

Abstract
Water reabsorption in the kidney represents a critical physiological event in the maintenance of body water homeostasis. This highly regulated process relies largely on vasopressin (VP) action and on the VP-sensitive water channel that is AQP2 expressed in principal cells of the kidney collecting duct (CD). Defects in the VP signaling pathway and/or in AQP2 cell surface expression can lead to an inappropriate reduction in renal water reabsorption and the development of nephrogenic diabetes insipidus (NDI), a disease characterized by polyuria and polydypsia. This review focuses on the major regulatory steps that are involved in AQP2 trafficking and function. Specifically, we begin with a discussion on VP-receptor (V2R)-independent mechanisms of AQP2 trafficking, with special emphasis on the NO-cGMP signaling pathway, followed by a review of the mechanisms that govern AQP2 endocytosis and exocytosis. We then discuss emerging data illustrating roles played by the actin cytoskeleton on AQP2 trafficking, and lastly we consider elements that affect AQP2 protein expression in cells. Recent advances in each topic are summarized and are presented in the context of their potential to serve as a basis for the development of novel therapies that may ultimately improve life quality of NDI patients.
Keywords: vasopressin receptor, AQP2, endocytosis, exocytosis, cGMP
Water homeostasis and urine concentration via water reabsorption in the urinary tubule are integral functions of the kidney. In normal human subjects, the glomerular system can filter 180 l/day of fluid, of which 90% is reabsorbed back into the circulating system in the proximal tubule and descending limb of Henle’s loop. The remaining 10% is reabsorbed under the regulation of vasopressin (VP) at the level of the collecting duct (CD). Nephrogenic diabetes insipidus (NDI) is a disease characterized by massive water loss (up to 20 l/day) via the kidneys, and the disease can either be acquired or inherited. Acquired NDI is often observed in patients suffering from disorders such as hypokalemia, hypercalcemia, ureteral obstruction and secondary aldosteronism. Additionally, at least 20% of bipolar patients treated with lithium acquire NDI. In inherited forms of NDI, early symptoms include fever, vomiting, anorexia, growth retardation and developmental delay, while later in life polyuria, polydypsia and even mental retardation can ensue in untreated cases. Both acquired and congenital forms of NDI have been linked to defects in the VP hormone signaling system which, under normal conditions, increases both apical cell surface expression and whole-cell abundance of the aquaporin-2 (AQP2) water channel in CD principal cells. The most severe forms of NDI are observed in congenital cases, where patients most often harbor mutations in the vasopressin type-2 receptor (V2R) gene, although a small percentage (10%) bear recessive or autosomal-dominant mutations in the AQP2 gene (1).
The release of VP, a cyclic nonapeptide hormone secreted by the posterior pituitary gland, is regulated in the brain in response to serum osmolarity and body volume status. In the presence of high serum osmolarity or hypovolemia, VP is released into the blood stream where it binds to V2R expressed on the basolateral surface of CD principal cells. V2R is a G-protein coupled receptor, and VP binding initiates the V2R signaling cascade, inducing a conformational change that promotes Gsα dissociation, adenylyl cyclase activation and consequently a rise in intracellular cAMP. The classical view of AQP2 trafficking, based on the so-called “shuttle hypothesis” originally proposed to explain water channel trafficking in the toad urinary bladder (2), postulates that a rise in cAMP concentration and ensuing activation of phosphokinase type A (PKA) leads to the phosphorylation of AQP2 at S256 located in its C-terminal domain, promoting exocytosis and cell-surface accumulation of AQP2 (3). In addition to cAMP elevation, an increase in intracellular Ca2+ concentration and Ca2+ oscillations are also a part of the VP response (4, 5). While it is likely that Ca2+ plays a role in AQP2 plasma membrane insertion in addition to cAMP, the exact mechanisms and relative contribution of each signal remain to be elucidated (see Fig. 1). Expression of AQP2 at the apical membrane leads to an influx of water into the cell driven by the interstitial osmotic gradient generated by urea and sodium. Water then exits the cell via AQP3 and AQP4 water channels located at the basolateral side of the cell, allowing its re-entry into the interstitium and the circulatory system.
Figure 1. Schematic representation of AQP2 trafficking in principal cells.
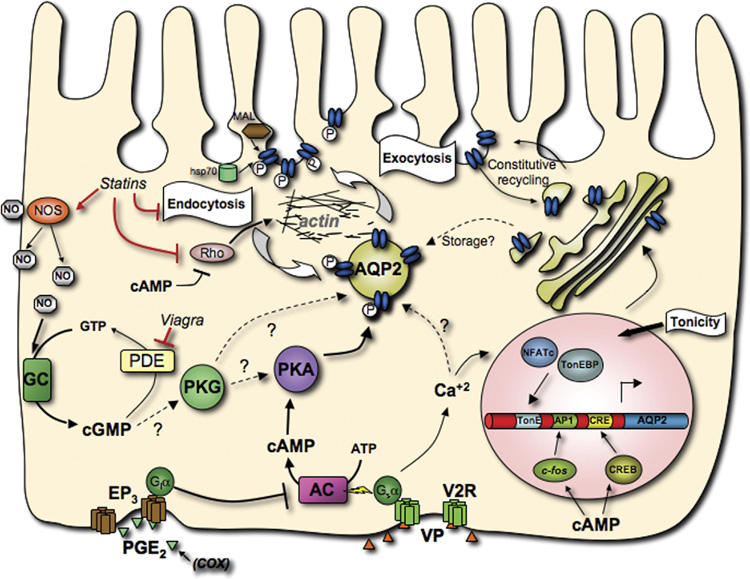
This model shows the interactions between the components of some of the major pathways that affect AQP2, and summarizes most of the points outlined in this review. The canonical V2R signaling pathway is depicted, with VP stimulation of V2R leading to the phosphorylation of AQP2 by PKA and subsequently altering the balance between exocytosis and endocytosis, leading to AQP2 accumulation at the apical plasma membrane. Also shown are the NO-cGMP pathway and the PGE2 receptor EP3 that can also positively and negatively modulate AQP2 trafficking respectively, as well as factors that affect AQP2 abundance and the elements and transcription factors that mediate this regulation. (activator protein-1 element (AP1), adenylate cyclase (AC), aquaporin-2 (AQP2), cAMP response element (CRE), CRE binding protein (CREB), cyclooxygenase (COX), G-protein i α subunit (Giα), G-protein s α subunit (Gsα), guanylate cyclase (GC), heat-shock protein of 70kDa (hsp70), myelin and lymphocyte-associated protein (MAL), nitric oxide (NO), nitric oxide synthase (NOS), nuclear factor of activated T-cells c (NFATc), phosphodiesterase (PDE), prostaglandin E2 (PGE2), prostaglandin receptor (EP3), protein kinase G (PKG), protein kinse A (PKA), Rho family small GTPase (Rho), tonicity response element (TonE), TonE binding protein (TonEBP), vasopressin (VP), vasopressin receptor type-2 (V2R))
Polyuria in acquired NDI can be partially reduced by a combination of treatments such as adequate hydration, low salt and/or low protein diet, diuretics and non-steroidal anti-inflammatory drugs (6). However, congenital patients often respond poorly to such therapies. Some studies have focused on rescuing misfolded V2R by developing non-peptidic lipid-soluble vasopressin ligands that cross the plasma membrane and reach misfolded receptors trapped in the ER. The ligand acts like a molecular chaperone, helping V2R refold, escape the ER quality control and reach the plasma membrane where endogenous VP can subsequently displace the VP analogue and activate the receptor (7). These compounds have shown some positive effects on patients bearing specific missense mutations or small insertion/deletion V2R mutations, but are not effective against truncated proteins, and furthermore, they did not completely alleviate polyuria. A second strategy has employed aminoglycoside antibiotics such as gentamicin. This class of antibiotics causes read-through of some nonsense V2 mutations in vitro and in vivo (8), but the beneficial effect of aminoglycosides is unfortunately overshadowed by its toxicity to the kidneys. Another disadvantage of attempts at rescuing mutant receptors is that they are often heavily dependent on the nature of the mutation and, therefore, any such therapy may not be widely applicable. Finally, use of cAMP phosphodiesterase (PDE) inhibitors such as rolipram has so far been unsuccessful for the treatment of NDI. While beneficial effects have been observed in mouse models, they have not been reproduced in human subjects, perhaps reflecting a difference in cAMP PDE localization (9,10).
The search for more effective therapeutic strategies for both acquired and congenital NDI has, thus, motivated many advances in our understanding of the V2R signaling cascade that regulates AQP2 trafficking. In addition, various other physiological factors that modulate AQP2 cell-surface localization as well as its abundance are now beginning to be uncovered. In this review, we begin by describing the V2R-independent NO/cGMP pathway and recent studies that have linked this important signaling cascade to AQP2 shuttling. We then discuss the use of phosphodiesterase inhibitors such as Viagra as potential therapeutic agents. Next, we summarize novel developments on the role of endocytosis on AQP2 trafficking and consider plausible targets for bypassing V2R as well as the possibility of employing statins in NDI treatment. Finally, we provide a brief overview of other signaling pathways currently being investigated that regulate AQP2 abundance and that may be explored in the future in the search for specific targets that can be readily exploited in novel therapeutic strategies in the treatment of NDI, as well as other forms of water imbalances.
The NO-cGMP signaling pathway
Together with the canonical cAMP-induced pathway, the NO-cGMP signaling pathway has been shown to play a role in AQP2 trafficking, prompting investigation of this signaling pathway as a means to develop alternative therapies for treatment of NDI. Nitric oxide is a free radical resulting from the enzymatic conversion of L-arginine to L-citrulline by one of three isoforms of nitric oxide synthetase (NOS) expressed in the kidney. NO produced by endothelial NOS (eNOS), inducible NOS (iNOS) and neuronal NO (nNOS) can diffuse and, therefore, act in both autocrine and paracrine fashion in the kidney. The classical NO signaling pathway depends on activation of soluble guanylate cyclase (sGC) located in several segments of the nephron including the CD, where it is expressed in principal cells. The major effect of an increase of cyclic guanosine monophosphate (cGMP) concentration is cGMP-dependent protein kinase (PKG) activation. However, cGMP also affects PKA and p21ras kinase activity (11, 12) and can directly regulate Na-channel and glucose transporter (GLUT4) activity as well as trafficking of type 1 and 5 water channels (AQP1 and 5) (13–16). All components of the NO-cGMP signaling pathway are expressed in renal epithelial cells, supporting the notion that the NO-cGMP signaling pathway plays a key role in principal cell physiology, including fluid transport in the kidney.
The effect of NO-cGMP in CD water reabsorption is still controversial. In one study, NO donors were found to decrease VP-induced water reabsorption as a consequence of reduced osmotic water permeability and sodium reabsorption while another report failed to observe this inhibitory effect (17, 18). Another study showed that NO antagonizes the effect of VP by altering cAMP levels (19). In our hands, VP increased the conversion of L-arginine to L-citrulline. However, increased NOS activity appears to result from an indirect effect and VP may simply increase substrate availability. Nevertheless, increasing evidence supports the idea that NO-cGMP is involved in renal water reabsorption. VP increases nNOS and eNOS expression in water-deprived rats, and eNOS expression is accompanied by a reduction of urine output, suggesting that it plays a role in water homeostatic mechanisms (20, 21). Simultaneous disruption of all three NOS isoforms led to NDI in mice (22) and a reduction of AQP2 whole-cell abundance. The mechanism that regulates AQP2 abundance is still elusive, but may be related to low cAMP intracellular levels detected in knock-out mice due to increased prostacyclin activity (22). A reduction of basal intracellular cGMP concentration due to the absence of NOS may lead to decreased levels of functional PKG that may subsequently affect cAMP-response-element (CRE)-dependent transcription (23). Thus, AQP2 expression, which is chiefly regulated by VP, may be additionally stimulated by NOS basal activity. This may partially explain the significant amount of AQP2 expression in Brattleboro rats, which do not express circulating VP.
Our study provided the first evidence that both sodium nitroprusside (SNP), a NO donor, and L-arginine, a precursor of NO, are able to shift the localization of AQP2 from the cytoplasm to the apical side of rat CD principal cells (24). This increase of AQP2 membrane insertion was cGMP-dependent but cAMP-independent. The role of cGMP in AQP2 trafficking was confirmed by analysis of the atrial natriuretic peptide receptor, which has intrinsic guanylate cyclase activity, and by analysis of the effects produced by the cell-permeant dibutyryl cGMP analogue. Both agents induced translocation of AQP2 to the plasma membrane. In addition, atrial natriuretic peptide infusion in rat showed a marked increase in AQP2 apical targeting (25). The mechanism by which cGMP induces AQP2 trafficking is still unclear. Our study showed that AQP2 can be phosphorylated by PKG, but we cannot reasonably eliminate the possibility that PKG phosphorylates PKA, nor can we yet rule out that an increase of cGMP concentration activates PKA and subsequently induces an accumulation of AQP2 at the plasma membrane.
Since cGMP increases AQP2 membrane insertion in rat kidney, we investigated the effect of selective cyclic-3’,5’-nucleotide phosphodiesterase (PDE) inhibitors, which abolish cGMP catabolism and subsequently increase cGMP concentration, on AQP2 cell surface expression. Intracellular cAMP and cGMP levels are strongly regulated by one or more members of 11 PDE families that together account for over 60 isoforms. Several isoforms are expressed along the nephron, such as cAMP sensitive phosphodiesterase (PDE 3 and 4), cGMP sensitive phosphodiesterase (PDE 5) and cAMP/cGMP selective phosphodiesterase (PDE 1). We used sildenafil citrate (Viagra), a selective cGMP phosphodiesterase (PDE5) inhibitor as a means to increase intracellular cGMP concentration. Sildenafil has been successfully used in clinical treatment of erectile dysfunction. We studied the effect of PDE5 inhibition on AQP2 trafficking in LLC-PK1 cells expressing c-myc tagged AQP2. Western blot analysis showed that the presence of sildenafil or 3-isobutyl-1-methylxanthine (IBMX), a non-selective cAMP/cGMP PDE inhibitor, enhanced AQP2 expression at the plasma membrane (26) (Fig. 2A). We also observed that both PDE5 inhibitors sildenafil and 4-{(3′,4′-(Methylenedioxy)benzyl)amino}-6-methoxyquinazoline (MBMQ) modulate endogenous AQP2 trafficking in rat kidney (24) (Fig. 2B). Sildenafil increased insertion of AQP2 in the apical membrane of principal cells of outer medullary CD both in vitro and in vivo but did not have an effect on AQP2 localization in cortical CD, a major site of water reabsorption (26). While our studies suggest that PDE inhibition may offer a promising approach for X-linked NDI therapy, further studies need to be performed in order to determine whether prolonged cGMP inhibition, or a combined therapeutic approach, can improve water reabsorption in patients suffering from NDI who may express variable amounts of AQP2 in their principal cells.
Figure 2. Western blot detection of AQP2 in plasma membrane-enriched fractions from LLC-PK1 cells expressing c-myc-tagged AQP2 (A). Indirect immunofluorescence microscopy of tissue slices showing AQP2 redistribution in the inner stripe (outer medulla) of collecting duct principal cells in response to PDE V inhibition (B).
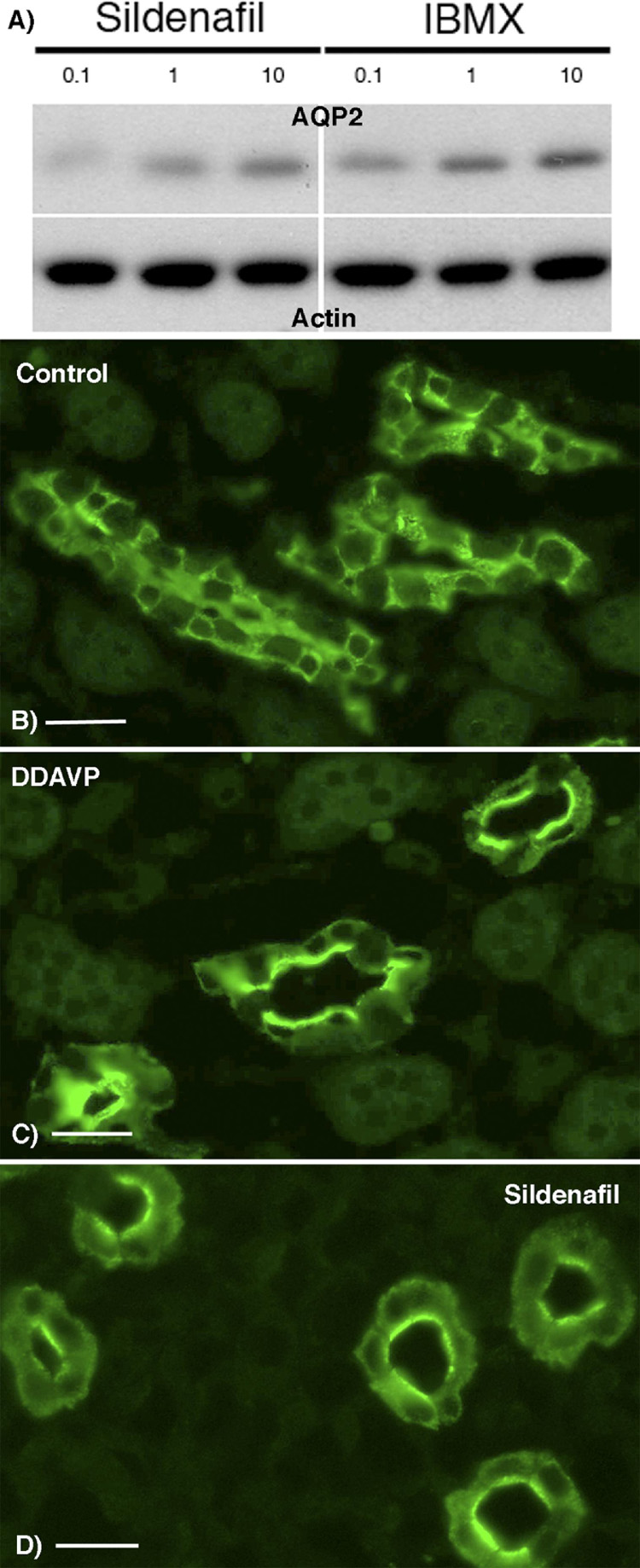
In panel A, cells were incubated 45 min in the presence of the selective PDE5 inhibitor (sildenafil) or with the non-selective PDE inhibitor (IBMX) at a 0.1, 1 or 10 fold higher dose than that corresponding to the EC50 of either chemical agent. A plasma membrane fraction was isolated from the cells and probed with anti-AQP2 antibodies. The same plot was reprobed with an anti-pan-actin monoclonal antibody as a loading control. Both sildenafil and IBMX induce the appearance of AQP2 in the plasma membrane fraction of the cells in a does-dependent manner.
In panel B kidney slices from a Sprague-Dawley rat were incubated for 10 min with (Deamino-Cys1, D-arg8)VP (DDAVP, 10 nM) or 45 min with sildenafil (0.5 µM) before fixation by immersion, sectioning and immunostaining for AQP2. Panel (A) shows a diffuse intracellular distribution of AQP2 in a control medullary collecting duct, whereas apical membrane accumulation (arrows) is induced in tissues treated with DDAVP (B) or sildenafil (C). Bar = 25 µm
AQP2 endocytosis and exocytosis
About 10% of congenital NDI cases are associated with AQP2 mutations rather than with defects of V2R signaling. Interestingly, the majority of these mutations are manifested as misrouting errors rather than as structural defects that affect the channel’s water permeability. Therefore, proper plasma membrane insertion/trafficking is integral to proper AQP2 functioning and correct water molecule transport. A reagent that affects this process may consequently represent a potential target for modulating water absorption. Several studies from our group have revealed that besides cAMP- and cGMP-stimulated trafficking, AQP2 rapidly recycles between an intracellular pool and the plasma membrane under baseline (non-stimulated) conditions. In light of this constitutive pathway, steady-state accumulation of AQP2 at the plasma membrane may be mediated not only by an increase in exocytosis, as originally postulated by the shuttle hypothesis, but also by a reduction in endocytosis.
The clathrin-mediated pathway is one of the major routes of endocytosis in eukaryotic cells, and is characterized by the selective internalization of proteins from the cell surface. An elaborate series of protein-protein interactions imparts selectivity on this highly dynamic process that involves the rapid assembly or disassembly of transient protein complexes. Both ATPases and GTPases modulate assembly of these complexes (27, 28), requiring multiple interactions between clathrin, dynamin, hsc70, the adaptor proteins AP2 and AP180, Esp15, and many other accessory proteins such as auxillin, endophilin and amphiphysin (28). The presence of molecules later identified as AQP2 in clathrin-coated pits was first observed over twenty years ago (29). More recently, endocytosis blockade achieved by transfecting a dominant-negative dynamin mutant into cultured cells was found to induce dramatic membrane accumulation of AQP2 (Fig. 3; (30)). Members of the heat-shock protein family (hsc70 and hsp70) were found to directly interact with AQP2 and regulate its trafficking. Immunogold EM showed that hsc70 co-localized with AQP2 in the plasma membrane. In addition, inhibition of endogenous hsc70 activity using a dominant-negative hsc70 mutant also caused dramatic membrane accumulation of AQP2 in cells (Fig. 3; (31)). This suggests that hsc70 is likely to be involved in AQP2 endocytosis, although it cannot be ruled that other biological functions are associated with the interaction of hsc/hsp70 and AQP2. A recent observation suggests that myelin and lymphocyte-associated protein (MAL) is involved in regulated AQP2 trafficking by physically interacting with AQP2. It increases AQP2 plasma membrane expression by attenuating its internalization (32).
Figure 3. AQP2 membrane accumulation can be induced by inhibiting endocytosis.
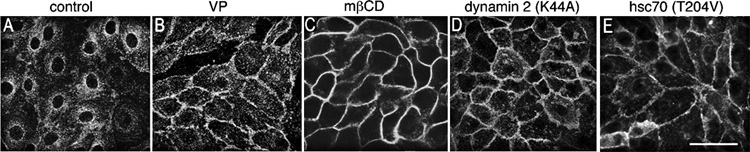
Control LLC-PK1 cells expressing AQP2 displayed baseline perinuclear AQP2 staining (A), whereas cells exposed to vasopressin (VP) showed strong AQP2 expression at the plasma membrane (B). Endocytosis was blocked in LLC-PK1 cells by methyl-β-cyclodextrin (mβCD) treatment (C), expressing a dominant interfering dynamin mutant (dynamin 2 DK44A) (D) or an ATPase deficient hsc70 mutant (T204V) (E). All three approaches to reduce endocytosis resulted in a dramatic increase of AQP2 expression at the plasma membrane. Immunostaining was performed using an anti-c-myc antibody to detect the c-myc tag of AQP2 in stably transfected LLC-PK1 cells. Bar = 20 µm.
An interesting aspect of AQP2 endocytosis is that AQP2 constitutive recycling is independent of S256 phosphorylation. AQP2-S256D mutants are localized in the plasma membrane in the absence of VP stimulation (33) while AQP2-S256A mutant expression is restricted to intracellular compartments (34). However, a cholesterol-depleting agent that inhibits endocytosis caused a large accumulation of AQP2 at the plasma membrane both in cell cultures (35) and in isolated perfused rat kidney (36). Even AQP2-S256A mutants rapidly accumulated at the cell surface under these conditions, indicating that AQP2 dephosphorylated at S256 can also accumulate at the plasma membrane. Other observations additionally indicate that S256 phosphorylation alone is not sufficient to induce translocation of AQP2 to the cell surface. In Brattleboro rats, which display decreased levels of AQP2 abundance due to the lack of circulating VP, AQP2 is mostly expressed in intracellular pools despite the fact that a significant amount of AQP2 is phosphorylated at S256. VP treatment increased AQP2 cell surface expression but did not appear to increase its phosphorylation (37).
While it was initially assumed that an increase of AQP2 exocytosis arises from AQP2 phosphorylation at S256, most assays to date measured only AQP2 cell surface accumulation rather than bona fide exocytosis. We have, however, recently demonstrated that a burst of AQP2 exocytosis occurs during the first 15–20 minutes of VP stimulation (Fig. 4). This observation was made using a novel fluorescence based assay that relies on expression of secreted soluble YFP (ssYFP) that passively labels biosynthetic/post-Golgi vesicles. This assay provides an indirect but quantitative means to measure AQP2 exocytosis. In addition, a recent study has shown that VP stimulation increases AQP2 expression at the cell surface by inducing its accumulation in “endocytosis-resistant” membrane domains (38), indicating that while VP enhances AQP2 exocytosis, it significantly reduces AQP2 endocytosis. Increased expression of water channels at the cell surface through altered exocytotic and endocytotic activity was already suggested by mathematical modeling over a decade ago prior to the molecular identification of aquaporins (39).
Figure 4. VP/FK treatment increases exocytosis in AQP2-expressing cells, but not in control cells.
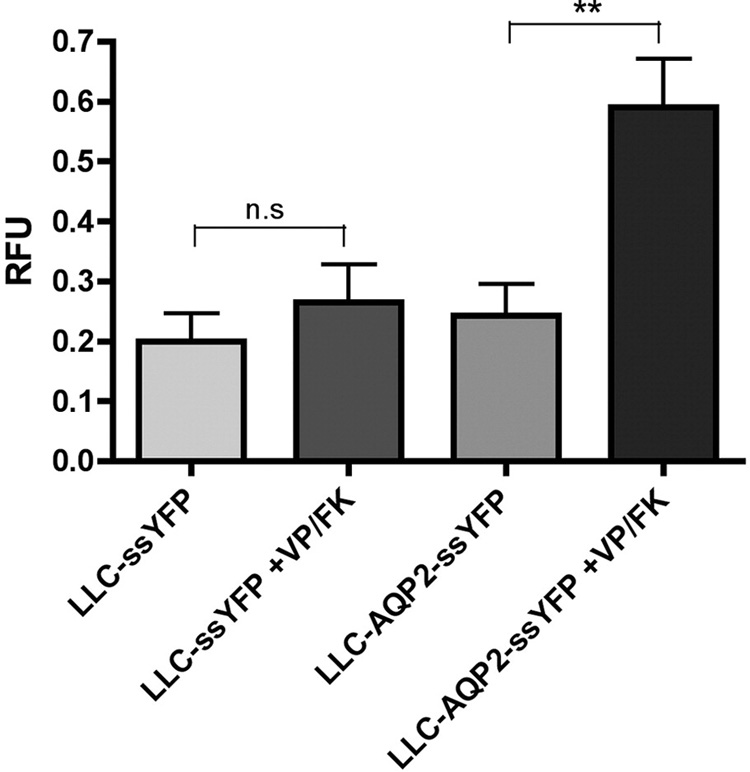
LLC-PK1 cells expressing AQP2 were transfected with a vector encoding a soluble, secreted form of yellow fluorescence protein, YFP (kindly provided by Jennifer Lippincott-Schwartz, NIH). The amount of ssYFP produced in LLC-ssYFP (which express YFP but not AQP2) and LLC-AQP2-ssYFP cells (which express AQP2 and YFP) and secreted in the extracellular medium after 15 min was measured by fluorimetry, and is similar between both cell lines under baseline conditions (bars 1 and 3 from left to right). When VP/FK is applied, AQP2-expressing cells show a large increase in ssYFP secretion within the first 15 min of stimulation, as compared to control cells (bars 2 and 4, respectively). These results are consistent with a large burst of exocytosis of AQP2-containing vesicles in response to VP/FK stimulation. Values were calculated as the relative increase from the 0 min baseline control and are expressed in relative fluorescence units (RFU). Each bar represents the average of 5 independent experiments performed in triplicate.
It is well known that protein phosphorylation and dephosphorylation markedly affect the biological activity of proteins. As discussed above regarding the S256 residue, AQP2 phosphorylation plays an important role in AQP2 trafficking/membrane accumulation. Analysis of potential AQP2 phosphorylation sites in addition to S256 suggests the presence of putative sites for at least four kinases, namely PKG, PKC, casein kinase II and Golgi casein kinase in addition to PKA. The potential role of these phosphorylation sites is currently under investigation by our group as well as others (33, 34, 37, 40–42). AQP2 targeting to the apical membrane may be achieved by manipulating its phosphorylation state, and pharmacological inhibition of phosphatase activity by okadeic acid is sufficient to increase expression of AQP2 at the plasma membrane of cultured cells (43). The events governing AQP2 phosphorylation and dephosphorylation will undoubtedly lead to the discovery of potential targets for the development of therapeutic reagents.
Potential role of statins in AQP2 trafficking
By reducing cholesterol-containing atherogenic lipoproteins (44) 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) dramatically improve cardiovascular outcome. Studies performed on cell cultures of proximal tubular cells have provided evidence that statins reduce receptor-mediated endocytosis (RME) and that this is a consequence of statin-induced impaired prenylation and, therefore, membrane association, of one or more GTP-binding proteins that play a key role in RME (45, 46). By reducing protein uptake, statins may even exert a renoprotective effect as suggested by animal models of kidney disease (47) and by a meta-analysis of randomized trials (48). Various observations point to the possibility that statins might be used as a means to increase AQP2 cell surface expression: 1) RME is a clathrin-mediated process which requires the participation of several GTP-binding proteins, such as Rho, Rac, and Rab. Consistent with this, prenylation of the GTP-binding protein Rap1A was found to be reduced by statins in cultured proximal cells (45). An effect similar to that induced by statins on RME in proximal cells may also occur in CD cells that could affect AQP2 endocytosis. In this respect, statins may increase AQP2 cell surface expression by reducing its endocytosis. 2) Low doses of statins were found to increase eNOS phosphorylation and activation in endothelial cells via increased Akt activation (49, 50), while high doses of statins were found to increase eNOS protein synthesis, through an increase of eNOS mRNA stability (51). Statins may consequently increase AQP2 cell surface expression by enhancing eNOS activity (see above). 3) High doses of statins were found to decrease Rho activity (50), a key player of actin reorganization that affects AQP2 trafficking, as discussed below. This effect may be attributed to inhibition of the geranylgeranylation and membrane localization of RhoA and by alterations in RhoA-dependent cell-signaling pathways, such as flk-1/KDR and Akt (52). Based on these observations, our laboratory is currently evaluating the role of statins in AQP2 trafficking with a view to developing potential novel therapies for the treatment of NDI.
Rearrangement of cytoskeletal components and regulation of small G-proteins
Actin polymerization and depolymerization is a dynamic and tightly regulated process that plays an important role in protein trafficking. Actin reorganization is controlled by the Rho family of small GTP binding proteins. This includes members of the RhoA-G, Cdc42 and Rac1 family that are activated after GDP is exchanged with GTP. The nucleotide exchange process is controlled by various proteins such as GTPase activating protein (GAP), GEFs and GDI. Depolymerization of the actin network results in an increase of AQP2 expression at the cell surface while blockade of VP-induced AQP2 translocation in response to Rho activation was shown to be associated with increased actin polymerization (53–55). Thus, modulation of the actin cytoskeleton might represent a therapeutic approach for NDI, despite the omnipresence of Rho that makes this protein difficult to specifically target in CD principal cells. At the very least, a better understanding of the mechanisms that regulate cytoskeletal reorganization and AQP2 trafficking will undoubtedly help identify therapeutic targets whose modified activities may provide the basis for future therapies.
A shift of the equilibrium between V2R and prostaglandin E2 (PGE2) receptor stimulation affects the polymerization state of the actin cytoskeleton and consequently affects AQP2 trafficking to the plasma membrane. An increase of cAMP concentration following V2R activation results in Rho inhibition (56) and the subsequent depolymerization of the actin cytoskeleton. PGE2, on the other hand, counteracts the VP-induced increase of osmotic water permeability in the renal CD. When PGE2 binds to the EP3 receptor, adenylate cyclase is inactivated resulting in an increase of actin polymerization via Rho activation. PGE2 may also counteract the intrinsic actin reorganization capability of AQP2 bearing vesicles, as suggested by a recent observation that shows that AQP2 can interact directly with actin and SPA-1, a specific Rap GAP (57).
PGE2 is abundantly expressed in the kidney. It derives from arachidonic acid via cyclooxygenase (58) and prostaglandin E synthetase (PGES) activities. Two cyclooxygenase isoforms, COX-1 and COX-2, are expressed in the kidney. Interestingly, COX-2 expression, which is known to be induced by physiological stress, is increased in NDI patients (59, 60). The development of selective COX inhibitors has raised several expectations. For example, rofecoxib (a COX-2 inhibitor) in combination with hydrochlorothiazide and a low salt formula reduced urine volume in a 1 month-old male infant (61). However, COX-2 inhibitors should be used with extreme caution because of the high risk of developing myocardial infarction (62). The adverse effects associated with this family of inhibitors suggest that more research should focus on the downstream effectors of the COX/PGE2 signaling pathway.
Three isomers of prostaglandin synthetase (PGES) have been recently described. Interestingly, the mPGE1 isoform is inducible and its expression is tightly related to COX-2 expression. mPGE1 is expressed in the CD and is increased in type 2 diabetes. The role of mPGES in NDI has not been fully investigated but the recent availability of selective mPGE1 inhibitors will allow us to investigate in-depth their potential therapeutic benefits (63). Several efforts have been made to develop PGE receptor antagonists. Three of four PGE receptor subtypes (prostaglandin E2 receptor type 1, 3 and 4) are expressed in different regions of the kidney. EP1 and EP4 are expressed in the glomerulus, whereas EP3 is undetectable in this region. However, two EP3 isoforms are expressed in the CD (64). Some inhibitors of the PGE receptor have been developed that show interesting effects. An EP1 selective antagonist has been shown to prevent the progression of nephropathy in streptozotocin-induced diabetic rats (65). In that study, Makino et al. showed that aspirin, a non selective COX inhibitor has more beneficial effects on urine volume than a COX-selective antagonist (65). This result indicates that selective PGE receptor antagonism may represent an efficient means of controlling water excretion and that every effort should be made to develop other PGE receptor inhibitors that target other PGE receptor isoforms such as EP3.
Other alternative mechanisms have recently been reported to regulate AQP2 trafficking that may provide potential targets for future NDI therapies. Both bradykinin and Epac have been shown to increase AQP2 membrane expression. Bradykinin binds to the B2 receptor and leads to Rho activation, subsequently attenuating AQP2 trafficking by stabilizing polymerized actin (66). Bradykinin binds two receptor subtypes, B1 and B2. B2 is constitutively expressed in the renal CD whereas B1 expression is inducible. Both receptors share similar signaling pathways (67). However, little information is available on the role that the B1 receptor plays in NDI pathophysiology. The B1 receptor is associated with the progression of insulin-dependent diabetes and has a protective role in renal ischemia. The development of selective antagonists may help us to better understand its possible link to NDI. AQP2 trafficking is additionally affected by cAMP-activation of the exchange protein (Epac) (68). Epac can be activated selectively and directly by a cAMP analogue (8-pCPT-2′-O-Me-cAMP). We speculate that activated Epac exchanges bound GDP with GTP in both Rap1 and Rap2 proteins, which play a role in cytoskeletal rearrangement.
Mechanisms that regulate AQP2 whole cell abundance
In addition to controlled AQP2 expression at the cell surface, an increase of AQP2 whole cell abundance represents an attractive approach for NDI therapy. Indeed, down-regulated AQP2 cell surface expression occurring in acquired NDI and in some cases of congenital NDI reflects down-regulated AQP2 abundance, which in some patients may limit the efficacy of strategies aimed simply at targeting pre-synthesized AQP2 to the cell surface. While reduced AQP2 abundance is associated with reduced V2R activity in some cases of NDI, such as hypercalcemia (69), other conditions of NDI appear to arise from VP-independent mechanisms. Recent evidence has shown that lithium-induced NDI is associated with an adenylyl cyclase-independent decrease of AQP2 mRNA expression, possibly resulting from decreased AQP2 transcription (70). In ureteral obstruction, VP-independent down-regulation of AQP2 abundance and cell surface expression was found to arise from increased cyclooxygenase-2 activity and PGE2 synthesis (71). VP-independent mechanisms that increase AQP2 abundance may, thus, prove to be extremely valuable for designing new therapeutic strategies to treat NDI, as illustrated below.
In the kidney, the expression of AQP2 is restricted to the renal collecting system (72,73) and is modulated by both VP and factors that act independently of VP. Several regulatory motifs that induce AQP2 transcriptional activity have been identified in the AQP2 promoter. The most well documented of these are AP1 and CRE sites that respectively bind cAMP-induced c-fos and the phosphorylated adenosine CRE binding protein (CREB) (74–76). In this respect, pCREB plays a dual role in regulating AQP2 by inducing its accumulation at the cell surface and by enhancing AQP2 transcription. AQP2 abundance increases with interstitial tonicity and recent findings have shown that this upregulation arises from increased transcription of the AQP2 gene. The tonicity-responsive enhancer binding protein (TonEBP) has been shown to play a key role in this event, most likely by binding to at least one TonE element present in the AQP2 promoter (77). Of particular interest to the present review, the stimulatory effect of TonEBP on AQP2 transcription was found to occur independently of VP (77). Moreover, a stimulatory effect of NFATc, a transcription factor that belongs to the same family as TonEBP, on AQP2 transcription was demonstrated in cultured renal cells together with cross-talk occurring between TonEBP and calcineurin-NFATc pathways that further enhances AQP2 transcription (78). Inhibition of TonEBP activity by calcineurin inhibitors, including cyclosporine A and its derivatives, has been shown to reduce AQP2 expression (78). Consequently, environmental signals that increase intracellular calcium, and calcineurin activation in particular, provide attractive targets for the promotion of AQP2 expression at the cell surface resulting from increased AQP2 whole cell abundance.
Several pieces of evidence have demonstrated that in addition to transcriptional regulation, AQP2 abundance is also modulated by post-transcriptional processing. AQP2 degradation is dependent on both lysosomal and proteasomal activity (79). Observations made from both in vitro and animal studies indicate that AQP2 protein degradation is inversely associated with changes in V2R activity (80). Moreover, both dihydrotachysterol-treated and fasted animals displayed a VP-independent decrease of AQP2 protein but not mRNA abundance indicating that AQP2 protein degradation is regulated by both VP and factors acting independently of VP (81, 82). In addition to controlled AQP2 protein degradation, enhanced translation of AQP2 mRNA may represent another means of increasing AQP2 whole cell abundance. In vitro studies revealed a feedback mechanism that is dependent on transcriptional activity, that acts independently of AQP2 degradation and that involves rapid synthesis of regulatory protein(s) that continuously reduce AQP2 mRNA translation (83). Aldosterone may enhance AQP2 protein abundance by alleviating such negative control on AQP2 mRNA translation (83). Further dissection of molecular elements involved in AQP2 degradation and mRNA translation may uncover potential targets delimiting therapies based on controlled AQP2 degradation/mRNA translation that would ultimately increase the expression of AQP2 at the cell surface.
Summary
Recent advances in our understanding of the cell biology of AQP2 recycling and the signaling pathways that lead to the membrane accumulation of AQP2 in principal cells have opened up several possible strategies for inducing this process in the absence of conventional vasopressin signaling via its G-protein coupled receptor, the V2R, which is defective in X-linked NDI. Furthermore, these strategies may also apply to other types of NDI, including some of the acquired forms. Superimposed on the need to stimulate AQP2 membrane trafficking is the requirement that sufficient AQP2 be expressed in principal cells of NDI patients to achieve effective therapy. We, therefore, also discuss some mechanisms that regulate AQP2 expression levels in target cells. Depending on the nature of the defect leading to NDI, it is likely that a combination of approaches, directed by the basic research endeavors that are ongoing in many labs, will be required to achieve a positive clinical outcome.
Acknowledgments
This work was supported by NIH grant DK38452. R. Bouley received a Young Investigator Award from the National Kidney Foundation. U. Hasler is supported by a Swiss FSBMB Fellowship and an ECOR Fellowship from MGH. H. A. J. Lu is supported by an NIH KO8 grant DK075940-01, and a Doctoral Level Postgraduate Scholarship from NSERC supports P. Nunes. The Microscopy Core facility of the MGH Program in Membrane Biology receives additional support from the Boston Area Diabetes and Endocrinology Research Center (DK57521) and the Center for the Study of Inflammatory Bowel Disease (DK43341).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deen PM. Mouse models for congenital nephrogenic diabetes insipidus: what can we learn from them? Nephrol Dial Transplant. 2007;22:1023–1026. doi: 10.1093/ndt/gfl787. [DOI] [PubMed] [Google Scholar]
- 2.Wade JB, Stetson DL, Lewis SA. ADH action: evidence for a membrane shuttle mechanism. Ann NY Acad Sci. 1981;372:107–117. doi: 10.1111/j.1749-6632.1981.tb15464.x. [DOI] [PubMed] [Google Scholar]
- 3.Kamsteeg EJ, Heijnen I, van Os CH, et al. The subcellular localization of an aquaporin-2 tetramer depends on the stoichiometry of phosphorylated and nonphosphorylated monomers. J Cell Biol. 2000;151:919–930. doi: 10.1083/jcb.151.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yip KP. Coupling of vasopressin-induced intracellular Ca2+ mobilization and apical exocytosis in perfused rat kidney collecting duct. J. Physiol. 2002;538:891–899. doi: 10.1113/jphysiol.2001.012606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickols HH, Shah VN, Chazin WJ, et al. Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. J Biol Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- 6.Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–F270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- 7.Morello JP, Salahpour A, Laperriere A, et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz A, Sangkuhl K, Lennert T, et al. Aminoglycoside pretreatment partially restores the function of truncated V(2) vasopressin receptors found in patients with nephrogenic diabetes insipidus. J Clin Endocrinol Metab. 2002;87:5247–5257. doi: 10.1210/jc.2002-020286. [DOI] [PubMed] [Google Scholar]
- 9.Deen PTP. Mouse models for congenital nephrogenic diabetes insipidus:what can we learn from them? Nephrol Dial Transplant. 2007;22:1023–1026. doi: 10.1093/ndt/gfl787. [DOI] [PubMed] [Google Scholar]
- 10.Bichet DG, Ruel N, Arthus MF, et al. Rolipram, a phosphodiesterase inhibitor, in the treatment of two male patients with congenital nephrogenic diabetes insipidus. doi: 10.1159/000186196. [DOI] [PubMed] [Google Scholar]
- 11.Oliveira CJ, Schindler F, Ventura AM, et al. Nitric oxide and cGMP activate the Ras-MAP kinase pathway-stimulating protein tyrosine phosphorylation in rabbit aortic endothelial cells. Free Radic Biol Med. 2003;35:381–396. doi: 10.1016/s0891-5849(03)00311-3. [DOI] [PubMed] [Google Scholar]
- 12.Yamada T, Matsuda K, Uchiyama M. Atrial natriuretic peptide and cGMP activate sodium transport through PKA-dependent pathway in the urinary bladder of the Japanese tree frog. J Comp Physiol (B) 2006;176:203–212. doi: 10.1007/s00360-005-0041-z. [DOI] [PubMed] [Google Scholar]
- 13.Anthony TL, Brooks HL, Boassa D, et al. Cloned human aquaporin-1 is a cyclic GMP-gated ion channel. Mol Pharmacol. 2000;57:576–588. doi: 10.1124/mol.57.3.576. [DOI] [PubMed] [Google Scholar]
- 14.Das S, Garepapaghi M, Palmer LG. Stimulation by cGMP of apical Na channels and cation channels in toad urinary bladder. Am J Physiol. 1991;260:C234–C241. doi: 10.1152/ajpcell.1991.260.2.C234. [DOI] [PubMed] [Google Scholar]
- 15.Etgen GJ, Jr, Fryburg DA, Gibbs EM. Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction- and phosphatidylinositol-3-kinase-independent pathway. Diabetes. 1997;46:1915–1919. doi: 10.2337/diab.46.11.1915. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa Y, Iida H, Ishida H. The muscarinic acetylcholine receptor-stimulated increase in aquaporin-5 levels in the apical plasma membrane in rat parotid acinar cells is coupled with activation of nitric oxide/cGMP signal transduction. Mol Pharmacol. 2002;61:1423–1434. doi: 10.1124/mol.61.6.1423. [DOI] [PubMed] [Google Scholar]
- 17.Garcia NH, Stoos BA, Carretero OA, et al. Mechanism of the nitric oxide-induced blockade of collecting duct water permeability. Hypertension. 1996;27:679–683. doi: 10.1161/01.hyp.27.3.679. [DOI] [PubMed] [Google Scholar]
- 18.Hirsch JR, Cermak R, Forssmann WG, et al. Effects of sodium nitroprusside in the rat cortical collecting duct are independent of the NO pathway. Kidney Int. 1997;51:473–476. doi: 10.1038/ki.1997.64. [DOI] [PubMed] [Google Scholar]
- 19.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol. 2002;282:F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- 20.Martin PY, Bianchi M, Roger F, et al. Arginine vasopressin modulates expression of neuronal NOS in rat renal medulla. Am J Physiol Renal Physiol. 2002;283:F559–F568. doi: 10.1152/ajprenal.00309.2001. [DOI] [PubMed] [Google Scholar]
- 21.Shin SJ, Lai FJ, Wen JD, et al. Increased nitric oxide synthase mRNA expression in the renal medulla of water-deprived rats. Kidney Int. 1999;56:2191–2202. doi: 10.1046/j.1523-1755.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 22.Morishita T, Tsutsui M, Shimokawa H, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 2005;102:10616–10621. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilz RB, Casteel DE. Regulation of gene expression by cyclic GMP. Circ Res. 2003;93:1034–1046. doi: 10.1161/01.RES.0000103311.52853.48. [DOI] [PubMed] [Google Scholar]
- 24.Bouley R, Breton S, Sun T, et al. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest. 2000;106:1115–1126. doi: 10.1172/JCI9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Li C, Nejsum LN, et al. Biphasic effects of ANP infusion in conscious, euvolumic rats: roles of AQP2 and ENaC trafficking. Am. J. Physiol. Renal Physiol. 2006;290:F530–F541. doi: 10.1152/ajprenal.00070.2005. [DOI] [PubMed] [Google Scholar]
- 26.Bouley R, Pastor-Soler N, Cohen O, et al. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra) Am J Physiol Renal Physiol. 2005;288:F1103–F1112. doi: 10.1152/ajprenal.00337.2004. [DOI] [PubMed] [Google Scholar]
- 27.Brodsky FM, Chen CY, Knuehl C, et al. Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. 2001;17:517–568. doi: 10.1146/annurev.cellbio.17.1.517. [DOI] [PubMed] [Google Scholar]
- 28.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 29.Brown D, Orci L. Vasopressin stimulates formation of coated pits in rat kidney collecting ducts. Nature. 1983;302:253–255. doi: 10.1038/302253a0. [DOI] [PubMed] [Google Scholar]
- 30.Sun TX, Van Hoek A, Huang Y, et al. Aquaporin-2 localization in clathrin-coated pits: inhibition of endocytosis by dominant-negative dynamin. Am J Physiol Renal Physiol. 2002;282:F998–F1011. doi: 10.1152/ajprenal.00257.2001. [DOI] [PubMed] [Google Scholar]
- 31.Lu HA, Sun TX, Matsuzaki T, et al. Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J Biol Chem. 2007;282:28721–28732. doi: 10.1074/jbc.M611101200. [DOI] [PubMed] [Google Scholar]
- 32.Kamsteeg EJ, Duffield AS, Konings IB, et al. MAL decreases the internalization of the aquaporin-2 water channel. Proc Natl Acad Sci U S A. 2007;104:16696–16701. doi: 10.1073/pnas.0708023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nejsum LN, Zelenina M, Aperia A, et al. Bidirectional regulation of AQP2 trafficking and recycling: involvement of AQP2-S256 phosphorylation. Am J Physiol Renal Physiol. 2005;288:F930–F938. doi: 10.1152/ajprenal.00291.2004. [DOI] [PubMed] [Google Scholar]
- 34.Procino G, Carmosino M, Marin O, et al. Ser-256 phosphorylation dynamics of Aquaporin 2 during maturation from the ER to the vesicular compartment in renal cells. Faseb J. 2003;17:1886–1888. doi: 10.1096/fj.02-0870fje. [DOI] [PubMed] [Google Scholar]
- 35.Lu H, Sun TX, Bouley R, et al. Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am J Physiol Renal Physiol. 2004;286:F233–F243. doi: 10.1152/ajprenal.00179.2003. [DOI] [PubMed] [Google Scholar]
- 36.Russo LM, McKee M, Brown D. Methyl-beta-cyclodextrin induces vasopressin-independent apical accumulation of aquaporin-2 in the isolated, perfused rat kidney. Am J Physiol Renal Physiol. 2006;291:F246–F253. doi: 10.1152/ajprenal.00437.2005. [DOI] [PubMed] [Google Scholar]
- 37.Christensen BM, Zelenina M, Aperia A, et al. Localization and regulation of PKA-phosphorylated AQP2 in response to V(2)-receptor agonist/antagonist treatment. Am J Physiol Renal Physiol. 2000;278:F29–F42. doi: 10.1152/ajprenal.2000.278.1.F29. [DOI] [PubMed] [Google Scholar]
- 38.Bouley R, Hawthorn G, Russo LM, et al. Aquaporin 2 (AQP2) and vasopressin type 2 receptor (V2R) endocytosis in kidney epithelial cells: AQP2 is located in 'endocytosis-resistant' membrane domains after vasopressin treatment. Biol Cell. 2006;98:215–232. doi: 10.1042/BC20040054. [DOI] [PubMed] [Google Scholar]
- 39.Knepper MA, Nielsen S. Kinetic model of water and urea permeability regulation by vasopressin in collecting duct. Am J Physiol. 1993;265:F214–F224. doi: 10.1152/ajprenal.1993.265.2.F214. [DOI] [PubMed] [Google Scholar]
- 40.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J. Biol. Chem. 1997;272:14800–14804. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- 41.Katsura T, Gustafson CE, Ausiello DA. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am. J. Physiol. 1997;272:F817–F822. [PubMed] [Google Scholar]
- 42.Stefan E, Wiesner B, Baillie GS, et al. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol. 2007;18:199–212. doi: 10.1681/ASN.2006020132. [DOI] [PubMed] [Google Scholar]
- 43.Brown D. The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol. 2003;284:F893–F901. doi: 10.1152/ajprenal.00387.2002. [DOI] [PubMed] [Google Scholar]
- 44.Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 45.Sidaway JE, Davidson RG, McTaggart F, et al. Inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase reduce receptor-mediated endocytosis in opossum kidney cells. J Am Soc Nephrol. 2004;15:2258–2265. doi: 10.1097/01.ASN.0000138236.82706.EE. [DOI] [PubMed] [Google Scholar]
- 46.Verhulst A, D'Haese PC, De Broe ME. Inhibitors of HMG-CoA reductase reduce receptor-mediated endocytosis in human kidney proximal tubular cells. J Am Soc Nephrol. 2004;15:2249–2257. doi: 10.1097/01.ASN.0000136778.32499.05. [DOI] [PubMed] [Google Scholar]
- 47.McFarlane SI, Muniyappa R, Francisco R, et al. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002;87:1451–1458. doi: 10.1210/jcem.87.4.8412. [DOI] [PubMed] [Google Scholar]
- 48.Fried LF, Orchard TJ, Kasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59:260–269. doi: 10.1046/j.1523-1755.2001.00487.x. [DOI] [PubMed] [Google Scholar]
- 49.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urbich C, Dernbach E, Zeiher AM, et al. Double-edged role of statins in angiogenesis signaling. Circ Res. 2002;90:737–744. doi: 10.1161/01.res.0000014081.30867.f8. [DOI] [PubMed] [Google Scholar]
- 51.Laufs U, La Fata V, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 52.Park HJ, Kong D, Iruela-Arispe L, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors interfere with angiogenesis by inhibiting the geranylgeranylation of RhoA. Circ Res. 2002;91:143–150. doi: 10.1161/01.res.0000028149.15986.4c. [DOI] [PubMed] [Google Scholar]
- 53.Tamma G, Klussmann E, Maric K, et al. Rho inhibits cAMP-induced translocation of aquaporin-2 into the apical membrane of renal cells. Am J Physiol Renal Physiol. 2001;281:F1092–F1101. doi: 10.1152/ajprenal.0091.2001. [DOI] [PubMed] [Google Scholar]
- 54.Klussmann E, Maric K, Wiesner B, et al. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem. 1999;274:4934–4938. doi: 10.1074/jbc.274.8.4934. [DOI] [PubMed] [Google Scholar]
- 55.Klussmann E, Tamma G, Lorenz D, et al. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem. 2001;276:20451–20457. doi: 10.1074/jbc.M010270200. [DOI] [PubMed] [Google Scholar]
- 56.Tamma G, Klussmann E, Procino G, et al. cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI. J Cell Sci. 2003;116:1519–1525. doi: 10.1242/jcs.00355. [DOI] [PubMed] [Google Scholar]
- 57.Noda Y, Sasaki S. Regulation of aquaporin-2 trafficking and its binding protein complex. Biochim Biophys Acta. 2006;1758:1117–1125. doi: 10.1016/j.bbamem.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Cox RA, Kanagalingam K. A spectrophotometric study of the denaturation of deoxyribonucleic acid in the presence of urea or formaldehyde and its relevance to the secondary structure of single-stranded polynucleotides. Biochem. J. 1968;108:599–610. doi: 10.1042/bj1080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotnik P, Nielsen J, Kwon TH, et al. Altered expression of COX-1, COX-2, and mPGES in rats with nephrogenic and central diabetes insipidus. Am J Physiol Renal Physiol. 2005;288:F1053–F1068. doi: 10.1152/ajprenal.00114.2004. [DOI] [PubMed] [Google Scholar]
- 60.Rao R, Zhang MZ, Zhao M, et al. Lithium treatment inhibits renal GSK-3 activity and promotes cyclooxygenase 2-dependent polyuria. Am J Physiol Renal Physiol. 2005;288:F642–F649. doi: 10.1152/ajprenal.00287.2004. [DOI] [PubMed] [Google Scholar]
- 61.Pattaragarn A, Alon US. Treatment of congenital nephrogenic diabetes insipidus by hydrochlorothiazide and cyclooxygenase-2 inhibitor. Pediatr Nephrol. 2003;18:1073–1076. doi: 10.1007/s00467-003-1195-0. [DOI] [PubMed] [Google Scholar]
- 62.Johnsen SP, Larsson H, Tarone RE, et al. Risk of hospitalization for myocardial infarction among users of rofecoxib, celecoxib, and other NSAIDs: a population-based case-control study. Arch Intern Med. 2005;165:978–984. doi: 10.1001/archinte.165.9.978. [DOI] [PubMed] [Google Scholar]
- 63.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi K, Takahashi N, Abe T, et al. Two isoforms of the rat kidney EP3 receptor derived by alternative RNA splicing: intrarenal expression co-localization. Biochem Biophys Res Commun. 1994;199:834–840. doi: 10.1006/bbrc.1994.1304. [DOI] [PubMed] [Google Scholar]
- 65.Makino H, Tanaka I, Mukoyama M, et al. Prevention of diabetic nephropathy in rats by prostaglandin E receptor EP1-selective antagonist. J Am Soc Nephrol. 2002;13:1757–1765. doi: 10.1097/01.asn.0000019782.37851.bf. [DOI] [PubMed] [Google Scholar]
- 66.Tamma G, Carmosino M, Svelto M, et al. Bradykinin signaling counteracts cAMP-elicited aquaporin 2 translocation in renal cells. J Am Soc Nephrol. 2005;16:2881–2889. doi: 10.1681/ASN.2005020190. [DOI] [PubMed] [Google Scholar]
- 67.Leeb-Lundberg LM, Marceau F, Muller-Esterl W, et al. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 68.Fukuhara S, Sakurai A, Sano H, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Procino G, Carmosino M, Tamma G, et al. Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int. 2004;66:2245–2255. doi: 10.1111/j.1523-1755.2004.66036.x. [DOI] [PubMed] [Google Scholar]
- 70.Li Y, Shaw S, Kamsteeg EJ, et al. Development of Lithium-Induced Nephrogenic Diabetes Insipidus Is Dissociated from Adenylyl Cyclase Activity. J Am Soc Nephrol. 2006;17:1063–1072. doi: 10.1681/ASN.2005080884. [DOI] [PubMed] [Google Scholar]
- 71.Norregaard R, Jensen BL, Li C, et al. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am J Physiol Renal Physiol. 2005 doi: 10.1152/ajprenal.00061.2005. [DOI] [PubMed] [Google Scholar]
- 72.Fushimi K, Uchida S, Hara Y, et al. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- 73.Nielsen S, DiGiovanni SR, Christensen EI, et al. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yasui M, Zelenin SM, Celsi G, et al. Adenylate cyclase-coupled vasopressin receptor activates AQP2 promoter via a dual effect on CRE and AP1 elements. Am. J. Physiol. 1997;272:F443–F450. doi: 10.1152/ajprenal.1997.272.4.F443. [DOI] [PubMed] [Google Scholar]
- 75.Hozawa S, Holtzman EJ, Ausiello DA. cAMP motifs regulating transcription in the aquaporin-2 gene. Am. J. Physiol. 1996;270:C1695–C1702. doi: 10.1152/ajpcell.1996.270.6.C1695. [DOI] [PubMed] [Google Scholar]
- 76.Frokiaer J, Marples D, Valtin H, et al. Low aquaporin-2 levels in polyuric DI +/+ severe mice with constitutively high cAMP-phosphodiesterase activity. Am. J. Physiol. 1999;276:F179–F190. doi: 10.1152/ajprenal.1999.276.2.F179. [DOI] [PubMed] [Google Scholar]
- 77.Hasler U, Jeon US, Kim JA, et al. Tonicity-responsive enhancer binding protein is an essential regulator of aquaporin-2 expression in renal collecting duct principal cells. J Am Soc Nephrol. 2006;17:1521–1531. doi: 10.1681/ASN.2005121317. [DOI] [PubMed] [Google Scholar]
- 78.Li SZ, McDill BW, Kovach PA, et al. Calcineurin-NFATc signaling pathway regulates AQP2 expression in response to calcium signals and osmotic stress. Am J Physiol Cell Physiol. 2007;292:C1606–C1616. doi: 10.1152/ajpcell.00588.2005. [DOI] [PubMed] [Google Scholar]
- 79.Hasler U, Mordasini D, Bens M, et al. Long Term Regulation of Aquaporin-2 Expression in Vasopressin-Responsive Renal Collecting Duct Principal Cells. J. Biol. Chem. 2002;277:10379–10386. doi: 10.1074/jbc.M111880200. [DOI] [PubMed] [Google Scholar]
- 80.Hasler U, Nielsen S, Feraille E, et al. Posttranscriptional control of aquaporin-2 abundance by vasopressin in renal collecting duct principal cells. Am J Physiol Renal Physiol. 2006;290:F177–F187. doi: 10.1152/ajprenal.00056.2005. [DOI] [PubMed] [Google Scholar]
- 81.Puliyanda DP, Ward DT, Baum MA, et al. Calpain-mediated AQP2 proteolysis in inner medullary collecting duct. Biochem Biophys Res Commun. 2003;303:52–58. doi: 10.1016/s0006-291x(03)00215-8. [DOI] [PubMed] [Google Scholar]
- 82.Wilke C, Sheriff S, Soleimani M, et al. Vasopressin-independent regulation of collecting duct aquaporin-2 in food deprivation. Kidney Int. 2005;67:201–216. doi: 10.1111/j.1523-1755.2005.00071.x. [DOI] [PubMed] [Google Scholar]
- 83.Hasler U, Mordasini D, Bianchi M, et al. Dual influence of aldosterone on AQP2 expression in cultured renal collecting duct principal cells. J. Biol. Chem. 2003;278:21639–21648. doi: 10.1074/jbc.M212388200. [DOI] [PubMed] [Google Scholar]