

Abstract
The identification and study of adaptively important genes in forest trees represents a formidable challenge because of their long generation spans. In annual or perennial herbs, formal genetic studies can be employed to identify the quantitative trait loci (QTLs) and/or candidate genes that underlie important traits, and the segregating populations can be transplanted into natural populations to measure the strength and direction of selection. However, the application of these methods to forest trees is difficult, because the creation of appropriate genetic material is extremely time-consuming in long-lived, woody plants, and lifetime fitness estimates are difficult or impossible to obtain. Although QTL mapping should in principle be feasible in wild intraspecific populations (as an alternative to artificial crosses), this approach is less likely to be successful in trees because LD (linkage disequilibrium) will decay quickly in large outbreeding plant populations. Within the present paper, we discuss a modified approach based on natural hybrid zones. We describe the use of wild annual sunflowers (Helianthus spp.) as a model for exploring the hybrid zone approach. Transplanted experimental hybrids allowed us to assess the adaptive value of individual chromosomal blocks in nature, and data on natural Helianthus hybrids suggest that similar approaches are possible in natural hybrid zones. Our results allowed us to test the role of hybridization in the origin of ecological divergence in wild sunflowers. In addition, they have practical implications for identifying adaptively important genes or QTLs in trees. This is exemplified by three temperate forest taxa, Populus (poplars, aspens, cottonwoods), Fraxinus (ash), and Quercus (oak). All three are diploid and important genomic tools are under development. Moreover, all three offer extensive hybrid zones whose likely age can be inferred from fossil data. Age data enables estimates of the size and frequency of chromosomal blocks in hybrids, thereby providing guidance in designing marker-based experiments. We predict that natural hybrid zones will be valuable tools for identifying the QTLs and/or candidate genes responsible for adaptive traits in forest trees.
Keywords: Hybrid zones, Admixture, Introgression, Adaptation, QTL, Species barriers
1. Introduction
The importance of adaptive genetic variation is widely recognized in forest ecology and management. The longevity and wide geographic distributions of tree species place a premium on genomic diversity because of the need to adapt to a wide range of site conditions (Hamrick and Godt, 1989; Isabel et al., 1995; Hamrick and Nason, 2000). In addition to spatial and temporal niche heterogeneity, anthropogenic pressure imposes a further stress on forest tree populations, often challenging their adaptive abilities (Mueller-Starck and Schubert, 2001). Hence, governments throughout the world are increasingly aware of the need for forest reproductive material that is genetically diverse and locally adapted (Geburek and Heinze, 1998; Kanowski, 2000). Unfortunately, the actual genes involved in adaptation to specific habitats or niches have very rarely been identified in trees (but see Frewen et al. (2000)), and, to our knowledge, molecular variation in such genes has never been tracked across the distribution range for any species. This unfortunate lack of information can be attributed to difficulties in identifying the relevant genes, as outlined below.
For many tree species, variation in ecologically important traits has been studied by provenance trials, that is, by comparing the performance of plants from different origins in homogeneous environments (Wright, 1976). Provenance trials, similar to reciprocal transplantation experiments, may be useful for testing the adaptive potential (or evolutionary constraints) of plant species in response to global warming, as demonstrated by a recent study on annual species (Etterson and Shaw, 2001). However, the power of provenance trials is rather limited if the goal is to identify the actual genes or quantitative trait loci (QTLs) involved in local adaptation. This is the case because in provenance trials, genotypes from geographically separated populations are mixed. Therefore, any association between traits and markers, potentially indicative of physical proximity along the chromosomes, will be confounded with the statistical interdependence caused by admixing alleles from different populations across all loci in the genome (Weir, 1996; Lynch and Walsh, 1998). Also, provenance trials may be too slow and time-consuming in forest trees, given the probable pace of climate change.
Many researchers have attempted to draw inferences about adaptive processes by population genetic surveys with markers that are either neutral, or whose adaptive significance is unknown. Indeed, indirect evidence suggests that sampling across ecological barriers to gene flow (e.g. across elevational gradients) can disrupt patterns of isolation by distance at neutral markers, thereby revealing past selection pressures and/or restricted gene flow (Williams and Arnold, 2001). However, it is unlikely that this approach will allow direct inferences about adaptively important genes, since recombination will quickly erase associations between the markers assayed and the genes responsible for fitness differences (Weir, 1996; Hedrick, 2000). In fact, most studies testing for concordance in patterns of variation at adaptive traits and neutral markers either report negative results (e.g. Savolainen and Hedrick, 1995; Karhu et al., 1995; McKay and Latta, 2002), or similarities due to the same historical events rather than selection (Lagercrantz and Ryman, 1990).
Maternally inherited chloroplast (cp) DNA markers yielded valuable information about genetic variability associated with local populations or provenances (e.g. Petit et al., 1997; Ferris et al., 1998). However, as pointed out by Kremer et al. (2002), the chloroplast genome is unlikely to contain genes sufficient to account for variation in complex adaptive characters. Indeed, in a large survey of oak stands (Quercus robur and Q. petraea) and provenance trials throughout Europe, no association was detected between chloroplastic divergence and phenotypic traits (Kremer et al., 2002).
An alternative method for identifying the molecular genetic basis of adaptive phenotypic variation involves the use of quantitative trait locus (QTL) mapping (Tanksley, 1993; Zeng, 1994; Kao et al., 1999). The QTL approach allows the identification of molecular markers that are physically linked to the genes of interest, and colocalization of QTLs and candidate gene sequences may lead to the identification of exactly those genes that account for variation in adaptive traits (Frewen et al., 2000; review by Mauricio, 2001).
Unfortunately, the application of the QTL methodology to forest trees poses several difficulties. First, standard QTL analysis (Tanksley, 1993; Mauricio, 2001) requires experimental crosses that are time-consuming and often impossible to obtain for trees (but see Hurme et al. (2000) for an interesting solution). Second, both QTL and candidate gene analysis require sufficient phenotypic variance and marker polymorphism in the “mapping population” (Karp et al., 1997; Lynch and Walsh, 1998), two requirements that reduce the choice of suitable crosses further. Third, identifying adaptive variation requires that the adaptive value of traits, QTLs, and candidate genes be assessed in the wild (Lexer et al., 2003c), that is, directly in the forest stand where fitness differences really matter. A logical conclusion from these difficulties would be to study QTLs in natural intraspecific populations, an approach that should theoretically be feasible in many animal or plant species (Luo et al., 2000; Wu and Zeng, 2001). However, such analyses may be difficult in forest trees, because linkage disequilibrium (LD) between markers and traits will decay quickly in large outbreeding plant populations (Weir, 1996; Hedrick, 2000).
In the present paper, we discuss alternative experimental designs based on natural hybrid zones. We outline the use of wild annual sunflowers (Helianthus spp.) as a model system for exploring the hybrid zone approach. More specifically, we summarize a series of studies involving transplanted experimental sunflower hybrids (Lexer et al., 2003a,b) and natural hybrid zones (Rieseberg et al., 1999a; Rieseberg and Buerkle, 2002). The main goal of these studies was to assess the role of hybridization in the origin of ecological divergence in annual sunflowers. We discuss the evolutionary implications of this work, as well as practical implications for comparable studies in forest trees, such as assays of the adaptive value of individual chromosome blocks or candidate genes directly in natural hybrid zones. We close by discussing the potential of the approach in selected forest tree genera.
2. Materials and methods
2.1. Study system Helianthus
The wild, annual sunflowers (Helianthus section Helianthus) include 12 self-incompatible, diploid (n = 17) species (Schilling and Heiser, 1981). The two most widespread species, Helianthus annuus and H. petiolaris, are abundant in the central and western US and have been the focus of our studies. They are easily distinguished by several morphological and chromosomal features (Heiser, 1947; Chandler et al., 1986), belong to divergent clades based on chloroplast DNA (Rieseberg et al., 1991) and nuclear ribosomal DNA variation (Rieseberg, 1991), and have different ecological requirements. In general, H. annuus is restricted to heavy, clay soils and H. petiolaris to dry, sandy soils. Nonetheless, these two habitats often are found in close proximity throughout the central and western US, resulting in the production of innumerable hybrid swarms or “mosaic” hybrid zones (Harrison and Rand, 1989). In addition, molecular data indicate that these two species gave rise to at least three diploid hybrid species in nature, H. anomalus, H. deserticola, and H. paradoxus (Rieseberg et al., 1990; Rieseberg, 1991). All three are adapted to extreme and novel habitats (sand dunes, desert floors, and salt marshes, respectively). It appears that the novel trait combinations required to colonize these extreme habitats originated by hybridization between the two parental species, H. annuus and H. petiolaris.
2.2. Plant materials
Two kinds of plant materials were used throughout this study: (i) experimental BC2 hybrids between H. annuus and H. petiolaris, (ii) natural hybrids sampled in hybrid zones between the same two parental species. The BC2 hybrids were used for transplantation experiments with the aim of studying selection on individual adaptive traits and QTLs. The natural hybrids were used for studying introgression of individual chromosome blocks across four replicate hybrid zones, and test for associations among markers and phenotypic traits.
2.2.1. Experimental BC2 hybrids
Experimental BC2 hybrids for the transplantation experiment were obtained by crossing two wild accessions of H. annuus and H. petiolaris and backcrossing a single interspecific F1 to a second individual of H. petiolaris. Because of the near sterility of the F1, only 38 BC1 plants could be generated. These 38 plants were subjected to a second round of backcrossing toward a single, third individual of H. petiolaris to obtain the BC2. Two-hundred-and-fifty-four BC2 hybrids were transplanted into salt marsh habitat of the ancient hybrid species H. paradoxus at the seedling stage, as part of a selection experiment to study the role of hybridization in the origin of salt adaptation (Lexer et al., 2003a,b). For 172 of these plants, DNA could be extracted for molecular marker genotyping (Lexer et al., 2003b).
2.2.2. Natural hybrids from sunflower hybrid zones
The four natural hybrid zones (H. annuus × H. petiolaris) discussed here have been described in detail elsewhere (Rieseberg et al., 1999a; Buerkle and Rieseberg, 2002). Each zone is less than 50 m in width, occurs in human-disturbed sites, and is distributed along a habitat gradient. For each zone, 4–5 seeds were collected from 10 to 20 plants sampled along a transect from the center to the H. annuus edge of the zone, and seeds were propagated in the Indiana University greenhouses (total sample size N = 228). Also, reference populations of each parental species were sampled for comparative purposes (N ~ 35 per species).
2.3. Field and laboratory methods
2.3.1. Experimental BC2 hybrids
The following characters were measured for the transplanted BC2 hybrids: sodium content (Na), sulfur content (S), magnesium content (Mg), boron content (B), calcium content (Ca), potassium content (K), leaf shape (LFSHAP), and leaf succulence (LFSUC) as potentially adaptive traits, and survivorship in days as well as growth rate in the field as potential fitness proxies (described in detail in Lexer et al. (2003a)).
Total genomic DNA was isolated from ~100 mg of dried leaf tissue per plant using the DNeasy plant mini kit (QIAGEN, Valencia, CA), and microsatellites isolated from cultivated H. annuus (Tang et al., 2002) were used for genome-wide marker analysis. Microsatellite PCRs were performed in 96 well-format and analyzed on an ABI 3700 automated sequencer as described by Burke et al. (2002) and Lexer et al. (2003b). A total of 71 microsatellite loci were typed in the experimental BC2.
2.3.2. Natural hybrids
The following characters were measured for plants from natural hybrid zones: 10 morphological characters including leaf ratio (length/width), the length of flowering stems, the length of disk flowers, phyllary width/shape/pubescence, stem pubescence, leaf serration, as well as chaff pubescence and color. Also, pollen viability was assayed for 100 pollen grains per plant using enzymatic staining (described in detail in Rieseberg and Buerkle (2002)).
DNA for molecular analyses was extracted as described above for BC2 hybrids. Random amplified polymorphic DNAs (RAPDs; Williams et al., 1990) were used for studying introgression patterns in the four replicate hybrid zones. RAPD reactions were analyzed using 1.5% TBE agarose gels and ethidium bromide staining as described by Rieseberg et al. (1999a). In total, 88 RAPD bands were analyzed.
2.4. Data analysis
2.4.1. Experimental BC2 hybrids
The transplantation experiment with BC2 hybrids was used for assaying the strength of selection on candidate adaptive traits and on the underlying QTLs. Selection on candidate traits was calculated as directional selection (affecting the mean of phenotypic traits), stabilizing/disruptive selection (affecting trait variances), and correlational selection (affecting trait covariances), following the methods of Lande and Arnold (1983). Briefly, directional selection gradients were estimated as partial regression coefficients using multiple regression, directional selection differentials were estimated as the covariances between traits and fitness, stabilizing/disruptive selection differentials as the covariances between relative fitness and the variance of each trait, and correlational selection differentials as the covariances between relative fitness and the pairwise products of character deviations from the trait means, following Lande and Arnold (1983) and Lynch and Walsh (1998). All analyses were run on standardized trait values. This multiple regression-based approach is outlined in more detail in Lexer et al. (2003a). Potassium (K) uptake was excluded from these analyses because it violated basic assumptions of regression analysis. The four mineral uptake characters Na, B, Mg, and S were replaced by a principal component in selection analyses, because they were highly correlated based on Pearson’s correlation coefficients.
QTL analyses in experimental hybrids employed a BC2 model. Briefly, marker alleles segregating from H. annuus were scored in a H. petiolaris genetic background. The QTLs for five candidate adaptive traits (Na, Ca, S, Mg, and K content in leaves) and one fitness character (survivorship in the salt marsh, measured in days) were mapped on an interspecific linkage map constructed for the same BC2 cross, using composite interval mapping (CIM; Zeng, 1993, 1994) as described in detail by Lexer et al. (2003b). Genome-wide threshold values for declaring the presence of QTLs were determined by 1000 permutations for each trait (Churchill and Doerge, 1994), and QTL magnitudes were expressed as the percent phenotypic variation explained (PVE) in the BC2.
Selection coefficients (s) for individual QTLs were calculated using the nearest molecular marker for each QTL (the marker closest to the LR peak) as a surrogate and survivorship in days as a fitness measure. Selection estimates were obtained for heterozygous BC2 genotypes carrying a marker allele derived from H. annuus, as described by Lexer et al. (2003b). Because the degree of dominance cannot be tested in a back-cross breeding design, gene action was assumed to be purely additive (h = 0.5).
2.4.2. Natural hybrids
The hybrid zone data were subjected to an analysis of marker introgression as described in detail in Rieseberg et al. (1999a). Briefly, maximum likelihood (ML) estimates of hybrid indices were obtained for each plant collected in the hybrid zone, making use of marker loci with diagnostic or frequency differences between the two parental species. These hybrid indices estimate the “hybridity” of each individual and vary from 0 to 1. Next, estimates of over-all marker introgression were obtained for each individual on the basis of hybrid indices, and deviations from expected introgression rates were then calculated for each locus using likelihood ratio tests. Associations between pollen viability and molecular markers were tested using permutation procedures, and fertility reduction scores at each locus were scaled for the effects of neighboring loci in the genome, as described in Rieseberg et al. (1999a). In order to explore the potential of the hybrid zone approach further, product-moment correlations were calculated as a measure of association between markers, and phenotypic traits in natural hybrids were analyzed using descriptive statistics (described by Rieseberg and Buerkle (2002)).
3. Results
3.1. Transplanted BC2 hybrids—adaptive value of individual chromosome blocks
3.1.1. Selection on candidate adaptive traits
By studying associations between candidate adaptive traits and fitness in experimental BC2 sunflower hybrids transplanted into the wild we were able to detect significant directional selection on three phenotypic traits: (1) leaf succulence, (2) Ca uptake, and (3) a principal component including Na and other elemental uptake characters. Directional selection on these traits was calculated in the form of directional selection gradients (Fig. 1) and directional selection differentials (Table 1, directional selection). The relationships among candidate adaptive traits expressed in this extreme salt marsh habitat is depicted in Table 2. Trait correlations (lower triangular matrix) are particularly strong between Na, S, Mg, and B uptake, which is why these traits were summarized by a principal component. Significant pairwise correlations were also observed between Ca uptake or leaf succulence and other characters (Table 2), however, these traits were not included in a composite variable, because additional multivariate analyses suggested they were largely independent (Lexer et al., 2003a).
Fig. 1.

Partial regression plot of directional selection gradients on four candidate adaptive traits: (A) leaf succulence; (B) Ca content; (C) the first principal component (PC1) of Na, S, Mg, and B content; (D) leafshape. For each plot, the respective candidate trait and relative fitness (survivorship in days) were regressed on all other traits to obtain residuals from each regression. The relative fitness residuals were then regressed on the candidate adaptive trait residuals. The selection gradients (beta) given in the upper left or right corner of each graph correspond to regression coefficients estimated from the full regression model (*: P < 0.05; ***: P < 0.005). Residuals of candidate adaptive traits were measured in standard deviation units. The entire multiple regression model, including all four traits as well as blocks to account for environmental variation, accounted for 38% of the fitness variation in the BC2.
Table 1.
Directional and correlational selection differentials in transplanted BC2 sunflower hybrids
| Na | S | Mg | B | Ca | LFSHAP | LFSUC | |
|---|---|---|---|---|---|---|---|
| Directional selectiona | −0.250*** | −0.250*** | −0.250*** | −0.250*** | +0.062 | −0.003 | +0.132*** |
| Correlational selectionb | |||||||
| Na | |||||||
| S | +0.058** | ||||||
| Mg | +0.079 | −0.009 | |||||
| B | −0.032 | −0.081 | −0.050 | ||||
| Ca | −0.229*** | −0.157** | −0.224*** | −0.163*** | |||
| LFSHAP | −0.064 | −0.037 | +0.001 | −0.020 | +0.004 | ||
| LFSUC | +0.009 | +0.020 | +0.117 | +0.047 | +0.123** | +0.077 | |
Directional selection differentials: significance levels for individual tests,
P < 0.005. Directional selection differentials for Na, S, Mg, and B are for the first principal component of these variables.
Correlational selection differentials:
: P < 0.01,
P < 0.005 for individual tests. Bold type: P < 0.05 after sequential Bonferroni correction. For trait abbreviations see text.
Table 2.
Correlations and covariances between phenotypic traits in transplanted BC2 sunflower hybrids
| Na | S | Mg | B | Ca | LFSHAP | LFSUC | |
|---|---|---|---|---|---|---|---|
| Na | +0.492 | +0.452 | +82.560 | +0.300 | −0.113 | −1.522 | |
| S | +0.607*** | +0.045 | +9.612 | +0.087 | −0.020 | −0.101 | |
| Mg | +0.675*** | +0.441*** | +12.938 | +0.096 | +0.017 | −0.135 | |
| B | +0.397*** | +0.301*** | +0.490*** | +25.405 | +3.269 | −41.734 | |
| Ca | +0.165* | +0.312*** | +0.415*** | +0.354*** | +0.111 | −0.244 | |
| LFSHAP | −0.070 | −0.074 | +0.085 | +0.059 | +0.193* | −0.001 | |
| LFSUC | −0.362*** | −0.145 | −0.257** | −0.291*** | −0.163* | −0.001 |
Upper triangular matrix: covariances. Lower triangular matrix: Pearson’s correlation coefficients.
: P < 0.05,
: P < 0:01,
P < 0:005 for individual tests. Bold type: P < 0:05 after sequential Bonferroni correction. For trait abbreviations see text.
The direction of selection on phenotypic traits (Fig. 1) contains important information about their possible functional significance. Positive directional selection on leaf succulence (salt succulence) and Ca uptake from the field soil confirms an important role for these traits in salt stress response in wild sunflowers (Yeo, 1998; Hasegawa et al., 2000; Welch and Rieseberg, 2002), and negative directional selection on Na uptake (and correlated minerals) indicates that net exclusion of Na from leaves provides a fitness advantage in the salt marsh. Surprisingly, no significant selection was detected for leaf shape (leaf length/width), a diagnostic morphological character in sunflowers (Fig. 1; Table 1).
While no stabilizing or disruptive selection was detected, an interesting result was obtained regarding the change in trait correlations due to selection (Table 1, correlational selection). Only one pair of traits experienced a significant change in trait correlations after correction for multiple tests, namely Ca and Na uptake (Table 1). Also, Ca/Na ratios were under strong positive directional selection in this habitat (s = +0.294). This indicates an adaptive role for increased Ca uptake coupled with a greater capacity for Na exclusion in the salt marsh, a scenario that is well supported by the molecular salt tolerance literature (Hasegawa et al., 2000). In conclusion, although this experiment was conducted to study the origin of a wild sunflower hybrid species (see Section 4), our dataset also provides an example for the usefulness of phenotypic selection experiments in choosing candidate adaptive traits for QTL analyses.
3.1.2. Selection coefficients for adaptive QTLs
Assaying the transplanted BC2 hybrids for 71 mapped microsatellite markers allowed us to conduct a genome-wide scan for quantitative trait loci (QTLs) expressed in extremely saline “hybrid” habitat. In total, 14 elemental uptake QTLs and 3 fitness QTLs, controlling survivorship in the salt marsh, were detected in the BC2. The three survivorship QTLs cumulatively explained 38% of the temporal variation in survivorship in the wild, and the elemental uptake QTLs cumulatively explained between 21% (S uptake) and 78% (Ca uptake) of the variation in each elemental uptake trait (detailed presentation in Lexer et al. (2003b)). Notably, all three survivorship QTLs were closely correlated with QTLs for Ca and K uptake (Fig. 2A), Na and Mg uptake (Fig. 2B), or Na uptake (Fig. 2C). In all three cases, the one-LOD support intervals of elemental uptake and survivorship QTLs overlapped, and the microsatellite markers closest to the QTL likelihood peaks were the same for both classes of QTLs (Fig. 2). Our results suggest that we have identified three chromosomal blocks associated with ecological selection in the wild.
Fig. 2.

Selected linkage groups of a genetic map derived from a second generation backcross population (BC2) of H. annuus × H. petiolaris, and QTL positions for survivorship and elemental uptake traits measured in the wild. Marker positions are shown by horizontal lines, and map distances between markers by numerals to the left of each group. Linkage groups were assigned according to microsatellite linkage maps for H. annuus (Burke et al., 2002; Tang et al., 2002). Marker groupings that differed between the intra- and interspecific maps (presumably due to pseudolinkage and/or fragmentation in the interspecific BC2) are indicated by thick black lines at the left of the groups. QTL positions with one-LOD support intervals, additive effects (±), and QTL magnitudes are indicated by vertical bars to the right of each group. Marker names are listed according to order below each group. Marker names starting with ORS refer to microsatellites isolated from H. annuus (Tang et al., 2002). Two additional AFLP markers of the original linkage map were not assayed in the present field study. Selection coefficients for individual chromosome blocks are indicated to the right of each survivorship QTL. Redrawn from Lexer et al. (2003b).
The strength of natural selection for elemental uptake QTLs was examined by calculating selection coefficients for heterozygous BC2 plants carrying molecular marker alleles derived from H. annuus. The selection coefficient was +0.126 for the tightly linked or pleiotropic QTLs controlling Ca and K uptake on linkage group 1 (Fig. 2A), −0.084 for the correlated QTLs controlling Mg and Na uptake on linkage group 4 (Fig. 2B), and −0.094 for the Na uptake QTL on linkage group 17b (Fig. 2C). The results indicate how the increased phenotypic variance and marker polymorphism present in a fairly complex hybrid pedigree can be used to map adaptively important QTLs directly in natural habitat. Although the present experiment was based on experimental hybrids, similar studies are conceivable for natural hybrid zones.
3.2. Introgression in natural hybrid zones
Studying introgression patterns of mapped molecular markers in replicate hybrid zones allowed the identification of chromosomal blocks that introgressed significantly more frequently or less frequently than expected, as exemplified by Fig. 3. Introgression patterns across replicate hybrid zones were largely concordant, thereby excluding genetic drift as being an important factor in generating the observed deviations from neutral expectations (presented in detail in Rieseberg et al. (1999b) and Buerkle and Rieseberg (2001)). For 26 chromosomal segments, introgression was significantly reduced, indicating that the genetic basis of the species’ barrier is complex. Notably, many of the negatively selected chromosomal segments were from linkage groups that are known to be collinear between the two parental species (e.g. Fig. 3). These collinear chromosomal segments, equivalent to QTLs, likely contain one or more genes that contribute to reproductive isolation between these wild sunflower species. For chromosomal segments close to rearrangements, it is difficult to disentangle the effects of the chromosomal rearrangement from those of linked genes.
Fig. 3.
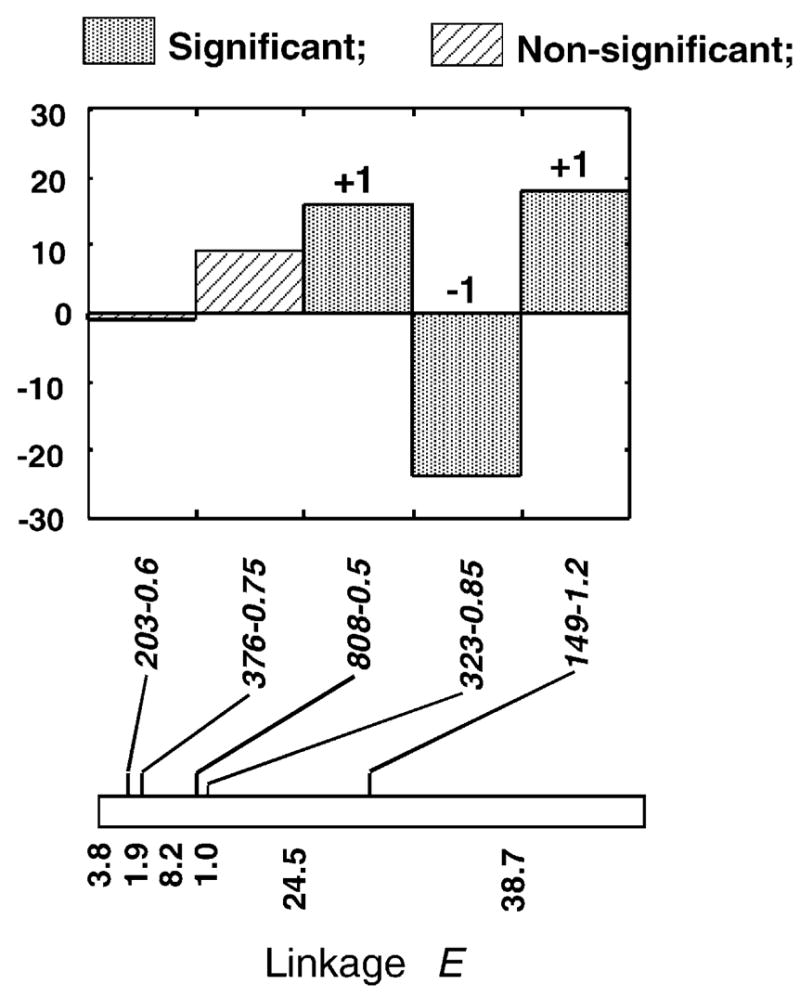
Direct count deviations from the expected number of introgressed marker alleles in three natural hybrid zones between H. annuus and H. petiolaris. The linkage group shown is collinear between the genomes of H. annuus and H. petiolaris. Mapped molecular markers are given above and map distances below the linkage group. Independently selected chromosome blocks are indicated by +1 or −1, depending on the direction of selection. Redrawn from Rieseberg et al. (1999b).
To determine the likely cause of the reduced rates of introgression for these chromosomal segments, we searched for correlations with an important reproductive isolating mechanism, pollen sterility. Significant associations were found with 16 of the 26 segments, providing a straightforward explanation of why this subset of chromosomal segments is negatively selected in hybrids (Rieseberg et al., 1999a). In an attempt to understand why the remaining 10 segments were negatively selected, we assayed all plants for numerous morphological differences between the species that we thought might contribute to the ecological divergence between them (Rieseberg and Buerkle, 2002). Unfortunately, the phenotypic data were too skewed toward one of the parental species, H. annuus, to allow meaningful analyses (Fig. 4). Although this was a disappointing result, the results did inform us regarding molecular marker requirements for hybrid zone QTL studies (Section 4.2.1) and sampling strategies for obtaining the ideal distribution of phenotypic variance in QTL analyses (Section 4.2.2).
Fig. 4.
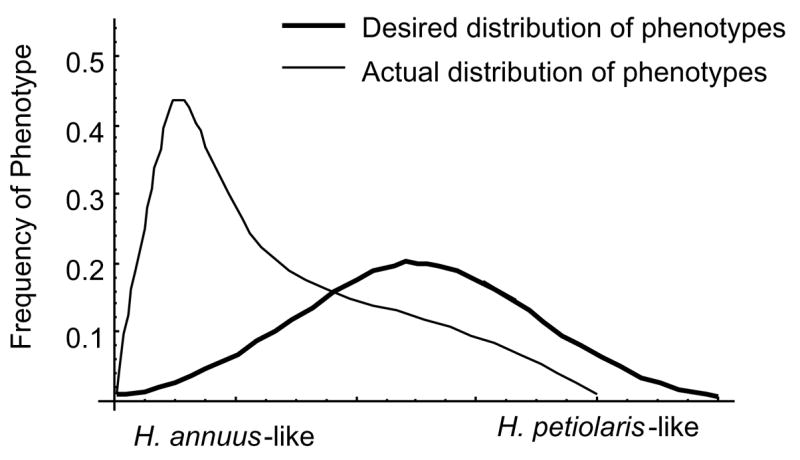
Observed vs. desired distributions of phenotypes for QTL mapping in wild sunflower hybrid zones. Redrawn from Rieseberg and Buerkle (2002).
4. Discussion
4.1. Selection on individual chromosome blocks in the wild—implications for hybrid speciation in sunflowers
The transplantation experiment presented here was designed to study the role of hybridization in the origin of novel adaptation in a wild diploid sunflower hybrid species, Helianthus paradoxus. By generating an experimental backcross population between the two parental species, H. annuus and H. petiolaris, and transplanting them into the extremely saline habitat of the natural hybrid species, we simulated what may have been the earliest steps in the speciation process that gave rise to H. paradoxus in nature. Our experiment allowed us to test the hypothesis that salinity adaptation in the hybrid neospecies resulted from the complementary action of additive QTL alleles dispersed between the parental species. This process is thought to be responsible for the generation of extreme (transgressive) phenotypes in segregating hybrid populations (Tanksley, 1993; Rieseberg et al., 1999b). Transgressive segregation through complementary gene action provides a plausible and simple explanation for the rapid evolution of habitat or niche divergence often observed in plant hybrid lineages (Rieseberg et al., 1999b; Lexer et al., 2003b).
Our results from this transplantation experiment concord with the “complementary gene action” hypothesis. First, for all of the candidate adaptive traits (Welch and Rieseberg, 2002), one or more back-cross hybrids had phenotypes that exceeded the range of the parental species and the mean phenotype of the natural hybrid species, H. paradoxus. Second, the candidate adaptive traits were found to be under strong directional selection in the habitat of the natural hybrid species (Fig. 1, Table 1). Third, three chromosomal blocks were detected in this study that were associated with QTLs conferring differences in survivorship in the salt marsh, and all three of them also had a significant effect on elemental uptake from the soil (Fig. 2; Lexer et al., 2003b). The fitness effects of these genomic blocks derived from the donor parent in the BC2 design, H. annuus, were in opposing directions (Fig. 2). QTL alleles with opposing effects were also detected for each of the elemental uptake characters (not shown; Lexer et al., 2003b). It is easy to see how these QTL alleles with opposing effects may be re-shuffled by recombination in early generation hybrids to generate a small number of transgressive genotypes with all QTL alleles in one direction, and this is exactly what is predicted by the “complementary gene action” hypothesis (Rieseberg et al., 1999b). It is therefore likely that selection for extreme (transgressive) genotypes in early hybrid generations did indeed facilitate the colonization of an extreme salt marsh habitat by H. paradoxus (Lexer et al., 2003a,b). More generally, our data provide support for the long-standing view that hybridization may provide the necessary genetic variation for natural selection to act upon (Anderson, 1949; Stebbins, 1959; Lewontin and Birch, 1966; Arnold, 1997; Barton, 2001).
4.2. Estimating the selective value of adaptive QTLs in hybrid zones—a possible approach for forest trees?
As outlined earlier in this paper, a major impediment to identify adaptive genetic variation in forest trees is the difficulty of estimating the adaptive value of QTLs or candidate genes in “conventional” crosses. Transplantation experiments like the one presented here for sunflowers are not feasible in forest trees, if the aim is to measure adaptive traits and fitness characters on adult plants. Association studies in natural hybrid zones may help to circumvent this problem, since genetic mapping of QTLs and selection assays can be combined directly in natural populations. Also, natural hybrid zones contain increased phenotypic variance and marker polymorphism that can be exploited for genetic analyses (Barton and Hewitt, 1985; Harrison, 1990; Barton and Gale, 1993). In particular, many of the traits or QTLs that are most important to adaptation may be invariant within populations or species (Orr, 2001), while the same traits (QTLs) are often variable in hybrids (reviews by Rieseberg et al. (2002) and Lexer et al. (2003c)). An important notion is that the two approaches presented here, selection experiments at the QTL level and introgression studies in hybrid zones, can potentially be combined. This may allow an assessment of the adaptive value of individual QTLs in natural forest stands, and our data on sunflower hybrid zones may inform such studies.
4.2.1. Marker requirements
A graphical result that emerges from our hybrid zone data in sunflowers is that the number of informative markers must be large enough to allow for genome-wide introgression studies. Analyzing introgression patterns as in Fig. 3 requires diagnostic markers, or markers with allele frequency differences between the two parental taxa (Rieseberg et al., 1998; 1999a). Note that the potential for identifying sufficient numbers of markers not only depends on the degree of genetic differentiation between the hybridizing populations, e.g. rather high differentiation in sunflowers (Rieseberg et al., 1999a), intermediate differentiation in North American Populus species (Martinsen et al., 2001), rather low differentiation in European oaks (Bodénès et al., 1997), but also on the molecular marker systems employed. Large-scale sequencing of expressed sequences (ESTs) in forest trees (http://dendrome.ucdavis.edu) as well as development of Bayesian approaches to hybrid identification (Anderson and Thompson, 2002) hold the promise to extend the applicability of this approach further in the future.
Fortunately, results from human populations (Stephens et al., 1994) indicate that in order to be informative, alternative allelic forms need not be close to fixation in the pure parental populations, as was the case in the present sunflower study. Rather, loci with smaller allele frequency differences between the parental populations (=0.3) should also produce sufficient linkage disequilibrium (LD) for association studies when admixed in hybrid populations. This may allow the detection of marker-trait associations in species pairs with little genetic differentiation, as suggested by preliminary results in European oaks (Saintagne et al., 2004).
4.2.2. Sampling strategies
As shown in Fig. 4, phenotypic distributions in the hybrid zone dataset were highly skewed towards H. annuus like phenotypes. This is not surprising, since these samples were collected to estimate introgression from Helianthus petiolaris into H. annuus, and not primarily for QTL analyses. However, the phenotypic distributions in Fig. 4 also illustrate an important consideration in sampling hybrid zones in trees.
In many forest taxa, introgression has been shown to occur preferentially in one direction (e.g. Bacilieri et al., 1996; Keim et al., 1989). Therefore, the hybrid individuals that are most informative for QTL analyses from a genotypic perspective may not be the most informative ones with respect to their phenotypes. This is because, from a genotypic perspective, the ideal dataset for QTL mapping consists of 5th to 12th generation hybrids. In these plants, recombination should have had sufficient time to break up the parental species’ genomes (Briscoe et al., 1994; Rieseberg and Buerkle, 2002). However, with respect to phenotype, the most informative hybrids will have a hybrid index near 0.5 (Fig. 4) and will therefore be early generation hybrids or even F1’s.
An optimal sampling strategy to resolve this paradox might involve screening a large number of hybrids with a moderate number of nuclear markers (20–30 loci) in order to identify the most recombinant genotypes (later generation hybrids). In these plants, linkage disequilibrium (LD) between loosely linked genes should still be detectable, while LD between unlinked genes should have decayed by then (Briscoe et al., 1994). Including only plants that are highly recombinant, and that contain genetic material from both parental species in a balanced distribution, will provide the basis for sampling a broad phenotypic distribution as outlined in Fig. 4. This may require sampling several hundred hybrids in the initial screening step, while sample sizes for the genome-wide QTL analysis will depend on the actual size of genome blocks in hybrids (below). Plants that were screened with markers but not included in the QTL analysis could be of interest elsewhere, e.g. for analyzing hybrid zone structure and inferring management and conservation measures, or simply for identifying and maintaining interesting genotypes for breeding and selection programs.
4.2.3. Estimating selection in natural hybrid zones
Perhaps the single most important difference between our selection study with transplanted experimental hybrids (Section 3.1) and selection assays in natural hybrid zones is the fact that hybrid zones contain substantial environmental variability that may confound marker-trait associations, particularly for adaptive traits strongly affected by environmental variance. In our transplantation experiment, environmental heterogeneity was taken into account by dividing the field site into “blocks”, and including them as random factors in ANOVA models (Lexer et al., 2003a). However, this procedure may not be applicable to hybrid populations in forest trees because of the increased environmental heterogeneity found in natural forest stands, and because of the size of the geographical areas studied. A more fruitful approach—other than the establishment of common garden trials—may be to record key habitat factors for every tree sampled, and include them as covariates in selection models (Sokal and Rohlf, 1995).
A second major difference between selection assays in hybrid zones and transplantation studies with experimental hybrids is that a priori pedigree information is generally not available for hybrid zones. However, this is less likely to be a problem, because diagnostic molecular markers will reveal the exact species origin of each chromosomal segment (e.g. Rieseberg et al., 1999a; Martinsen et al., 2001; Rieseberg and Buerkle, 2002). Therefore, fitness differences at codominant markers can be assigned to QTL alleles derived from each parental species. This will yield reliable estimates of the strength of selection, as long as a major proportion of the phenotypic variation in the trait of interest can be attributed to interspecific differences (Orr, 2001). In addition, the use of highly polymorphic codominant markers holds the promise to reconstruct even complex pedigree structures in wild populations (Queller and Goodnight, 1989; Blouin et al., 1996; Lynch and Ritland, 1999), potentially reducing the problem to an analysis of hybrid crosses between individual trees.
4.3. Potential of the hybrid zone approach in selected forest tree genera
4.3.1. Populus
The genus Populus (poplars, cottonwoods, and aspens) is certainly the most advanced genetic model species in forest trees, with its relatively small genome size (550 Mb; 2n = 38), and its complete genomic sequence becoming available soon (Bradshaw et al., 2001; Wullschleger et al., 2002). Species barriers in Populus are known to be porous, and hybridization is frequent wherever two related species have overlapping ranges (Keim et al., 1989; Rajora and Dancik, 1992; Eckenwalder, 1996; Martinsen et al., 2001).
One aspect renders Populus particularly suitable for QTL studies in natural hybrid zones: species barriers appear to be genic rather than chromosomal. This is expected because interspecific hybrids in Populus are generally diploid (Eckenwalder, 1996), and homologies between different species’ linkage maps are widespread (Cervera et al., 2001). Hence, species barriers appear to be composed primarily of pairs of complementary genes conferring intrinsic incompatibilities (reduced hybrid fertility) or genes involved in habitat or niche divergence, i.e. exactly those groups of genes that are so important to forest ecology and management.
Molecular gene introgression studies have been conducted for hybrid zones among North American cottonwoods (Populus fremontii × P. angustifolia; Keim et al., 1989; Martinsen et al., 2001). The results suggest that different genes or chromosomal blocks do introgress at different rates, or vary in their potential to spread within the recipient species’ populations (Martinsen et al., 2001). In addition, the increased genetic variance present in hybrids P. fremontii × P. angustifolia has been utilized for examining interactions with herbivores and pathogens, and some of the best evidence for the importance of hybrid zones for biodiversity in the plant kingdom have been conducted in these species (Whitham, 1989; Whitham et al., 1999).
In other hybrid systems, such as P. alba × P. tremula (Rajora and Dancik, 1992), divergent habitat preferences related to flooding or other disturbance regimes along river floodplains (Karrenberg et al., 2002) may provide a venue for introgression of adaptive traits. Hybrid zones between P. alba and P. tremula occur along several major European river systems (Rajora and Dancik, 1992), thus providing a “replicated natural hybridization experiment” for comparing QTLs as well as fitness effects of traits, QTLs, and candidate genes along parallel evolutionary trajectories.
With respect to possible applications in forestry, it is important to note that in poplar breeding, adaptive traits like abiotic stress tolerance or disease resistance can often only be obtained from other species (Stettler et al., 1996). Insights into the function of adaptively important genes in this genus may thus have direct implications for the breeding of new cultivars, either by ‘gene-assisted’ traditional crossings (i.e. supported by molecular analyses of offspring for suitable recombinants), or by transformation-based gene transfer between closely related genomes (Bradshaw et al., 2001; Wullschleger et al., 2002).
4.3.2. Fraxinus
Fraxinus (ash) is another diploid genus (2n = 46) that may be useful as a model for hybrid zone studies among temperate forest trees. Fraxinus is represented by F. excelsior, the common ash, throughout most of central Europe. The Mediterranean F. angustifolia, narrowleaf ash, extends into the Pannonian basin (Hungary and neighboring regions), but has only recently been recognized as a true species—both are very similar morphologically (Jelem, 1974; Fukarek, 1971). Fraxinus angustifolia is a floodplain species, with considerable tolerance to prolonged flooding. It appears that through introgression of F. angustifolia alleles, flooding tolerance finds its way into the F. excelsior genome, enabling this upland species to colonize frequently flooded sites more effectively further upstream where F. angustifolia meets its lower temperature limit (Volk, 2002; Jelem, 1974).
Little is known about the genetic make-up of hybrid populations of F. excelsior × F. angustifolia, e.g. about the frequency of different hybrid generations or genotypic classes. However, since both species reached their present distribution soon after the retreat of the glacial ice sheets approximately 100–200 tree generations ago (Huntley and Birks, 1983; Tinner and Lotter, 2001), later generation hybrids should be frequent. Although the hybrid zones of F. excelsior × F. angustifolia are considerably older than those studied in wild sunflowers, the number of generations of hybridization is roughly equivalent. This may provide ideal conditions for QTL mapping, since a large number of recombination events should have accumulated in these hybrids.
Unfortunately, Fraxinus species are comparatively poorly characterized genetically. Although a moderate number of molecular markers have been developed for ash (e.g. Jeandroz et al., 1996; Morand-Prieur et al., 2002), no genetic linkage map is available. However, development of such tools might benefit from research on another member of the Oleaceae, the cultivated olive Olea europaea (e.g. Rallo et al., 2000; Sefc et al., 2000; De La Rosa et al., 2002). The development of genomic tools in ash and olive, in combination with the fact that Fraxinus spp. and their hybrids are still comparatively easy to classify in the field (compared to, say, willows), all make ash a highly interesting candidate for hybrid zone-based QTL mapping approaches.
4.3.3. Quercus
Oak (Quercus) is a particularly complex genus (Burger, 1975; van Valen, 1976), and its porous species boundaries and numerous intermediate forms attracted the curiosity of early evolutionists (Darwin, 1859). Clearly, hybridization and introgression among diploid (2n = 24) species have played an important role during oak evolution (Petit et al., 1997; Belahbib et al., 2001; Howard et al., 1997). Despite relatively high levels of interspecific gene exchange (Whittemore and Schaal, 1991; Petit et al., 1997; Belahbib et al., 2001), divergent ecological selection has generated substantial interspecific differentiation in adaptive traits. This ecological divergence appears to account for the coexistence of closely related sympatric species within extensive hybrid zones, e.g. in the European white oaks Q. robur and Q. petraea (Kremer et al., 1993). These hybrid populations may be suitable for QTL mapping approaches as outlined in this paper, although the setting provided by these two species may be more difficult than those for other genera.
Extensive molecular marker surveys in Q. robur and Q. petraea indicate that only a small proportion of markers differ in allele frequencies between the species (Bodénès et al., 1997). Analysis of the genomic locations of these markers suggests that only a limited number of genomic regions or “hot spots”, equivalent to QTLs, may separate two genomes that are otherwise poorly protected from interspecific gene flow (Saintagne et al., 2004). Although the porosity of oak genomes may not permit genome-wide QTL scans in oak hybrid zones, the important adaptive traits differentiating oak species are likely to be controlled by the segments differentiating their genomes. So only some additional fine-mapping may be required. Of course, it will be interesting to see if candidate genes involved in divergent soil preferences are located within genomic regions that differ between the two species. It also should be noted that diagnostic differences are not required for admixture linkage disequilibrium mapping (Stephens et al., 1994), so the approaches outlined in the present paper may be more applicable to oaks than is superficially apparent.
4.4. Conclusions
Natural hybrid zones are promising tools for identifying adaptive genetic variation in organisms that are long-lived or otherwise of limited genetic tractability. They potentially allow forest geneticists to circumvent the need for experimental multi-generation crosses, offer increased marker polymorphism and phenotypic variance in adaptive traits, and hold the promise of genetically mapping QTLs or candidate genes for phenotypic traits and simultaneously assessing their adaptive value directly in natural forest stands.
In the present paper, we attempted to draw first conclusions about the applicability of this approach using existing data from an annual plant genus: wild annual sunflowers (Helianthus spp.). By combining QTL mapping and selection assays on transplanted sunflower hybrids, we were able to assess the adaptive value of individual chromosome blocks in the habitat of a wild sunflower hybrid species. Our results indicate that QTL alleles from both parental species are required to provide a selective advantage in the wild, thereby indicating an important role for hybridization in the rapid evolution of novel adaptation in this study system. Additional data from natural hybrids indicate that similar studies are possible directly in natural hybrid zones, provided that sufficient numbers of diagnostic markers are available, and that large enough number of recombinant hybrid genotypes can be identified.
The approaches outlined in this paper may be directly applicable to hybrid zones in forest trees, as outlined by a short literature review of three forest tree genera: Populus (poplars, aspens, cottonwoods), Fraxinus (ash), and Quercus (oak). Temperate tree genera are particularly amenable to this kind of study, because molecular markers and genetic linkage maps are available, as are extensive molecular and fossil datasets on their postglacial history (e.g. Huntley and Birks, 1983; Petit et al., 1997; Kremer et al., 2002). This allows forest geneticists to date the approximate age of hybrid zones, thereby providing an upper bound for the number of hybrid generations present. Such estimates will be extremely valuable in designing QTL experiments, since the age of individual hybrid crosses will dictate the size and frequency of parental genome blocks in hybrids (Martinsen et al., 2001) and the marker densities required.
Acknowledgments
We thank Jacob Malcom and Gordon Warrick (US Fish and Wildlife Service) for their assistance during field work on transplanted sunflower hybrids in New Mexico, USA, as well as Jeanette Whitton, Keith Gardner, and Alex Buerkle for their pioneering work on sunflower hybrid zones. Thanks to Jennifer L. Durphy for skilled technical assistance in the lab. This research was supported by NSF grants BSR-9419206 and DEB-9806290 as well as NIH award GM59065 to LHR. Financial support for CL came from the Austrian Science Foundation (Erwin-Schroedinger grant no. J 2148).
References
- Anderson E. Introgressive Hybridization. Wiley; New York: 1949. [Google Scholar]
- Anderson EC, Thompson EA. A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 2002;160:1217–1229. doi: 10.1093/genetics/160.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold ML. Natural Hybridization and Evolution. Oxford University Press; New York: 1997. [Google Scholar]
- Bacilieri R, Ducousso A, Petit RJ, Kremer A. Mating system and asymmetric hybridization in a mixed stand of European oaks. Evolution. 1996;50:900–908. doi: 10.1111/j.1558-5646.1996.tb03898.x. [DOI] [PubMed] [Google Scholar]
- Barton NH. The role of hybridization in evolution. Mol Ecol. 2001;10:551–568. doi: 10.1046/j.1365-294x.2001.01216.x. [DOI] [PubMed] [Google Scholar]
- Barton NH, Gale KS. Genetic analysis of hybrid zones. In: Harrison RG, editor. Hybrid Zones and the Evolutionary Process. Oxford University Press; Oxford: 1993. pp. 13–45. [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annu Rev Ecol Syst. 1985;16:113–148. [Google Scholar]
- Belahbib N, Pemonge MH, Ouassou A, Sbay H, Kremer A, Petit RJ. Frequent cytoplasmic exchanges between oak species that are not closely related: Quercus suber and Q ilex in Morocco. Mol Ecol. 2001;10:2003–2012. doi: 10.1046/j.0962-1083.2001.01330.x. [DOI] [PubMed] [Google Scholar]
- Blouin MS, Parsons M, Lacaille V, Lotz S. Use of microsatellite loci to classify individuals by relatedness. Mol Ecol. 1996;5:393–401. doi: 10.1111/j.1365-294x.1996.tb00329.x. [DOI] [PubMed] [Google Scholar]
- Bodénès C, Joandet S, Laigret F, Kremer A. Detection of genomic regions differentiating two closely related oak species Quercus petraea (Matt) Liebl and Quercus robur L. Heredity. 1997;78:433–444. [Google Scholar]
- Bradshaw HD, Ceulemans R, Davis J, Stettler R. Emerging model systems in plant biology: poplar (Populus) as a model forest tree. J Plant Growth Regul. 2001;19:306–313. [Google Scholar]
- Briscoe D, Stephens JC, O’Brien SJ. Linkage disequilibrium in admixed populations: applications in gene mapping. J Hered. 1994;85:59–63. [PubMed] [Google Scholar]
- Buerkle CA, Rieseberg LH. Low intraspecific variation for genomic isolation between hybridizing sunflower species. Evolution. 2001;55:684–691. doi: 10.1554/0014-3820(2001)055[0684:livfgi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Burger W. The species concept in Quercus. Taxon. 1975;24:45–50. [Google Scholar]
- Burke JM, Tang S, Knapp SJ, Rieseberg LH. Genetic analysis of sunflower domestication. Genetics. 2002;161:1257–1267. doi: 10.1093/genetics/161.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera MT, Storme V, Ivens B, Gusmao J, Liu BH, Hostyn V, Slycken JV, Montagu MV, Boerjan W. Dense genetic linkage maps of three Populus species (Populus deltoides, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics. 2001;158:787–809. doi: 10.1093/genetics/158.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler JM, Jan C, Beard BH. Chromosomal differentiation among the annual Helianthus species. Syst Bot. 1986;11:353–371. [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. The origin of species. In: Hutchins RM, editor. Great Books of the Western World. Encyclopaedia Britannica; London: 1859. [Google Scholar]
- De La Rosa R, James CM, Tobutt KR. Isolation and characterization of polymorphic microsatellites in olive (Olea europaea L.) and their transferability to other genera in the Oleaceae. Mol Ecol Notes. 2002;2:265–267. [Google Scholar]
- Eckenwalder JE. Systematics and evolution of Populus. In: Stettler RF, Bradshaw HD, Heilman PE, Hinckley TM, editors. Biology of Populus, and Its Implications for Management and Conservation. NRC Research Press; Ottawa: 1996. pp. 7–33. [Google Scholar]
- Etterson JR, Shaw RG. Constraint to Adaptive evolution in response to global warming. Science. 2001;294:151–154. doi: 10.1126/science.1063656. [DOI] [PubMed] [Google Scholar]
- Ferris C, King RA, Vainola R, Hewitt GM. Chloroplast DNA recognizes three refugial sources of European oaks and suggests independent eastern and western immigrations to Finland. Heredity. 1998;80:584–593. doi: 10.1046/j.1365-2540.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- Frewen BE, Chen THH, Howe GT, Davis J, Rohde A, Boerjan W, Bradshaw HD. Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics. 2000;154:837–845. doi: 10.1093/genetics/154.2.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukarek P. The latest results of studies of the range of Fraxinus angustifolia. Gozd Vestn. 1971;29:193–201. [Google Scholar]
- Geburek TH, Heinze B. Erhaltung genetischer Ressourcen im Wald-Normen, Programme, Maßnahmen. Ecomed-Verlagsgesellschaft; Landsberg: 1998. [Google Scholar]
- Hamrick JL, Godt MJW. Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS, editors. Plant Population Genetics, Breeding and Genetic Resources. Sinauer Associates; Sunderland, MA: 1989. pp. 43–63. [Google Scholar]
- Hamrick JL, Nason JD. Gene flow in forest trees. In: Young A, Boshier D, Boyle T, editors. Forest Conservation Genetics—Principles and Practice. CSIRO Publishing; Collingwood: 2000. pp. 81–90. [Google Scholar]
- Harrison RG. Hybrid zones: windows on evolutionary processes. Oxford Surv Evol Biol. 1990;7:69–128. [Google Scholar]
- Harrison RG, Rand DM. Mosaic hybrid zones and the nature of species boundaries. In: Otte D, Endler JA, editors. Speciation and Its Consequences. Sinauer Associates; Sunder-land, MA: 1989. pp. 111–133. [Google Scholar]
- Hasegawa PM, Bressan RA, Zhu JK, Bohnert HJ. Plant cellular and molecular responses to high salinity. Annu Rev Plant Physiol Plant Mol Biol. 2000;51:463–499. doi: 10.1146/annurev.arplant.51.1.463. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Genetics of Populations. Jones and Bartlett Publishers; Sudbury, MA: 2000. [Google Scholar]
- Heiser CB. Hybridization between the sunflower species Helianthus annuus and H. petiolaris. Evolution. 1947;1:249–262. [Google Scholar]
- Howard DJ, Preszler RW, Williams J, Fenchel S, Boecklen WJ. How discrete are oak species? Insights from a hybrid zone between Quercus grisea and Quercus gambellii. Evolution. 1997;51:747–755. doi: 10.1111/j.1558-5646.1997.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Huntley B, Birks H. An Atlas of Past and Present Pollen Maps of Europe: 0–13 000 Years Ago. Cambridge University Press; Cambridge: 1983. [Google Scholar]
- Hurme P, Sillanpaa MJ, Arjas E, Repo T, Savolainen O. Genetic basis of climatic adaptation in Scots pine by Bayesian quantitative trait locus analysis. Genetics. 2000;156:1309–1322. doi: 10.1093/genetics/156.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabel N, Beaulieu J, Bousquet J. Complete congruence between gene diversity estimates derived from genotypic data at enzyme and random amplified polymorphic DNA loci in black spruce. Proc Natl Acad Sci USA. 1995;92:6369–6373. doi: 10.1073/pnas.92.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeandroz S, Frascaria-Lacoste N, Bousquet J. Molecular recognition of the closely related Fraxinus excelsior and F. oxyphylla (Oleaceae) by RAPD markers. For Genet. 1996;3:237–242. [Google Scholar]
- Jelem H. Die Auwälder der Donau in Österreich. Mitteilungen der Forstlichen Bundesversuchsanstalt. 1974;109 [Google Scholar]
- Kanowski PJ. Politics, policies and the conservation of forest genetic diversity. In: Young A, Boshier D, Boyle T, editors. Forest Conservation Genetics—Principles and Practice. CSIRO Publishing; Collingwood: 2000. pp. 275–289. [Google Scholar]
- Kao CH, Zeng ZB, Teasdale RD. Multiple interval mapping for quantitative trait loci. Genetics. 1999;152:1203–1216. doi: 10.1093/genetics/152.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhu A, Hurme P, Karjalainen M, Karvonen P, Karkkainen K, Neale D, Savolainen O. Do molecular markers reflect patterns of differentiation in adaptive traits of conifers? Genetics. 1995;93:215–221. doi: 10.1007/BF00225748. [DOI] [PubMed] [Google Scholar]
- Karp A, Edwards KJ, Bruford M, Funk S, Vosman B, Morgante M, Seberg O, Kremer A, Boursot P, Arctander P, Tautz D, Hewitt GM. Molecular technologies for biodiversity evaluation: opportunities and challenges. Nat Biotechnol. 1997;15:625–629. doi: 10.1038/nbt0797-625. [DOI] [PubMed] [Google Scholar]
- Karrenberg S, Edwards PJ, Kollmann J. The life history of Salicaceae living in the active zone of floodplains. Freshw Biol. 2002;47:733–748. [Google Scholar]
- Keim P, Paige KN, Whitham TG, Lark KG. Genetic analysis of an interspecific hybrid swarm of Populus: occurrence of unidirectional introgression. Genetics. 1989;123:565–575. doi: 10.1093/genetics/123.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer A, Kleinschmit J, Cottrell J, Cundall EP, Deans JD, Ducousso A, Koenig AO, Lowe AJ, Munro RC, Petit RJ, Stephan BR. Is there a correlation between chloroplastic and nuclear divergence, or what are the roles of history and selection on genetic diversity in European oaks? For Ecol Manage. 2002;156:75–87. [Google Scholar]
- Kremer A, Savill PS, Steiner KC. Genetics of oaks. Ann Sci For. 1993;50:1–472. [Google Scholar]
- Lagercrantz U, Ryman N. Genetic structure of Norway spruce (Picea abies): concordance of morphological and allozyme variation. Evolution. 1990;44:38–53. doi: 10.1111/j.1558-5646.1990.tb04278.x. [DOI] [PubMed] [Google Scholar]
- Lande R, Arnold SJ. The measurement of selection on correlated characters. Evolution. 1983;37:1210–1226. doi: 10.1111/j.1558-5646.1983.tb00236.x. [DOI] [PubMed] [Google Scholar]
- Lewontin RC, Birch LC. Hybridization as a source of variation for adaptation to new environments. Evolution. 1966;20:315–336. doi: 10.1111/j.1558-5646.1966.tb03369.x. [DOI] [PubMed] [Google Scholar]
- Lexer C, Welch ME, Raymond O, Rieseberg LH. The origin of ecological divergence in Helianthus paradoxus (Asteraceae): selection on transgressive characters in a novel hybrid habitat. Evolution. 2003a;57:1989–2000. doi: 10.1111/j.0014-3820.2003.tb00379.x. [DOI] [PubMed] [Google Scholar]
- Lexer C, Welch ME, Raymond O, Rieseberg LH. Natural selection for salt tolerance QTLs in wild sunflower hybrids: implications for the origin of Helianthus paradoxus, a diploid hybrid species. Mol Ecol (special feature: genes in ecology) 2003b;12:1225–1235. doi: 10.1046/j.1365-294x.2003.01803.x. [DOI] [PubMed] [Google Scholar]
- Lexer C, Randell RA, Rieseberg LH. Experimental hybridization as a tool for studying selection in the wild. Ecology (special feature: selection experiments for ecologists—concepts, methods, and directions) 2003c;84:1688–1699. [Google Scholar]
- Luo ZW, Tao SH, Zeng ZB. Inferring linkage disequilibrium between a polymorphic marker locus and a trait locus in natural populations. Genetics. 2000;156:457–467. doi: 10.1093/genetics/156.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ritland K. Estimation of pairwise relatedness with molecular markers. Genetics. 1999;152:1753–1766. doi: 10.1093/genetics/152.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh JB. Genetics and Analysis of Quantitative Traits. Sinauer Associates; Sunderland, MA: 1998. [Google Scholar]
- Martinsen GD, Whitham TG, Turek RJ, Keim P. Hybrid populations selectively filter gene introgression between species. Evolution. 2001;55:1325–1335. doi: 10.1111/j.0014-3820.2001.tb00655.x. [DOI] [PubMed] [Google Scholar]
- Mauricio R. Mapping quantitative trait loci in plants: uses and caveats for evolutionary biology. Nat Rev. 2001;2:370–381. doi: 10.1038/35072085. [DOI] [PubMed] [Google Scholar]
- McKay JK, Latta RG. Adaptive population divergence: markers QTL and traits. Trends Ecol Evol. 2002;17:285–292. [Google Scholar]
- Morand-Prieur ME, Vedel F, Raquin C, Brachet S, Sihachakr D, Frascaria-Lacoste N. Maternal inheritance of a chloroplast microsatellite marker in controlled hybrids between Fraxinus excelsior and Fraxinus angustifolia. Mol Ecol. 2002;11:613–617. doi: 10.1046/j.1365-294x.2002.01453.x. [DOI] [PubMed] [Google Scholar]
- Mueller-Starck G, Schubert R, editors. Genetic Response of Forest Systems to Changing Environmental Conditions. Kluwer Academic Publishers; Dordrecht: 2001. Preface. [Google Scholar]
- Orr HA. The genetics of species differences. Trends Ecol Evol. 2001;16:343–350. doi: 10.1016/s0169-5347(01)02167-x. [DOI] [PubMed] [Google Scholar]
- Petit RJ, Pineau E, Demesure B, Bacilieri R, Ducousso A, Kremer A. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci USA. 1997;94:9996–10001. doi: 10.1073/pnas.94.18.9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queller DC, Goodnight KF. Estimating relatedness using genetic markers. Evolution. 1989;43:258–275. doi: 10.1111/j.1558-5646.1989.tb04226.x. [DOI] [PubMed] [Google Scholar]
- Rajora OP, Dancik BP. Genetic characterization and relationships of Populus alba, P. tremula and P. x canescens and their clones. Theor Appl Genet. 1992;84:291–298. doi: 10.1007/BF00229485. [DOI] [PubMed] [Google Scholar]
- Rallo P, Dorado G, Martin A. Development of simple sequence repeats (SSRs) in olive tree (Olea europaea L.) Theor Appl Genet. 2000;101:984–989. [Google Scholar]
- Rieseberg LH. Homoploid reticulate evolution in Helianthus: evidence from ribosomal genes. Am J Bot. 1991;78:1218–1237. [Google Scholar]
- Rieseberg LH, Buerkle CA. Genetic mapping in hybrid zones. Am Nat. 2002;159:S37–S49. doi: 10.1086/338371. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Carter R, Zona S. Molecular tests of the hypothesized hybrid origin of two diploid Helianthus species (Asteraceae) Evolution. 1990;44:1498–1511. doi: 10.1111/j.1558-5646.1990.tb03841.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Beckstrom-Sternberg S, Liston A, Arias D. Phylogenetic and systematic inferences from chloroplast DNA and isozyme variation in Helianthus sect Helianthus. Syst Bot. 1991;16:50–76. [Google Scholar]
- Rieseberg LH, Baird SJE, Desrochers AM. Patterns of mating in wild sunflower hybrid zones. Evolution. 1998;52:713–726. doi: 10.1111/j.1558-5646.1998.tb03696.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics. 1999a;152:713–727. doi: 10.1093/genetics/152.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Archer MA, Wayne RK. Transgressive segregation adaptation and speciation. Heredity. 1999b;83:363–372. doi: 10.1038/sj.hdy.6886170. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Widmer A, Arntz MA, Burke JM. Directional selection is the primary cause of phenotypic diversification. Proc Natl Acad Sci USA. 2002;99:12242–12245. doi: 10.1073/pnas.192360899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintagne C, Bodenes C, Barrenche T, Pot D, Plomion C, Kremer A. Distribution of genomic regions differentiating oak species assessed by QTL detection. Heredity. 2004;92:20–30. doi: 10.1038/sj.hdy.6800358. [DOI] [PubMed] [Google Scholar]
- Savolainen O, Hedrick PW. Heterozygosity and fitness: no association in Scots pine. Genetics. 1995;140:755–766. doi: 10.1093/genetics/140.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling EE, Heiser CB. An infrageneric classification of Helianthus section Helianthus (Compositae) Taxon. 1981;30:393–403. [Google Scholar]
- Sefc KM, Lopes S, Mendonca D, Dos Santos MR, Machado MLD, Machado AD. Identification of microsatellite loci in olive (Olea europaea) and their characterization in Italian and Iberian olive trees. Mol Ecol. 2000;9:1171–1173. doi: 10.1046/j.1365-294x.2000.00954.x. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. The Principles and Practice of Statistics in Biological Research. State University of New York at Stony Brook; New York, NY: 1995. Biometry. [Google Scholar]
- Stebbins GL. The role of hybridization in evolution. P Am Philos Soc. 1959;103:231–251. [Google Scholar]
- Stephens JC, Briscoe D, O’Brien SJ. Mapping by linkage disequilibrium in human populations—limits and guidelines. Am J Hum Genet. 1994;55:809–824. [PMC free article] [PubMed] [Google Scholar]
- Stettler RF, Zsuffa L, Wu R. The role of hybridization in the genetic manipulation of Populus. In: Young A, Boshier D, Boyle T, editors. Forest Conservation Genetics—Principles and Practice. CSIRO Publishing; Collingwood: 1996. [Google Scholar]
- Tang S, Yu KJ, Slabaugh MB, Shintani DK, Knapp SJ. Simple sequence repeat map of the sunflower genome. Theor Appl Genet. 2002;105:1124–1136. doi: 10.1007/s00122-002-0989-y. [DOI] [PubMed] [Google Scholar]
- Tanksley SD. Mapping polygenes. Ann Rev Genet. 1993;27:205–233. doi: 10.1146/annurev.ge.27.120193.001225. [DOI] [PubMed] [Google Scholar]
- Tinner W, Lotter AF. Central European vegetation response to abrupt climate change at 82 ka. Geology. 2001;29:551–554. [Google Scholar]
- van Valen L. Ecological species, multispecies and oaks. Taxon. 1976;25:233–239. [Google Scholar]
- Volk VH. Is ash (Fraxinus Excelsior L.) native to central European flood plains? Forstwiss. Centralbl. 2002;121:128–137. [Google Scholar]
- Weir BS. Genetic Data Analysis II. Sinauer Associates; Sunderland, MA: 1996. [Google Scholar]
- Welch ME, Rieseberg LH. Habitat divergence between a homoploid hybrid sunflower species Helianthus paradoxus (Asteraceae) and its progenitors. Am J Bot. 2002;89:472–479. doi: 10.3732/ajb.89.3.472. [DOI] [PubMed] [Google Scholar]
- Whitham TG. Plant hybrid zones as sinks for pests. Science. 1989;244:1490–1493. [Google Scholar]
- Whitham TG, Martinsen GD, Floate KD, Dungey HS, Pott BM, Keim P. Plant hybrid zones affect biodiversity: tools for a genetic-based understanding of community structure. Ecology. 1999;80:416–428. [Google Scholar]
- Whittemore AT, Schaal BA. Interspecific gene flow in sympatric oaks. Proc Natl Acad Sci USA. 1991;88:2540–2544. doi: 10.1073/pnas.88.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JGK, Kubelik AR, Kenneth JL, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acid Res. 1990;18:6531–6535. doi: 10.1093/nar/18.22.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JH, Arnold ML. Sources of genetic structure in the woody perennial Betula occidentalis. Int J Plant Sci. 2001;162:1097–1109. [Google Scholar]
- Wright JW. Introduction to Forest Genetics. Academic Press; San Diego, CA: 1976. [Google Scholar]
- Wu RL, Zeng ZB. Joint linkage and linkage disequilibrium mapping in natural populations. Genetics. 2001;157:899–909. doi: 10.1093/genetics/157.2.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger SD, Jansson S, Taylor G. Genomics and forest biology: Populus emerges as the perennial favorite. The Plant Cell. 2002;14:2651–2655. doi: 10.1105/tpc.141120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo A. Molecular biology of salt tolerance in the context of whole-plant physiology. J Exp Bot. 1998;49:915–929. [Google Scholar]
- Zeng ZB. Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc Natl Acad Sci USA. 1993;90:10972–10976. doi: 10.1073/pnas.90.23.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng ZB. Precision mapping of quantitative trait loci. Genetics. 1994;136:1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]