

Abstract
The earliest step in creating the cerebral cortex is the specification of neuroepithelium to a cortical fate. Using mouse genetic mosaics and timed inactivations, we demonstrate that Lhx2 acts as a classic selector gene and essential intrinsic determinant of cortical identity. Lhx2 selector activity is restricted to an early critical period when stem cells comprise the cortical neuroepithelium, where it acts cell-autonomously to specify cortical identity and suppress alternative fates in a spatially dependent manner. Laterally, Lhx2 null cells adopt antihem identity, whereas medially they become cortical hem cells, which can induce and organize ectopic hippocampal fields. In addition to providing functional evidence for Lhx2 selector activity, these findings show that the cortical hem is a hippocampal organizer.
Introduction
Classic genetic analyses in Drosophila have described the roles of ‘selector’ genes (1-3), which drive developmental patterning events by cell-autonomously specifying cell identity, suppressing alternative fates, regulating cell affinity, and positioning developmental borders that often serve as secondary signaling centers. The LIM homeobox gene Lhx2 - a vertebrate orthologue of the well-described Drosophila selector gene Apterous (Ap) (1) - has been postulated to act as a selector gene in the developing mouse cerebral cortex (4). Lhx2 is expressed in cortical precursor cells, but not in the adjacent telencephalic dorsal midline, which consists of choroid plexus epithelium (CPe) and the intervening cortical hem, a secondary source of Bone morphogenetic protein (Bmp) and Wingless-int (Wnt) signals (Figs. S1, S4) (4, 5). Previous studies indicate that the hem is required for hippocampal induction and/or expansion (6, 7), but evidence that the hem is sufficient to induce and organize hippocampal tissue has been lacking. Conventional Lhx2 null embryos (“standard” knockout, or sKO) (8) possess excessive hem and CPe at the expense of hippocampus and neocortex (4, 5). While consistent with a selector gene phenotype, the basic issue of cell autonomy could not be resolved, since a cell-autonomous fate transformation (cortex-to-hem/CPe) could not be distinguished from a nonautonomous hem/CPe expansion due to defects in cortical precursor proliferation (8).
Results
The preneurogenic critical period
Lhx2 expression arises in the forebrain prior to neurulation and is strong in the cortical neuroepithelium, but absent from the dorsal midline, after neural tube closure (Fig. S1A,B). At E10.5, Lhx2 and Lmx1a (hem marker) displayed significant spatial overlap, which became markedly reduced by E12.5 (Fig. 1A) (4, 5). These patterns suggest a cross-suppression mechanism (2) regulating cortex-hem cell fate and contributing to cortex-hem border (CHB) formation and refinement. Consistent with this, Lhx2 sKO embryos displayed expanded hem (Figs. 1B; S1C) and Lhx2-hem overlap domains (Fig. S1D) by E10.5.
Figure 1.
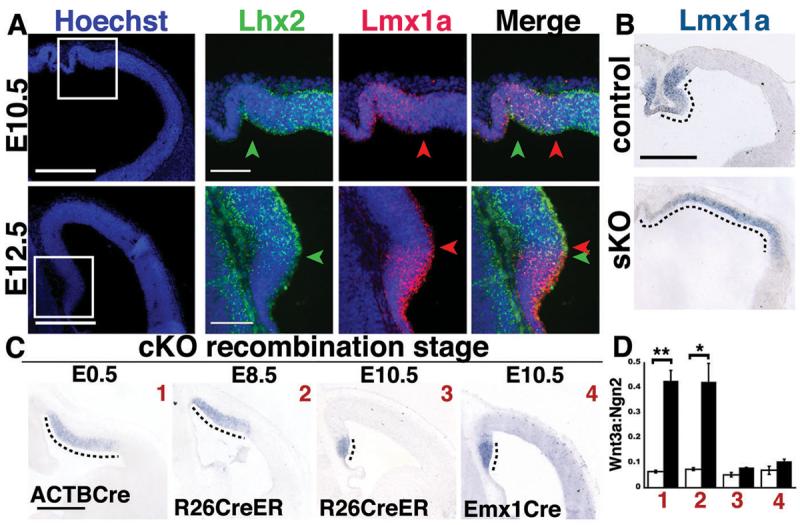
Hem fate suppression by Lhx2 during an E8.5-E10.5 critical period. (A) Double fluorescent ISH, boxed regions enlarged (right). At E10.5, cortical (Lhx2, green) and hem (Lmx1, red) markers overlap. By E12.5, this overlap is reduced (arrowheads). Scale bars: 100 μm (Hoechst panels, 300 μm). (B) Lmx1a ISH. By E10.5, the excessive hem phenotype (dotted lines) is present in Lhx2 sKO mutants. Scale bar: 300 μm. (C) Temporal inactivation studies; Wnt3a ISH. E12.5 cKO hem expansion (dotted lines) is similar following E0.5 (ACTBCre) or E8.5 inactivations (R26CreER), but inapparent after E10.5 inactivations (R26CreER or Emx1Cre). Scale bar: 300 μm. (D) Quantification of hem:dorsal telencephalon area ratios (Wnt3a:Ngn2 areas). White bars, littermate controls; black bars, Lhx2 cKO mutants; error bars, SEM; t-test, * p<0.05; **p<0.005. Unless otherwise noted, all images henceforth are coronal sections, medial left.
To better define the critical period for hem fate suppression by Lhx2, we performed timed inactivations with an Lhx2 conditional knockout (cKO) mouse (Fig. S2) (9). Lhx2 inactivation at E0.5 (ACTBCre driver) (10) or E8.5 (tamoxifen-inducible R26CreER driver) (11) resulted in E12.5 hem expansion or diminished dorsal telencephalic phenotypes that were qualitatively and quantitatively indistinguishable (Figs. 1C,D; S3A) (12). This indicated a critical period starting on or after E8.5, and no essential Lhx2 functions in the forebrain prior to this stage.
Tamoxifen (TM)-mediated recombination typically occurs within 48 hours of administration (11, 13). To test E10.5 as the critical period endpoint, we used E10.5 TM injections or Emx1Cre, which drives dorsal telencephalon-specific Cre recombination by E10.5 (14). Both strategies resulted in subtle to inapparent E12.5 phenotypes (Figs. 1C,D; S3A). This defines E8.5-E10.5 as the critical period for hem suppression by Lhx2, a period when the cortical neuroepithelium is composed almost exclusively of stem cells (15) and is not yet producing definitive cortical plate neurons (16).
Lhx2 selector activity at the molecular level
To examine cell-autonomy unambiguously, we generated Lhx2 null mosaics using two complementary methods: (1) mouse embryonic stem cell (ESC) aggregation chimeras (17, 18) using GFP-expressing Lhx2 null ESCs derived from Lhx2+/− matings (Fig. S4), and (2) low-dose TM injections at E5.5 in Lhx2 cKO mice (12). To identify null cells in Lhx2 cKO mosaics, we developed and validated an affinity-purified Lhx2-specific polyclonal antiserum (12).
While control ESC chimeras had random distributions of GFP-expressing cells and normal dorsal telencephalic patterning (Fig. 2A), Lhx2 null cells in mutant ESC chimeras and cKO mosaics clustered into patches. Null patches within medial cortex displayed downregulation or absence of cortically expressed Emx1, Foxg1 or Pax6 (Figs. 2B,C; S5C), and ectopic induction of hem markers Lmx1a, Wnt2b, or Wnt3a (Fig. 2D,E). These results establish a cell-autonomous role for Lhx2 in the molecular specification of cortical identity and suppression of hem fate. Interestingly, while Lhx2 null embryos have excessive CPe in addition to hem (4), no molecular (Ttr) or morphologic (epithelial simplification) evidence of ectopic CPe was detected in E12.5 or E14.5 mosaic embryos (Fig. 2; data not shown). Thus, Lhx2 activity in cortical stem cells specifically regulates a cortex-hem rather than cortex-CPe fate decision.
Figure 2.
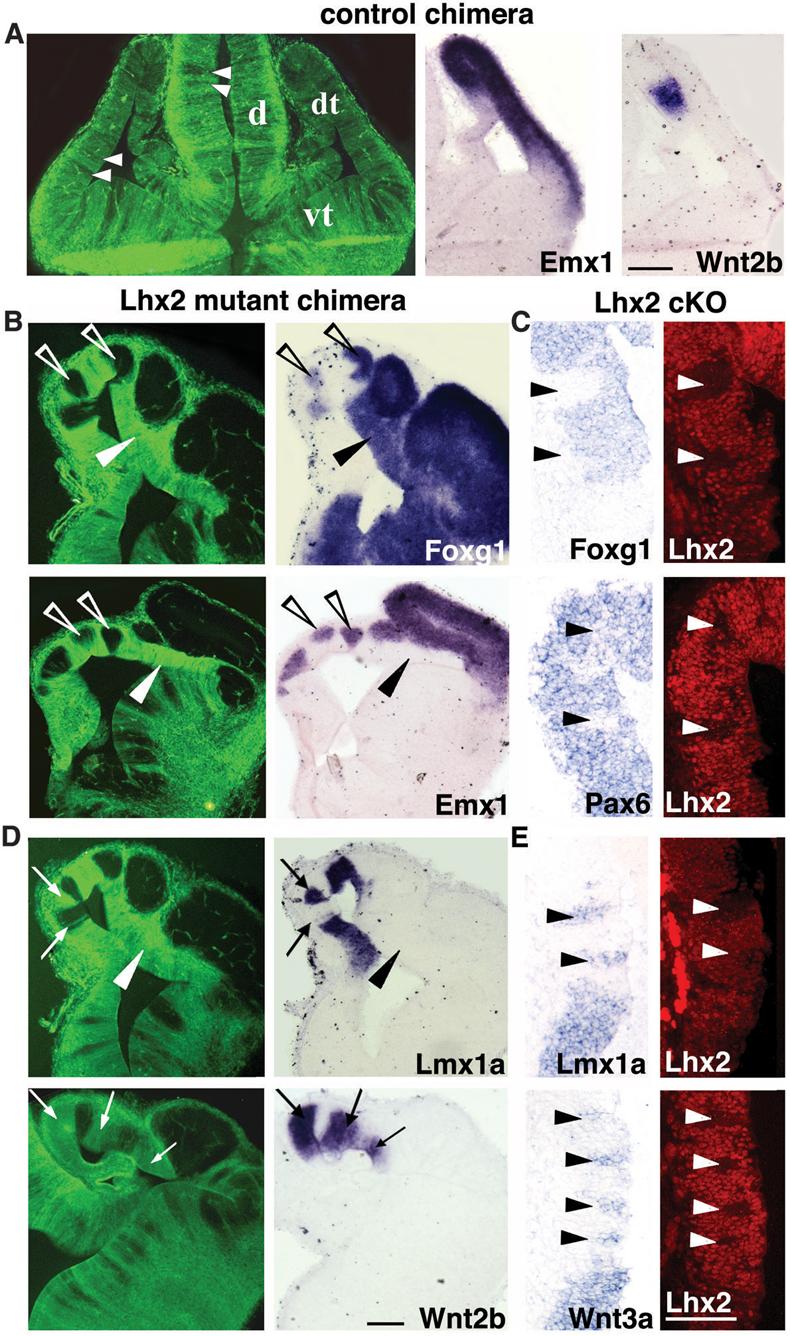
Cell-autonomous cortex-to-hem fate transformation in Lhx2 null mosaics. (A) In control ESC chimeras, GFP-expressing cells intermix randomly with host cells, and dorsal telencephalic patterning is normal. (B,C) Lhx2 null cells in mutant ESC chimeras (GFP-positive cells in B) or in tamoxifen-generated cKO mosaics (Lhx2 negative patches in C) do not express Foxg1, Emx1, or Pax6 in medial cortical regions (arrowheads in C). Surrounding wild-type cells express these markers (open arrowheads in B). Laterally, Lhx2 null cells express Foxg1, but not Emx1 (black arrowheads in B). (D,E) Lhx2 null cells express cortical hem markers Lmx1a, Wnt2b, and Wnt3a medially (arrows in D, arrowheads in E), but not laterally in the dorsal telencephalon (arrowheads in D). Scale bars: 100 μm. d, diencephalon; dt, dorsal telencephalon; vt, ventral telencephalon.
The cortex-to-hem transformation in Lhx2 null cells was spatially dependent (Figs. 2B,D; S5). Similar to Lhx2 sKO mutants (4, 5), the “hem competent zone” in mutant ESC chimeras and cKO mosaics comprised the medial telencephalic wall and extended some distance laterally, but did not include the entire pallium. Lateral Lhx2 null patches did not express hem markers or the definitive cortical marker Emx1, but had reduced Pax6 and unaffected Foxg1 levels (Figs. 2; S5). This marker profile characterizes the subpallium; however, like Lhx2 sKO mutants (4, 5), subpallial genes Dlx1, Dlx2 and Mash1 were not ectopically induced in lateral Lhx2 null patches (Fig. S4; data not shown).
Emx1 absence, with Pax6 and Foxg1 presence, also characterizes the antihem, a putative secondary signaling center at the lateral extreme of the pallium (19, 20). Strikingly, the antihem-specific marker Dbx1 was induced in lateral Lhx2 null patches in mutant ESC chimeras (Fig. 3D) and in lateral regions of the Lhx2 sKO dorsal telencephalon (Figs. 3A,B; S6). Adjacent sections demonstrated an Lhx2 sKO pallium composed almost exclusively of antihem and hem fates (Fig. 3B). Lhx2 activity in cortical precursors therefore suppresses two alternative fates (hem and antihem) at the edges of cortex and results in an Lhx2 null pallium devoid of hippocampal and neocortical identity at the molecular level (5), a distinct phenotype among cortical transcription factor mutants (Fig. 3C).
Figure 3.
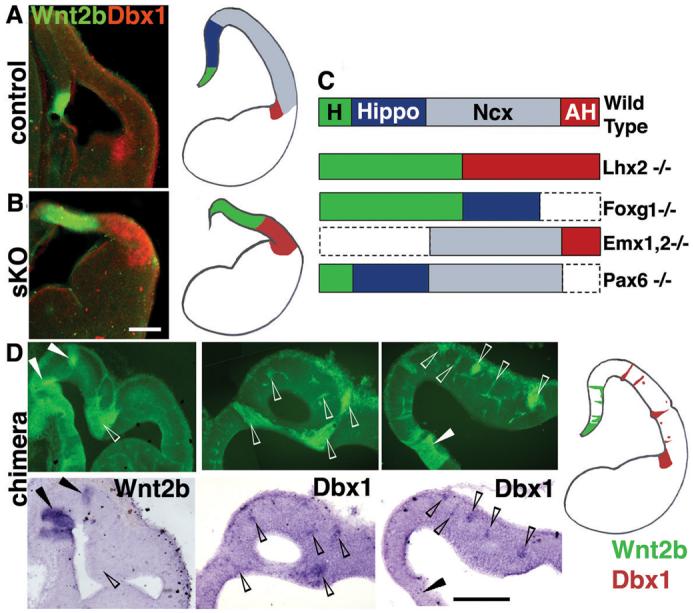
Hem and antihem fate expansion in Lhx2 sKO and ESC chimeras. (A) In E12.5 controls, a false-color overlay of Wnt2b (green) and Dbx1 (red) expression shows hem and antihem domains separated widely by the relatively large cortical (neocortical and hippocampal) neuroepithelium. (B) In E12.5 Lhx2 sKO brains, hem (green) and antihem (red) are both expanded and meet with no intervening cortical neuroepithelium. (C) Schematics of mutant dorsal telencephalic phenotypes illustrating how the complete absence of hippocampus and neocortex is unique to the Lhx2 mutant (4, 5, 28-30). AH, antihem; H, hem; Hippo, hippocampus; Ncx, neocortex. (D) Lhx2 null cells in ESC chimeras express Wnt2b medially (arrowheads) and Dbx1 laterally (open arrowheads), confirming ectopic cell-autonomous adoption of hem and antihem identity, respectively. GFP Images in D are assembled from multiple frames. Scale bars: 200 μm.
Lhx2 selector activity at the cellular level
In mosaic embryos, a significant degree of Lhx2 null cell clustering was apparent in dorsal telencephalon, lesser degrees in ventral telencephalon, and no obvious clustering elsewhere in the forebrain (Figs. 2,4). Conversely, wild-type (Lhx2 positive) dorsal telencephalic cells also aggregated in mutant ESC chimeras and often formed true neural rosettes (Figs. 2B,D; S7), a morphology not displayed by any Lhx2 null cell aggregate. To examine clustering further, we compared E12.5 distributions of cells recombined for Lhx2 or an unlinked locus (rosa26) using the low-dose TM strategy. Both qualitatively and quantitatively, Lhx2 null cells displayed significant clustering throughout the dorsal telencephalon, while rosa26 recombined cells did not (Figs. 4A,B; S3B,C). Taken together, these findings indicate that both Lhx2 null and positive cells each have significant homophilic affinity preferences.
Figure 4.

Lhx2 on-off state confers differential cell affinity. E12.5 mosaic (Lhx2cKO/sKO;R26CreER/fl) and control (Lhx2cKO/+;R26CreER/fl) littermates after 5 μg/gm TM injection on E5.5. (A) Xgal (blue) and Lhx2 IHC (red), with corresponding dot panels. Xgal-positive cells in mosaic animals are scattered, whereas Lhx2 negative cells in mosaics form contiguous patches. Dotted lines designate pial surfaces. Scale bar: 50 μm. (B) Quantification of recombined cells (black circles) completely isolated from non-recombined cells. The percentage of completely isolated Xgal+ cells in Lhx2 cKO mosaic embryos (9.8%) was similar to that calculated in R26R embryos lacking the Lhx2 cKO allele (8.5%). (C) Lhx2 IHC. Compared to controls, the medial Lhx2 negative domain (between arrowhead and asterisk) in mosaic embryos is enlarged. Scale bar: 100 μm. (D) Three hour in vitro aggregation (21) of E14.5 Emx1Cre;Lhx2 cKO mutant and control littermate cells. Cells in mutant-mutant mixes distribute randomly, whereas cells in mutant-control mixes segregate into discrete clusters. Scale bar: 25 μm.
In mutant ESC chimeras, including those containing a very low percentage of dorsal telencephalic ESC incorporation, the endogenous cortical hem invariably contained Lhx2 null cells (Fig. S4), consistent with Lhx2 negativity conferring hem identity. Lhx2 null cells in these chimeras also preferentially colonized the dorsomedial cortex. To examine this further, we inspected Lhx2 and rosa26 recombined cells in low-dose TM embryos. Whereas the rosa26 recombination or accumulation did not display significant dorsomedial bias (Fig. S3C; 20 sections from 3 embryos), the dorsomedial telencephalon in the cKO almost invariably displayed an enlarged Lhx2-negative domain (Fig. 4C; 50/54 sections from 4 embryos). Lhx2 null cells therefore preferentially colonize, sort and/or migrate to the endogenous hem and dorsomedial cortex.
To determine whether cell surface properties account for differential clustering in vivo, we performed short-term in vitro aggregation studies (21). Mixes of Lhx2 mutant and control littermate cells displayed significant segregation compared to mutant-mutant mixes within three hours (Fig. 4D; 82% vs. 37% segregation ratios, χ2 p<0.01). The Lhx2 mutant-mutant segregation ratio (37%) was similar to control ratios described previously for embryonic cortical cells (∼36%) (21, 22), while the Lhx2 control-mutant ratio (82%) appeared greater than control-Paxsey/sey mixes (∼50%) (22). Thus, in addition to molecular markers, Lhx2 specifies cortex vs. hem fates at the level of cell surface properties that drive differential cell clustering in vivo.
Lhx2 selector activity at the functional level
Since definitive evidence for hem activity as an organizer has been lacking (23), we examined whether ectopic hem cells exerted cell-nonautonomous effects on neighboring wild-type (Lhx2 positive) cortical tissue. In E15.5 mutant ESC chimeras, Wnt2b expression in Lhx2 null patches became restricted to the ventricular surface (Fig. 5B,J), as occurs in the normal hem (Fig. 5I). Juxtaposed to the ectopic Wnt2b domains were Prox1-expressing regions, (dentate granule cell marker; Fig. 5C,E) (24), which in turn were adjacent to KA1-expressing clusters (hippocampal CA3 field marker; Fig. 5C,G) (25), thus recapitulating the normal hem-dentate-CA3 spatial relationships. Multiple Prox1/KA1 patches were located significant distances away from their endogenous counterparts (Fig. 5D,F) and at several rostrocaudal levels (data not shown). Within each patch, Prox1 staining was typically strongest adjacent to the ectopic hem tissue (arrowheads in Fig. 5E), suggesting recapitulation of normal dentate cell migration patterns (Fig. 5A). Notably, all Prox1 and KA1 patches were limited to wild-type tissue, consistent with a cell-nonautonomous effect by ectopic hem cells and an inability of Lhx2 null cells to take on hippocampal identities.
Figure 5.
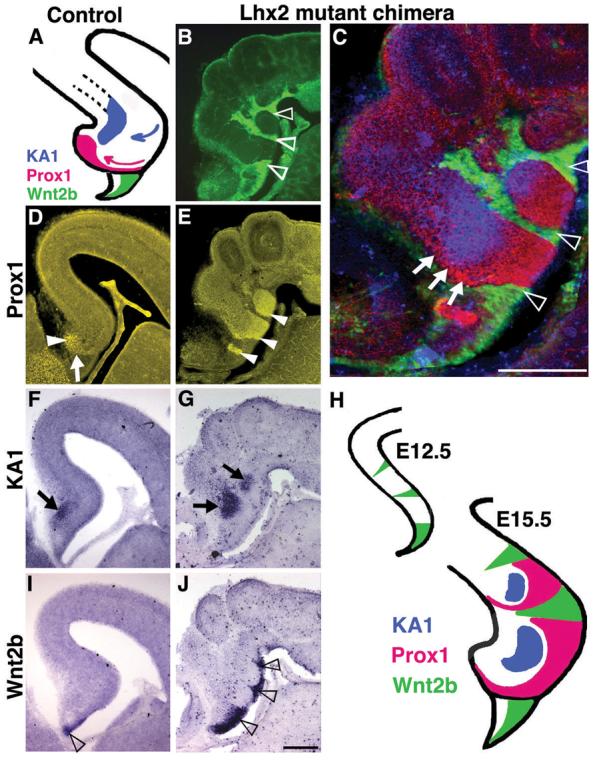
Multiple hippocampal fields induced by ectopic hem cells in Lhx2 null ESC chimeras. (A,D,F,I) At E15.5, control Prox1 positive dentate granule cells (arrowhead in D) have accumulated after migrating along a curved trajectory (pink arrow in A, arrow in D). KA1 expression (arrow in F) identifies the adjacent hippocampal field CA3. Wnt2b-expressing cortical hem is reduced to the fimbrial ventricular surface (open arrowhead in I). (B,E,G,J) In mutant chimeras, GFP expression and Prox1 staining (same section) show dentate cells (arrowheads in E) juxtaposed to Lhx2 mutant patches (open arrowheads in B,C) and an adjacent region of KA1 expression (arrows in G). Lhx2 mutant patches express Wnt2b in the ventricular zone (open arrowheads in J). (C,H) Overlay of (B,E,G) shows Prox1 expressing cells arranged around the KA1 patch, suggestive of a migrating stream (arrows, C). Images in B and C assembled from multiple frames. Scale bars: 300 μm.
By E17.5, hem size and Wnt expression are normally diminished; correspondingly, ectopic hem patches were often undetectable. Nonetheless, one chimeric hemisphere displayed two distinct Prox1-expressing dentate gyri oriented in the same direction (Fig. 6B). Strikingly, each gyrus was associated with its own presumptive Ammon's horn (Figs. 6C), as well as an independent radial glial palisade (Fig. 6E), a scaffolding that guides the migration of dentate cells (26). The two radial glial palisades originated from widely separated positions in the ventricular zone, with intervening regions corresponding to CA field origins (Lhx9, Fig. 6F, G), consistent with hippocampal duplication. Wnt signaling is also known to organize the radial glial palisade (27), providing a basis for both the specification and organization of ectopic hippocampi that form adjacent to ectopic hem patches.
Figure 6.
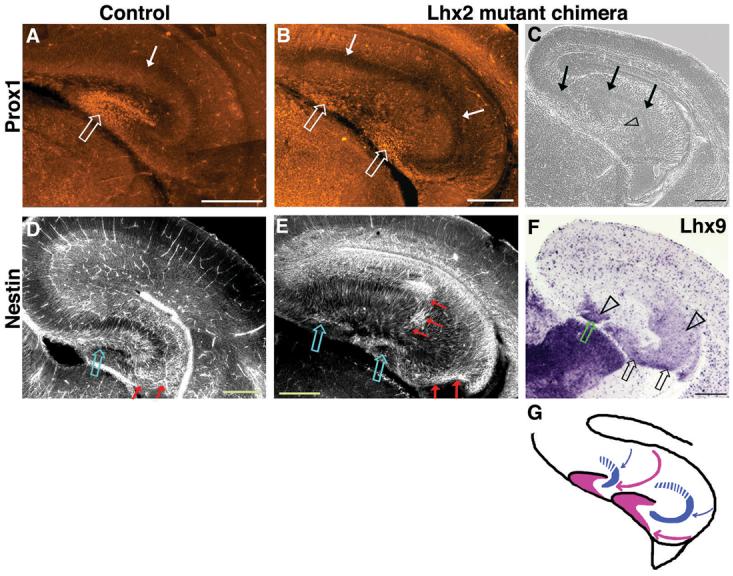
Structural organization of ectopic hippocampi in Lhx2 null ESC chimeras. (A-B) At E17.5, Prox1 immunostaining labels the control dentate gyrus and two morphologically distinct dentate gyri in the mutant chimeric brain (open arrows). (C) A phase contrast image of an adjacent section reveals that the normally continuous cell-dense layer of the CA fields has split in the chimeric brain (arrows). (F) This split layer corresponds to two distinct Ammon's horns, labeled by CA3+DG marker Lhx9. (D,E) Nestin immunostaining labels the control radial glial palisade and marks two such palisades in the mutant chimera, which take a curved trajectory (red arrows) from the ventricular zone, terminating adjacent to the two dentate gyri (open arrows). (G) Schematic of migration paths in chimeric brain (pink arrows, dentate; blue arrows, CA1/CA3). Images in A,B,D,E were assembled from multiple frames.
Ectopic induction of multiple hippocampal fields was also apparent in another E17.5 hemisphere that contained detectable GFP-positive cells distant from the endogenous hem (Fig. S8). These patches were associated with ectopic expression of dentate/CA3 marker Lhx9 adjacent to CA1/neocortex marker SCIP, which appeared between Lhx9-positive domains (Fig. S8), indicating the presence of distinct hippocampal fields at a significant distance from the normal hippocampus.
Discussion
The molecular, cellular (affinity), and functional evidence (organizer and responder activities) for classic selector functions (1) define Lhx2 as a cortical selector gene. Lhx2 selector activity is specifically required by cortical stem cells, without which these cells eventually adopt hem or antihem fates rather than hippocampal or neocortical identities. Lhx2 negative hem cells, in turn, act non-autonomously on Lhx2 positive cortical cells to induce and pattern hippocampal tissue. The absence of neocortex and hippocampus in Lhx2 null embryos contrasts with the preservation of one or both of these cortical structures in Pax6, Foxg1, and Emx1/2 null mutants (Fig. 3C) (28-30). These transcription factors are therefore likely to act after Lhx2, with Foxg1 being a mediator (28) of Lhx2-dependent hem fate suppression (Fig. S9). Lhx2 is itself downstream of Six3 in zebrafish (31), which is required to form the entire rostral prosencephalon (32), suggesting that Six3 creates a rostral forebrain field within which Lhx2 specifies cortical identity. The similarities between Lhx2 in the cortex and Ap in the Drosophila dorsal wing compartment – which specifies cell identity (dorsal), suppresses alternative fate (ventral), and confers differential affinity properties that position a signaling center (DV wing boundary) (33, 34) – are striking. Thus, conserved Ap/Lhx2 selector functions, as demonstrated in human-fly rescue experiments (35), apparently extend to the mammalian cerebral cortex.
Cross-suppression and differential affinity are typical mechanisms used by selector genes to make tissue borders (2), and our studies implicate Lhx2 selector activity in CHB formation and refinement. CHB refinement may also involve cell sorting and/or migration (Fig. 4C). Differential affinity is typically central to cell lineage restrictions at tissue borders (2), and previous studies provide evidence for lineage restrictions at the CHB. Wnt3a-Cre (7) and Gdf7-Cre lineage analyses (4, 36), as well as Frizzled10-lacZ studies based on evidence for lacZ perdurance (37), indicate strong confinement of hem precursors from E10.5-12.5, while D6-Cre lineage tracing provides similar evidence from the cortical side of the CHB (38). Together, these data imply that the CHB is an Lhx2-dependent lineage boundary. As both a developmental compartment and secondary signaling center, the hem may share similarities to the zona limitans intrathalamica (ZLI), a secondary Sonic hedgehog (Shh) production site that also displays lineage restriction (39).
While Lhx2 also regulates the cortex-antihem border (CAB), Lhx2 roles at the CAB and CHB probably differ, since Lhx2 is expressed in a continuous fashion across the lateral pallium-ventral pallium (antihem) border. Importantly, however, Lhx2 does not regulate the pallial-subpallial boundary (PSB), which instead relies on Pax6 selector gene activities (2, 5, 40). Moreover, Pax6 is required to generate antihem (19). Thus, reduced but maintained Pax6 expression in lateral Lhx2 null cells suffices to maintain antihem and suppress subpallial fates (40), allowing distinct CAB (Lhx2-dependent) and PSB (Pax6-dependent) formation mechanisms to be distinguished (Fig. S9).
That ectopic hem cells can induce and organize ectopic hippocampal tissue in a cell-nonautonomous fashion provides definitive functional evidence that the hem is a hippocampal organizer. These hem functions may occur prior to E12.5, since previous explant studies did not uncover hem organizer activity at this stage (41). Hem organizer activity is likely mediated by Wnt signals (6, 27, 42, 43), while Bmps may specify hem fate rather than hippocampal identity (4, 44). Fgf8, a rostromedially localized signaling molecule, may serve as an intermediary, since it is inhibited by Bmps and in turn suppresses Wnt expression (45). Our findings extend the known early interactions of the Bmp-Fgf8-Wnt signaling mechanisms at the telencephalic midline. Within this signaling framework, Lhx2 plays a dual role in suppressing hem fate while specifying the fate of hem-responsive tissue to allow for hippocampal specification within the Lhx2 positive cortical field.
Supplementary Material
Materials and Methods
Figs. S1, S2, S3, S4, S5, S6, S7, S8, S9
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank T. Boehm, V. Chizhikoz, E. Grove, E. Lai, Q. Ma, R. Maurer, K. Millen, and J. Rubenstein for gifts of plasmid DNA; F. D. Porter for Lhx2 mutant breeding pairs; the UCI Transgenic Mouse Facility for ES cell targeting and generation of the Lhx2 cKO mouse; the TIFR Animal House and UCI ULAR staff for excellent support; to C. Hsu, R. Vu, B. Cyrus, and G. Asuelime for technical support in cKO analyses; to Satish Kumar (CCMB), Shyamala Mani (NBRC), and Mitradas Panicker (NCBS) for training in ES cell and chimera techniques. This work was supported by a Swarnajayanti Fellowship (Dept. of Science and Technology, Govt. of India) and a Wellcome Trust Senior Fellowship (056684/Z/99/Z) to ST, NIH (MH02029) and the Whitehall and March of Dimes Birth Defects Foundations (ESM), NIH (AG23583) (LAF), a traveling fellowship from the journal Development and from the British Council to BM, postdoctoral fellowships from the Department of Biotechnology (Govt. of India) to VSM and NG, a Kanwal Rekhi Career Development award to LS, NIH (NS07444) (KEH).
Footnotes
References and Notes
- 1.Garcia-Bellido A. Ciba Found Symp. 1975;0:161. doi: 10.1002/9780470720110.ch8. [DOI] [PubMed] [Google Scholar]
- 2.Irvine KD, Rauskolb C. Annu Rev Cell Dev Biol. 2001;17:189. doi: 10.1146/annurev.cellbio.17.1.189. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence PA, Struhl G. Cell. 1996 Jun 28;85:951. doi: 10.1016/s0092-8674(00)81297-0. [DOI] [PubMed] [Google Scholar]
- 4.Monuki ES, Porter FD, Walsh CA. Neuron. 2001 Nov 20;32:591. doi: 10.1016/s0896-6273(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 5.Bulchand S, Grove EA, Porter FD, Tole S. Mech Dev. 2001 Feb;100:165. doi: 10.1016/s0925-4773(00)00515-3. [DOI] [PubMed] [Google Scholar]
- 6.Lee SM, Tole S, Grove E, McMahon AP. Development. 2000 Feb;127:457. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Development. 2006 Feb;133:537. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- 8.Porter FD, et al. Development. 1997 Aug;124:2935. doi: 10.1242/dev.124.15.2935. [DOI] [PubMed] [Google Scholar]
- 9.Materials and methods are available as supporting material on Science Online.
- 10.Lewandoski M, Meyers EN, Martin GR. Cold Spring Harb Symp Quant Biol. 1997;62:159. [PubMed] [Google Scholar]
- 11.Badea TC, Wang Y, Nathans J. J Neurosci. 2003 Mar 15;23:2314. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Additional supporting material available at http://www.ucihs.uci.edu/som/pathology/faculty/emonuki/index.html.
- 13.Hayashi S, McMahon AP. Dev Biol. 2002 Apr 15;244:305. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 14.Jin XL, et al. Biochem Biophys Res Commun. 2000 Apr 21;270:978. doi: 10.1006/bbrc.2000.2532. [DOI] [PubMed] [Google Scholar]
- 15.Gotz M, Huttner WB. Nat Rev Mol Cell Biol. 2005 Oct;6:777. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi T, Nowakowski RS, Caviness VS., Jr. J Neurosci. 1995 Sep;15:6046. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryja V, et al. Stem Cells. 2005 Aug;23:965. doi: 10.1634/stemcells.2004-0174. [DOI] [PubMed] [Google Scholar]
- 18.Nagy A, Gertsenstein M, Vintersten K. In: Manipulating the Mouse Embryo: A Laboratory Manual. Nagy A, Vintersten K, Behringer R, editors. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2003. pp. 453–506. e. al. [Google Scholar]
- 19.Assimacopoulos S, Grove EA, Ragsdale CW. J Neurosci. 2003 Jul 23;23:6399. doi: 10.1523/JNEUROSCI.23-16-06399.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puelles L, et al. J Comp Neurol. 2000 Aug 28;424:409. doi: 10.1002/1096-9861(20000828)424:3<409::aid-cne3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Gotz M, Wizenmann A, Reinhardt S, Lumsden A, Price J. Neuron. 1996 Mar;16:551. doi: 10.1016/s0896-6273(00)80074-4. [DOI] [PubMed] [Google Scholar]
- 22.Stoykova A, Gotz M, Gruss P, Price J. Development. 1997 Oct;124:3765. doi: 10.1242/dev.124.19.3765. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Pleasure SJ. Dev Neurosci. 2005 Mar-Aug;27:93. doi: 10.1159/000085980. [DOI] [PubMed] [Google Scholar]
- 24.Bagri A, et al. Development. 2002 Sep;129:4249. doi: 10.1242/dev.129.18.4249. [DOI] [PubMed] [Google Scholar]
- 25.Tole S, Christian C, Grove EA. Development. 1997 Dec;124:4959. doi: 10.1242/dev.124.24.4959. [DOI] [PubMed] [Google Scholar]
- 26.Rickmann M, Amaral DG, Cowan WM. J Comp Neurol. 1987 Oct 22;264:449. doi: 10.1002/cne.902640403. [DOI] [PubMed] [Google Scholar]
- 27.Zhou CJ, Zhao C, Pleasure SJ. J Neurosci. 2004 Jan 7;24:121. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzio L, Mallamaci A. J Neurosci. 2005 Apr 27;25:4435. doi: 10.1523/JNEUROSCI.4804-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinozaki K, Yoshida M, Nakamura M, Aizawa S, Suda Y. Mech Dev. 2004 May;121:475. doi: 10.1016/j.mod.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Stoykova A, Treichel D, Hallonet M, Gruss P. J Neurosci. 2000 Nov 1;20:8042. doi: 10.1523/JNEUROSCI.20-21-08042.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ando H, et al. Dev Biol. 2005 Nov 15;287:456. doi: 10.1016/j.ydbio.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Lagutin OV, et al. Genes Dev. 2003 Feb 1;17:368. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blair SS, Brower DL, Thomas JB, Zavortink M. Development. 1994 Jul;120:1805. doi: 10.1242/dev.120.7.1805. [DOI] [PubMed] [Google Scholar]
- 34.Diaz-Benjumea FJ, Cohen SM. Cell. 1993 Nov 19;75:741. doi: 10.1016/0092-8674(93)90494-b. [DOI] [PubMed] [Google Scholar]
- 35.Rincon-Limas DE, et al. Proc Natl Acad Sci U S A. 1999 Mar 2;96:2165. doi: 10.1073/pnas.96.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Currle DS, Cheng X, Hsu CM, Monuki ES. Development. 2005 Aug;132:3549. doi: 10.1242/dev.01915. [DOI] [PubMed] [Google Scholar]
- 37.Zhao C, Guan W, Pleasure SJ. Brain Res. 2006 Mar 10;1077:48. doi: 10.1016/j.brainres.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 38.Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Neuroscience. 2003;122:129. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- 39.Kiecker C, Lumsden A. Nat Rev Neurosci. 2005 Jul;6:553. doi: 10.1038/nrn1702. [DOI] [PubMed] [Google Scholar]
- 40.Kroll TT, O'Leary DD. Proc Natl Acad Sci U S A. 2005 May 17;102:7374. doi: 10.1073/pnas.0500819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tole S, Grove EA. J Neurosci. 2001 Mar 1;21:1580. doi: 10.1523/JNEUROSCI.21-05-01580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Development. 2000 Feb;127:469. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- 43.Machon O, et al. Dev Biol. 2007 Nov 1;311:223. doi: 10.1016/j.ydbio.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 44.Hebert JM, Mishina Y, McConnell SK. Neuron. 2002 Sep 12;35:1029. doi: 10.1016/s0896-6273(02)00900-5. [DOI] [PubMed] [Google Scholar]
- 45.Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. Development. 2004 Nov;131:5639. doi: 10.1242/dev.01428. [DOI] [PubMed] [Google Scholar]
- 46.Soriano P. Nat Genet. 1999 Jan;21:70. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 47.Cheng X, et al. J Neurosci. 2006 Jul 19;26:7640. doi: 10.1523/JNEUROSCI.0714-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flanagan LA, Rebaza LM, Derzic S, Schwartz PH, Monuki ES. J Neurosci Res. 2006 Apr;83:845. doi: 10.1002/jnr.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian X, Goderie SK, Shen Q, Stern JH, Temple S. Development. 1998 Aug;125:3143. doi: 10.1242/dev.125.16.3143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.