107, Found: 495.2110.
3-[(Diphenyl-p-tolyl-methyl)-aminooxy]-N-[(phenylmethoxy)carbonyl]-l-serine Methyl Ester (20)
Prepared from 9 (1.31 g, quantitative yield). [α]20D + 6.5 (c 1.08, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.33 - 7.16 (m, 15 H), 7.13 (AB, JAB = 8.4 Hz, 2 H), 7.05 (AB, JAB = 8.4 Hz, 2 H), 6.69 (s, 1 H), 5.40 (d, J = 8.8 Hz, 1 H), 5.06 (m, 2 H), 4.52 (m, 1 H), 3.91 (dd, J = 10.8, 4.8 Hz, 1 H), 3.84 (dd, J = 10.8, 3.6 Hz, 1 H), 3.59 (s, 3 H), 2.28 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 171.1, 156.0, 144.2, 141.1, 136.6, 136.3, 129.1, 129.0, 128.6, 128.5, 128.2, 128.1, 127.8, 127.0, 73.9, 67.0, 53.8, 52.5, 21.0. IR (KBr) νmax: 3402.8, 2949.6, 1717.3, 1509.0, 1208.2, 1058.7, 698.1. ESI-MS (+VE) m/z: 547.2 (M + Na)+. HR-ESI/APCI MS calcd for C32H32N2NaO5 (M +Na) +: 547.2209, Found: 547.2213.
General Procedure for the Preparation of Final Products 1a - 1c and 2a
To a solution of intermediates 17, 18 or 20 (1.0 equiv.) in THF (10 mL) with H2O (10 mL) at 0 °C, was added LiOH·H2O (1.2 equiv.) and the mixture was stirred at 0 °C (2 h). THF was removed by rotary evaporation and the residual aqueous phase was washed with Et2O then acidified to pH 3 - 4 by addition of 1 N HCl. (Note: For 20 saturated NH4Cl was used rather than 1 N HCl.) The mixtures were extracted with EtOAc (3 × 50 mL) and the combined EtOAc extracts were washed with H2O (50 mL) and brine (50 mL) and dried (Na2SO4). The organic phase was concentrated and the residue was dissolved in MeOH (20 mL) and stirred with 10% Pd·C under 1 atm H2 (2 h). (Note: The preparation of 1c began at this point by the direct hydrogenation of 19) The Pd·C was removed by filtration and the filtrate was concentrated and the residue was dissolved in dioxane (10 mL) and H2O (10 mL). To this was added 9-fluorenylmethyl succinimidyl carbonate (FmocOSu) (1.1 equiv.) and NaHCO3 (2.0 equiv.) and the mixture was stirred at room temperature (overnight). The reaction solution was acidified to pH 3 - 4 by addition of 1 N HCl. (Note: For 2a saturated NH4Cl was used rather than 1 N HCl.) The mixture was extracted with EtOAc (3 × 50 mL) and the combined EtOAc extract was washed with H2O (50 mL) and brine (50 mL) and dried (Na2SO4). Purificaiton by silica gel column chromatography (CH2Cl2:MeOH) provided final products 1a - 1c and 2a as viscous colorless oils. Lyophilization from CH3CN/H2O provided white powders, which were suitable for solid-phase applications.
(3S)-3-Carboxy-9,9-dimethyl-7-oxo-5,8-dioxa-2,6-diazadecanoic Acid 1-(9H-Fluoren-9-ylmethyl) Ester (1a)
Prepared from 17 (0.62 g, 58% yield over 3 steps). [α]20 + 4.4 (c 1.30, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.97 (brs, 1 H), 7.30 (d, J = 7.6 Hz, 2 H), 7.65 - 7.50 (m, 2 H), 7.37 (t, J = 7.6 Hz, 2 H), 7.30 - 7.17 (m, 2 H), 6.48 (brs, 1 H), 4.54 (m, 1 H), 4.37 (m, 2 H), 4.28 (dd, J = 11.4, 3.4 Hz, 1 H), 4.22 (d, J = 7.2 Hz, 1 H), 4.02 (dd, J = 11.4, 5.4 Hz, 1 H), 1.46 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 158.29, 156.56, 143.70, 143.56, 141.25, 127.72, 127.07, 125.14, 119.95, 83.81, 75.79, 67.45, 52.73, 46.98, 28.02, 27.55. IR (KBr) νmax: 3288.0, 2977.6, 2364.3, 1700.9, 1521.6, 1449.2, 1160.0, 734.7. ESI-MS (+VE) m/z: 465.1 (M + Na)+. HR-ESI/APCI MS calcd for C23H26N2NaO7(M +Na) +: 465.1638, Found: 465.1636.
(3S)-3-Carboxy-10,10-dimethyl-8-oxo-6,9-dioxa-2,7-diazadecanoic Acid, 1-(9H-fluoren-9-ylmethyl) Ester (1b)
Prepared from 18 (1.60 g, 81% yield over 3 steps). [α]20 + 9.0 (c 0.79, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.6 Hz, 2 H), 7.69 - 7.60 (m, 2 H), 7.52 (m, 1 H), 7.36 (t, J = 7.4 Hz, 2 H), 7.27 (t, J = 7.6 Hz, 2 H), 6.67 (brs, 1 H), 4.55 (m, 1 H), 4.43 - 4.28 (m, 2 H), 4.20 (t, J = 7.4 Hz, 1 H), 3.96 (m, 2 H), 2.15 (m, 2 H), 1.47 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 175.64, 157.92, 156.86, 143.81, 143.67, 141.23, 127.68, 127.07, 125.21, 119.91, 106.60, 82.81, 73.12, 67.37, 51.77, 47.00, 29.68, 28.12, 27.54. IR (KBr) νmax: 3296.7, 2977.6, 2355.6, 1700.9, 1523.5, 1160.9, 1106.9, 909.3, 735.7. ESI-MS (+VE) m/z: 479.1 (M + Na)+. HR-ESI/APCI MS calcd for C24H28N2NaO7 (M +Na) +: 479.1794, Found: 479.1808.
(3S)-3-Carboxy-11,11-dimethyl-9-oxo-7,10-dioxa-2,8-diazadecanoic Acid 1-(9H-Fluoren-9-ylmethyl) Ester (1c)
Prepared from 19 (0.82 g, 47% yield over 2 steps). [α]20 + 0.152 (c 0.77, CHCl3). mp: 55 - 65 °C. 1H NMR (400 MHz, CDCl3) δ 7.75 (2H, d, J = 7.6 Hz), 7.62-7.56 (2H, m), 7.39 (2H, t, J = 6.8 Hz), 7.30 (2H, t, J = 7.2 Hz), 5.74 (1H, s), 4.56-4.32 (3H, m), 4.22 (1H, t, J = 6.8 Hz), 3.91 (2H, s), 2.16-2.04 (1H, m), 1.96-1.84 (1H, m), 1.78-1.68 (2H, m), 1.47 (9H, s). 13C NMR (100 MHz, CDCl3) δ 157.44, 156.49, 143.85, 141.20, 127.63, 127.04, 125.14, 119.88, 82.09, 75.80, 67.12, 53.72, 47.05, 38.33, 30.89, 28.76, 28.17, 23.90. IR (neat) νmax = 3295, 2977, 2941, 2879, 1710, 1525, 1451, 1368, 1249, 1165, 1110, 1043, 758, 739, 699. ESI-MS m/z 493.1 (M + Na+). HR-ESI/APCI MS calcd for C25H30N2NaO7 (M + Na) +: 493.1951, Found: 493.1957.
3-[(Diphenyl-p-tolyl-methyl)-aminooxy]-N-[[(9H-fluoren-9-ylmethyl)oxy]carbonyl]-l-serine (2a)
Prepared from 20 (0.40 g, 58% yield over 3 steps). [α]20D - 9.4 (c 0.75, CHCl3). 1H NMR (400 MHz, DMSO-d6) δ 7.91 (dd, J = 7.4, 2.2 Hz, 1 H), 7.71 (t, J = 5.4 Hz, 1 H), 7.65 (m, 1 H), 7.45 - 7.16 (m, 16 H), 7.09 - 7.00 (m, 5 H), 5.02 (dd, J = 16.2, 12.6 Hz, 1 H), 4.32 - 4.18 (m, 3 H), 3.84 (m, 1 H), 3.60 (t, J = 9.6 Hz, 1 H), 2.25 (s, 1.6 H), 2.21 (s, 1.4 H). 13C NMR (100 MHz, DMSO-d6) δ 172.4, 156.6, 145.1, 144.3, 142.0, 141.2, 137.4, 136.0, 129.2, 128.8, 128.5, 128.2, 128.1, 127.9, 127.5, 127.0, 125.7, 120.5, 74.1, 73.4, 66.3, 65.9, 54.3, 47.0, 21.0. IR (KBr) νmax: 3404.7, 2365.3, 1706.7, 1508.1, 1229.4, 1056.8, 700.0. ESI-MS (+VE) m/z: 621.2 (M + Na)+. HR-ESI/APCI MS calcd for C38H34N2NaO5 (M +Na) +: 621.2365, Found: 621.2368.
Determination of Enatiomeric Purity of 1c
Phenylalanine dipeptides were prepared by solid-phase techniques (Figure 2), and the resulting diastereomers were separated by reverse-phase HPLC (Figure 3). Analysis of the racemic d, l-phenylalanine containing dipeptides (20) indicated good separation of diastereomers. The dipeptide 21 prepared using enantiomerically pure l-phenylalanine did not show detectable diastereomeric contamination, indicating that the ee value of 1c is > 98%.
Figure 2.
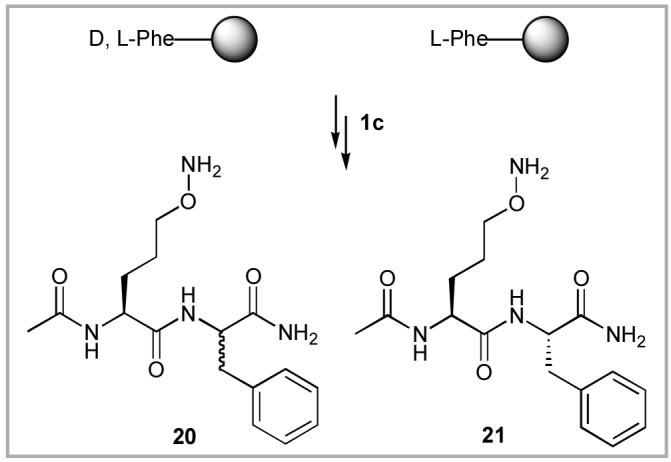
Determination of enantiomeric puriy of 1c.
Figure 3.

HPLC chromatograms of 20 (top) and 21 (bottom). Identity of the 3 major peaks were confirmed by ESI mass spectra [337.1(M+H)+ and 359.1 (M+Na)+].
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick.
References
- (1).Erlanson DA, MCDowell RS, O’Brien T. J. Med. Chem. 2004;47:3463. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- (2)(a).Maly DJ, Choong IC, Ellman JA. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2419–2424. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang YL, Krosky DJ, Seiple L, Stivers JT. J. Am. Chem. Soc. 2005;127:17412. doi: 10.1021/ja055846n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Spetzler JC, Hoeg-Jensen T. J. Peptide Sci. 1999;5:582. doi: 10.1002/(SICI)1099-1387(199912)5:12<582::AID-PSC228>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (4).Lang I, Donze N, Garrouste P, Dumy P, Mutter M. J. Peptide Sci. 1998;4:72. doi: 10.1002/(sici)1099-1387(199802)4:1<72::aid-psc130>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- (5).Adamczyk M, Reddy RE. Synth. Commun. 2001;31:579. [Google Scholar]
- (6)(a).McGarvey GJ, Benedum TE, Schmidtmann FW. Org. Lett. 2002;4:35914. doi: 10.1021/ol0264861. [DOI] [PubMed] [Google Scholar]; b) Pernerstorfer J, Schuster M, Blechert S. Chem. Commun. 1997:1949. [Google Scholar]; (c) Ryan SJ, Zhang Y, Kennan A. J. Org. Lett. 2005;7:4765. doi: 10.1021/ol0520222. [DOI] [PubMed] [Google Scholar]; (d) Xie W, Zou B, Pei D, Ma D. Org. Lett. 2005;7:2775. doi: 10.1021/ol050991r. [DOI] [PubMed] [Google Scholar]
- (7).Yang Y-K, Tae J. SynLett. 2003:2017. [Google Scholar]
- (8).Jones DS, Hammaker JR, Tedder ME. Tetrahedron Lett. 2000;41:1531. [Google Scholar]
- (9).Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- (10).Kelleher F, Proinsias KO. Tetrahedron Lett. 2007;48:4879. [Google Scholar]
- (11).Peluso S, Imperiali B. Tetrahedron Lett. 2001;42:2085. [Google Scholar]
- (12).Wolfe S, Wilson M-C, Cheng M-H, Shustov GV, Akuche CI. Can. J. Chem. 2003;81:937. [Google Scholar]
- (13). Available from Novabiochem, Inc.
- (14).Jiang S, Li P, Lai CC, Kelley JA, Roller PP. J. Org. Chem. 2006;71:7307. doi: 10.1021/jo061037q. [DOI] [PubMed] [Google Scholar]
- (15).Easmon J, Heinisch G, Holzer W, Matuszczak B. Archiv der Pharmazie. 1995;328:367. doi: 10.1002/ardp.19953280403. [DOI] [PubMed] [Google Scholar]
- (16)(a).Liu F, Stephen AG, Adamson CS, Gousset K, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr. Org. Lett. 2006;8:5165. doi: 10.1021/ol0622211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu F, Stephen AG, Fisher RJ, Burke TR. Bioorg. Med. Chem. Lett. 2008;18:1096. doi: 10.1016/j.bmcl.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). The major fraction obtained by HPLC purification of the peptide resulting using 1a to replace the Thr residue, provided a mass spectral molecular ion (1422.4) that was 31 amu lower than expected for the correct product (1453.5). This is consistent with beta elimination of HO-NH2 followed by reduction of the resulting double bond during under the reducing conditions of resin cleavage using triethylsilane.
- (18). The major fraction obtained by HPLC purification of the peptide resulting from the use of 2a to replace the Thr residue, provided mass spectral molecular ions consistent with the desired peptide product [m/z 1454.5, (M+H) and 1476.5 (M+Na)]