

Abstract
PURPOSE
To focus on the proteomic analysis of 14-3-3 proteins and to determine their cellular localization and functional role during glaucomatous neurodegeneration.
METHODS
Complementary proteomic approaches were used to identify phosphorylated proteins in a chronic pressure-induced rat model of glaucoma. To detect interacting proteins, specific protein complexes were eluted using coimmunoprecipitation and recombinant protein-based affinity pull-down for subsequent mass spectrometric analysis. Western blot analysis was performed for validation of the proteomic findings, and immunohistochemical analysis of rat eyes and human donor eyes determined the cellular localization of 14-3-3 proteins. In addition, in vivo treatment experiments were conducted using JNK and protein phosphatase inhibitors.
RESULTS
Findings of mass spectrometry, Western blotting, and tissue immunolabeling revealed the presence of different 14-3-3 isotopes in RGCs and their up-regulation and phosphorylation during glaucomatous neurodegeneration. Consecutive experiments through proteomic analysis identified various proteins interacting with 14-3-3, which included calmodulin and a proapoptotic member of the Bcl-2 family, Bad; 14-3-3 was found to keep phospho-Bad sequestered in the cytoplasm. However, this association was disrupted in ocular hypertensive eyes in correlation with Bad dephosphorylation and 14-3-3 phosphorylation, thereby leading to mitochondrial translocation of Bad for apoptotic function. Inhibition of JNK activity and of protein phosphatase activity complementarily secured the 14-3-3-scaffold of Bad in the cytoplasm and preserved optic nerve axons in ocular hypertensive eyes.
CONCLUSIONS
Findings of this in vivo study identify that an important protein family associated with checkpoint control pathways, 14-3-3, is involved in cellular signaling during glaucomatous neurodegeneration in a phosphorylation-dependent manner.
Progressive loss of optic nerve axons and apoptosis of retinal ganglion cells (RGCs) result in characteristic optic nerve atrophy and visual field defects in glaucoma. Although the initial site of glaucomatous injury is unclear, RGC survival and axon health are dependent on each other. Therefore, a treatment strategy targeting RGC rescue is a prerequisite to prevent further axon abnormalities and to achieve functional gain in glaucoma patients. Growing evidence supports that besides caspase activation through the receptor-mediated extrinsic pathway,1 the intrinsic pathway of apoptosis through mitochondria constitutes an important component of RGC death signaling during glaucomatous neurodegeneration.2–4 The proposed molecular pathways of mitochondria-mediated RGC death involve proapoptotic members of the Bcl-2 family, including Bax and Bad. For example, Bax and p53, a transcriptional activator of Bax, have been associated with neurodegeneration induced by different stimuli.5,6 Bax deficiency in DBA/2J mice exhibiting inherited glaucoma has been found to protect from RGC death, although it does not prevent axonal degeneration.7,8 Using an experimentally induced mouse model of glaucoma, Bax expression has been found to be higher in ocular hypertensive eyes than in control eyes and to be correlated with RGC apoptosis.9 In a study using a rat model of experimental glaucoma, intrinsic survival programs triggered at the early stage of injury have been associated with an upregulation of phospho-Bad.10 More recently, the mitochondrial apoptosis pathway induced by experimental elevation of intraocular pressure (IOP) in rat and mouse eyes has been linked to Bad dephosphorylation by calcineurin.11
Previous evidence supports the importance of phosphorylation cascades in RGC signaling during glaucomatous neurodegeneration,12,13 and the present study identified that the RGC proteins phosphorylated in a rat model of glaucoma include the 14-3-3 family. Among the most abundant proteins in the brain with preferential localization to neurons, including RGCs,14 14-3-3 proteins constitute an important protein family associated with checkpoint control pathways.15 This highly conserved family of small (28–33 kDa), acidic, dimeric proteins consists of at least seven distinct subunit isoforms (α/β, γ, δ/ζ, ε, η, τ, and σ, where α and δ are the phosphorylated forms of β and ζ, respectively). They bind to multiple protein ligands, mostly after their serine/threonine phosphorylation at a defined motif. Phosphorylation-dependent binding with 14-3-3 can alter the subcellular localization, stability, phosphorylation state, activity, and molecular interactions of many target proteins, thereby implicating 14-3-3 proteins as key regulators in diverse intracellular signal transduction pathways.16,17 Based on studies using transgenic mice that express dominant-negative 14-3-3 alleles, a primary function of mammalian 14-3-3 proteins is the inhibition of apoptosis.18
To determine the association of 14-3-3 with cell death signaling in experimental glaucoma, we used targeted proteomic approaches and in vivo treatment experiments for functional testing. Findings of these experiments support that the 14-3-3 family of proteins is involved in the regulation of protein trafficking in a phosphorylation-dependent manner with important functional implications associated with RGC death during glaucomatous neurodegeneration. Proteins interacting with 14-3-3 included a proapoptotic member of the Bcl-2 family, Bad. Although phosphorylated Bad normally remains sequestered in the cytoplasm by 14-3-3 scaffold, findings from proteomic analysis and tissue immunolabeling collectively supported Bad translocation to mitochondria after 14-3-3 phosphorylation and Bad dephosphorylation in ocular hypertensive eyes. Furthermore, neuronal damage in ocular hypertensive eyes was found to be decreased by maintenance of the 14-3-3/Bad interaction using treatments inhibiting 14-3-3 phosphorylation and Bad dephosphorylation. Thus, 14-3-3 proteins constitute an important regulatory pathway of cell death signaling during glaucomatous neurodegeneration, which controls the subcellular localization and function of Bad.
MATERIALS AND METHODS
Experimental Design
IOP elevation was induced in rats by hypertonic saline injections into episcleral veins, as detailed. All the animals were handled according to the regulations of the Institutional Animal Care and Use Committee, and all procedures adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The degree of cumulative IOP exposure was estimated by calculating the area under the pressure-time curve in the ocular hypertensive eye and then subtracting this IOP-time integral from that in the normotensive fellow eye (expressed in units of mm Hg/d).3 Axon loss was determined by comparing the axon count in the ocular hypertensive eye compared with the control fellow eye.3 Enriched RGC protein samples were collected by pooling from rat eyes matched for IOP exposure and axon loss. Specifically, the protein samples obtained from moderately damaged eyes with a cumulative IOP exposure of 200 to 400 mm Hg/d were used in this study and corresponded to a relative axon loss value no more than 50%.3
Complementary approaches of the targeted proteomics were used to identify phosphorylated proteins and their interacting proteins in ocular hypertensive eyes compared with controls. Phosphorylated proteins were initially identified using a two-dimensional (2-D) poly-acrylamide gel electrophoresis (2-D PAGE)-based approach, through which phosphoproteins were detected by phosphoprotein staining (Pro-Q Diamond; Invitrogen/Molecular Probes, Carlsbad, CA) of 2-D gels followed by protein gel stain (Sypro Ruby; Invitrogen/Molecular Probes) for total protein profiling. Phosphoproteins revealed by this specific staining were then identified through peptide mass fingerprinting and peptide sequencing using liquid chromatography-tandem mass spectrometry (LC-MS/MS). For large-scale quantification, differential display analysis was performed to compare the intensity of protein spots matched on 2-D gel images obtained from ocular hypertensive and control eyes. As a second approach, phosphoproteins in mixed protein samples were enriched using spin columns containing immobilized metal-affinity chromatography media, and enriched samples were subjected to gel-free protein identification using LC-MS/MS analysis. To identify interacting proteins, specific protein complexes were eluted using coimmunoprecipitation and recombinant protein-based affinity pull-down for subsequent LC-MS/MS analysis. For further validation of the proteomic findings, Western blot analysis was performed using specific antibodies, and immunohistochemistry determined cellular localization of the identified proteins in the retina and optic nerve.
To determine the functional importance of the identified protein interactions and their phosphorylation dependence, in vivo treatment experiments were performed using DJNKI and FK506 to inhibit c-jun amino(N)-terminal kinase (JNK) activity and protein phosphatase activity, respectively. To determine JNK-mediated protein phosphorylation, one group of animals received daily intraperitoneal injections of DJNKI19 or control D-TAT peptide (10 mg/kg; Alexis, San Diego, CA) for 4 weeks, started at the time of the first measured elevation of IOP. Another group of animals was treated with FK506 (Alexis) at a dose of 5 mg/kg (dissolved in 10% ethanol containing 2% Tween 20 in PBS) by daily intraperitoneal injections during the same treatment period. Similar injections of the vehicle served as an additional control. Another group of animals received combined treatment with DJNKI and FK506. There were at least five rats in each treatment group matched for IOP exposure.
Phosphorylation status of studied proteins was assessed in treated animals relative to untreated controls. A treatment effect was also determined by counting optic nerve axons. RGC apoptosis was evaluated in treated and untreated animals; however, given that retinal protein samples were used for proteomic analysis, in situ labeling for apoptosis was not quantified because of the limited number of slides for appropriate sampling. It should be noted that in addition to the advantage of saving retinas for proteomic analysis, optic nerve axon counts were considered the most appropriate for demonstrating cumulative neuronal damage, thereby providing more accurate information than counting RGC apoptosis, which could only detect a few cells at any given time.
Experimental Rat Glaucoma Model
As in previous studies,3 IOP elevation was induced in male Brown Norway rats (average weight, 300 g) by hypertonic saline injections into episcleral veins, as originally described by Morrison’s group.20 Briefly, under general and topical anesthesia and after placement of a plastic ring around the equator, approximately 0.1 mL of 1.75 M saline was unilaterally injected into the venous system. The injection was repeated 1 week later. Rats were housed in constant low-level light, and all IOP measurements were performed in conscious rats between 10:00 AM to 12:00 PM to minimize diurnal variability in IOP. Baseline IOP was obtained before the first saline injection, and the measurements were repeated twice per week using a calibrated tonometer (Tono-Pen; Medtronic Solan, Jacksonville, FL).
Optic Nerve Axon Counts
Optic nerve axons were counted using a protocol similar to that previously described.3 Briefly, excised optic nerves were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, overnight and postfixed in 2% osmium tetroxide for 1 hour. Dehydrated tissues were embedded in epoxy resin, and 1-µm cross-sections of the myelinated optic nerves were stained with 1% toluidine blue. Captured images were analyzed with a software (Axiovision; Carl Zeiss, Thornwood, NY) by counting the axon density in randomized regions of the optic nerve using a systematic sampling protocol2 that included the center, midperiphery, and peripheral margin of the optic nerve in four quadrants (at least 40% of the total axon number). The axon-counting procedure was performed in a masked fashion without knowledge of the experimental status of rat eyes. Total axon estimates were calculated by multiplying the mean axon density by the total area of the optic nerve. The total optic nerve area was measured by outlining its outer border, and the mean of three area measurements was used. The percentage of axon loss was then expressed by comparing the estimated total number of axons in the ocular hypertensive eye with that of the control fellow eye.
Protein Lysation
Retinas were mechanically dissected from enucleated eyes for protein lysation. Enriched samples of RGC proteins were obtained through the two-step immunomagnetic selection process previously described.1,2 Optimized conditions were maintained throughout the RGC purification procedure, which was completed in less than 2 hours, with an excellent recovery matching the high survival rate in culture.1,2 RGCs isolated by this procedure were originally identified based on retrograde labeling with a fluorescent tracer, cell morphology, and immunolabeling for specific markers.1 In addition, the purity of the isolated RGCs was reconfirmed by Western blot analysis using specific antibodies recognizing different retinal cell markers (data not shown). Briefly, retinas were dissociated in Eagle minimum essential medium containing 20 U/mL papain, 1 mM L-cysteine, 0.5 mM EDTA, and 0.005% DNase at 37°C for 40 minutes; this was followed by incubation in an inhibitor solution containing Eagle minimum essential medium, 0.2% ovomucoid, 0.04% DNase, and 0.1% bovine serum albumin. After trituration through a 1-mL pipette to yield a suspension of single cells, immunomagnetic selection of RGCs was performed through the two-step process. In the first step, an antibody to macrophage surface antigen was used. In the second step, the macrophage-depleted cell suspension was incubated with magnetic beads bound to a monoclonal antibody specific to Thy-1.1 (Millipore/Chemicon, Billerica, MA). Protein lysation used a urea/thiourea lysis buffer containing 9 mg/mL dithiothreitol, 40 mg/mL CHAPS, 0.42 g/mL urea, 0.15 g/mL thiourea, 4.45% carrier ampholytes (pH 3–10), and protease inhibitors, as previously described.3 In addition, to preserve phosphorylation states, a mixture of a broad spectrum of phosphatase inhibitors (Calbiochem, San Diego, CA) was added to the buffer solution. To isolate the mitochondrial protein fraction, differential centrifugation (at 600g followed by 11,000g) was used.
Two-Dimensional PAGE-Based Mass Spectrometric Analysis of Phosphoproteins
Two-dimensional PAGE was performed as previously described.3 Briefly, using an electrophoresis system (Zoom IPG Runner; Invitrogen, Carlsbad, CA), 7 cm pH 3–10 or pH 4.5–5.5 strips (Zoom; Invitrogen), and NuAGE Novex 4% to 12% Bis-Tris gels (Zoom; Invitrogen), isoelectric focusing was performed at 175 V for 15 minutes, 175–2000 V ramp for 45 minutes, and 2000 V for 30 minutes, followed by the 2-D separation at 200 V/200 mA for 45 minutes. Images were obtained by a gel documentation system. Phosphorylated proteins were directly stained on 2-D gels using a phosphoprotein staining kit (Pro-Q Diamond; Invitrogen/Molecular Probes)21,22 followed by protein gel stain for total protein profiling.
Protein content was determined based on spot intensity in a series of 2-D gels obtained using equally loaded protein samples from ocular hypertensive eyes and normotensive fellow eyes in a masked fashion. The 2-D gel image analysis software (Phoretix; Nonlinear Dynamics, Newcastle on Tyne, UK) was used to estimate Mr and pI coordinates of the proteins and to perform densitometric analysis and comparisons between different gels. Total spot volume was calculated to estimate the proportion of analyzed spot within the 2-D gel. Average mode of background subtraction was used to normalize the intensity before calculating spot volumes and fold changes between ocular hypertensive and control samples. For the analysis of phosphoprotein staining, after matching the corresponding spots on protein gel stain and phosphoprotein-stained gel images, the intensity of phosphoprotein staining (the integrated value after background subtraction on phosphoprotein-stained gels) was normalized to their protein content obtained by measuring the intensity of protein gel stain (the integrated value after background subtraction on 2-D gels). Fold change was expressed by comparing the integrated values obtained using at least four different samples.
For protein identification, spots were excised from 2-D gels and analyzed by mass spectrometry. Peptide mass fingerprinting and peptide sequencing from tryptic digests of the gel pieces and bioinformatic analysis from the mass spectra were performed using the previously described criteria.3 Briefly, gel samples were reduced with dithiothreitol, alkylated with iodoacetamide, and digested with modified trypsin (Promega, Madison, WI). Digests were mixed with the same volume of 4-hydroxy-α-cyano-cinnamic acid (4HCCA) solution, and 1 µL of the mixture was loaded on to a well coated with a thin layer of 4HCCA. The samples were then washed with 5% formic acid and analyzed with the use of a mass spectrometer (TofSpec 2E MALDI-TOF; Waters, Milford, MA). Peak lists from the samples were searched against NCBI and SwissProt databases with MassLynx and online with Mascot.
In addition, samples were analyzed using LC-MS/MS, as previously described.3 Briefly, peptides in digested samples were extracted by 15-minute incubations in 5% formic acid and acetonitrile (ACN). Diluted samples were injected onto a 300 µm × 5 mm C18 precolumn (PepMap; LC Packing, Sunnyvale, CA); after washing, peptides were separated on a 75 µm × 150 mm C18 analytical column (Symmetry; Waters) at a flow rate of 200 nL/min for more than 40 minutes using a gradient from 100% solvent A (5% ACN/0.1% formic acid) to 40% solvent B (95% ACN/0.1% formic acid) and were maintained at 40% solvent B for 20 minutes. The LC elute was then directed into a mass spectrometer (Q-TOF; Waters) in data-dependent scan mode. A software program (ProteinLynx 4.0; Waters) was used to obtain peptide sequences from raw MS/MS data. Protein identification was based on more than two manually confirmed peptides.
Phosphoprotein Enrichment and Gel-Free Protein Analysis
To improve the sensitivity of phosphoprotein identification, phosphoproteins were enriched using a specific kit (BD Biosciences/Clontech, San Jose, CA). After charging and equilibrating the spin columns containing immobilized metal-affinity chromatography media, samples were diluted in acidic buffer and were bound by maintaining the low pH. The phosphoprotein mixture was denatured in 8 M urea, reduced with dithiothreitol, alkylated with iodoacetamide, diluted with 100 mM NH4HCO3 (to dilute urea to 2 M), and digested with modified trypsin. Digested samples was desalted using C18 spin columns (Pierce, Rockford, IL), dried by speed vacuum, and dissolved in a loading buffer containing 20% ACN, 100 mM acetic acid (HAc), and 2 mM ammonium acetate (NH4Ac). Samples were then loaded on a preconditioned strong cation-exchange (SCX) cartridge, washed twice with 20 µL loading buffer, and eluted using 20 µL HAc/NH4Ac buffers (15 eluting buffers of increasing ionic strength and pH were used, and the final buffer contained 10 mM HAc and 615 mM NH4Ac with pH 6.5). The SCX fractions were concentrated to approximately 1 µL using a speed vacuum and were diluted to 7 µL with 5% ACN/0.1% formic acid. Five microliters of the samples was loaded on a C18 precolumn (LC Packing), eluted to a 75 µm × 100 mm C18 capillary column (Symmetry; Waters), and separated with a solvent gradient up to 50% ACN. The LC elute was coupled to a nano-LC sprayer, and MS/MS spectra were acquired with a mass spectrometer (Q-TOF; Waters) in data-dependent scan mode. Only ions with 2+, 3+, or 4+ charges were selected for MS/MS analysis. MS/MS spectra were searched against SwissProt data-base with ProteinLynx 4.0 (Waters). The mass error allowed was set to 25 ppm, and a minimum of three consecutive residues was required for a positive peptide match. All positive peptide matches were examined manually, and protein identification was based on more than two peptides showing a manually confirmed sequence match.
Coimmunoprecipitation
A specific kit (Pierce) was used for coimmunoprecipitation of interacting proteins. After direct covalent immobilization of the primary antibody (a monoclonal antibody to 14-3-3; Abcam, Cambridge, MA), immunoprecipitation of the antigen (bait protein), and coimmunoprecipitation of interacting proteins (prey proteins) were performed using the spin columns. Unspecific interactions were identified by using the provided control gel and quenched antibody coupling gel and substituting IgG for the specific antibody. Eluted fraction was also tested through immunoblotting for validation of the interacting protein identification.
Recombinant Protein-Based Affinity Pull-Down
The affinity pull-down was performed with a specific kit (Pierce) and used GST-tagged recombinant 14-3-3 isoforms (Abcam). To capture the prey protein, immobilized bait protein was mixed with protein lysate in a binding buffer. After washing steps, interacting proteins were eluted. To determine unspecific binding, parallel reactions were conducted using equimolar amounts of Flag tag-null proteins instead of the tested fusion proteins. In addition, agarose gel control, no bait-added control, and the immobilized bait control without the protein lysate were processed in parallel. We also performed immunoblotting of the pull-downed fraction to validate interacting proteins.
Quantitative Western Blot Analysis
Immunoblotting was performed as previously described.1,23 Briefly, electrophoresis was carried out as described, and the gels were transferred to a nitrocellulose membrane using a semidry transfer system (Bio-Rad, Hercules, CA). For immunolabeling, membranes were blocked in solution (50 mM Tris-HCl, 154 mM NaCl, 0.1% Tween-20, pH 7.5) containing 5% nonfat dry milk for 1 hour and were incubated overnight with the primary antibody at room temperature. Primary antibodies used included monoclonal antibodies to 14-3-3 (1:500), Bad (1:500), or calmodulin (1:1000) (Abcam). To determine phosphoproteins, we used phosphorylation site-specific antibodies to 14-3-3 (1:1000; PhosphoSolutions, Aurora, CO) or Bad (1:500; Cell Signaling, Danvers, MA) as the primary antibody. In addition, monoclonal antibodies to tubulin or cytochrome c oxidase (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) were used as primary antibodies to confirm the cytoplasmic and mitochondrial fractions of protein samples. Secondary antibody incubation was performed using a goat anti-mouse IgG conjugated with horseradish peroxidase (1:2000; Sigma-Aldrich, St. Louis, MO) for 1 hour at room temperature. Immunoreactivity was visualized by enhanced chemiluminescence using commercial reagents (Amersham, Piscataway, NJ). The primary antibody was omitted to serve as a control. Optical densities of protein bands on digitized immunoblotted membrane images were obtained using image analysis software (Axiovision; Carl Zeiss) and were normalized to the average mode of background subtraction. At least four immunoblots were obtained with new samples. The average value obtained using ocular hypertensive samples was compared with the average value obtained from controls to calculate the fold change in protein expression.
Immunohistochemistry
To determine the extent and cellular localization of identified proteins, histologic sections of the retina and optic nerve were immunolabeled using immunoperoxidase labeling and double-immunofluorescence labeling techniques. All procedures were similar to that previously described.12,24,25 Briefly, after fixation, posterior poles of the enucleated eyes were embedded in paraffin and processed for 6-µm longitudinal sections, as previously described.12,24,25 For immunoperoxidase labeling, slides were pretreated with 3% hydrogen peroxide in methanol to decrease endogenous peroxidase activity. After washing, slides were incubated with 20% inactivated serum (Millipore/Chemicon) for 30 minutes at room temperature to block background staining. Primary antibody incubation with monoclonal antibodies to 14-3-3 (Abcam) or phospho-14-3-3 (PhosphoSolutions) or monoclonal antibodies to different 14-3-3 isoforms (alpha/beta, delta/zeta, eta, and gamma; 1:100; Santa Cruz Biotechnology) for 16 hours at 4°C was followed by washing and secondary antibody incubation with a biotinylated mouse IgG (1:400; Millipore/Chemicon) for 1 hour at room temperature. Slides were then incubated with ABC solution (Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. After several washes, color was developed by incubation with 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) as cosubstrate for 5 to 7 minutes. Hematoxylin was used for counterstaining. Primary antibodies used for double-immunofluorescence labeling included the same 14-3-3 antibodies used for immunoperoxidase labeling. Phosphorylation site-specific antibodies to 14-3-3 (1:100; PhosphoSolutions) or Bad (1:100; Cell Signaling) were also used to determine phosphoproteins. We used a rabbit antibody against prohibitin as a mitochondrial marker (1:100; Abcam). In addition, a rabbit antibody to brn-3a (1:400; Millipore/Chemicon) was used as a marker for RGCs. Secondary antibodies used for double-immunofluorescence labeling were Alexa Fluor 488-conjugated anti-mouse or Alexa Fluor 568-conjugated anti-rabbit IgG (2 µg/mL; Invitrogen/Molecular Probes). The primary antibody was eliminated from the incubation medium, or serum was used to replace the primary antibody to serve as the negative control. During double-immunofluorescence labeling, slides were also incubated with each primary antibody, followed by the inappropriate secondary antibody to determine that each secondary antibody was specific to the species against which it was made. After washing and mounting, slides were examined using a phase-contrast/fluorescence microscope, and images were recorded by digital photomicrography (Axiovision; Carl Zeiss). Each specific immunolabeling was performed using at least six histologic slides from each eye after masking the slides for the experimental status of eyes. To control variations, histologic slides obtained from ocular hypertensive and control eyes and negative controls were simultaneously processed during each setting of the immunolabeling. Immunolabeling was quantitatively graded on digitized images in a masked fashion by measuring the specific immunolabeling areas using image analysis software (Axiovision), as recently described.12,24,25 A percentage value was expressed for each slide as the average ratio of the measurement to the total area analyzed, multiplied by 100. The mean extent of immunolabeling was calculated for each eye after subtracting the value obtained from the negative control slide.
In Situ Detection of Apoptosis
A DNA fragmentation detection kit (FragEL; Calbiochem) was used to detect apoptotic cells in retina sections using terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL). This kit contains terminal deoxynucleotidyl transferase (TdT) for nonisotopic labeling of the exposed 3′-OH ends of DNA fragments generated in response to apoptotic signals. After incubation in the presence of fluorescein-labeled nucleotides for 1 hour, the generated fluorescein signal was detected by fluorescence microscopy. Incubation with the fluorescein-labeled nucleotide mixture without the presence of TdT served as a negative control.
RESULTS
Phosphorylated RGC Proteins during Glaucomatous Neurodegeneration in Ocular Hypertensive Rat Eyes Include the 14-3-3 Family of Proteins
As an initial attempt to identify phosphorylated proteins during glaucomatous neurodegeneration through a 2-D PAGE-based proteomic approach, phosphoproteins were detected by phosphoprotein staining of 2-D gels followed by protein gel stain for total protein profiling. For large-scale quantification, we performed differential display analysis of protein spots revealed by this specific staining. We therefore compared the intensity of phosphoprotein-stained spots matched on 2-D gel images obtained using ocular hypertensive and control samples after normalization of each spot to protein content measured by the intensity of protein gel stain. This analysis revealed more than 100 spots exhibiting new or increased phosphorylation in ocular hypertensive eyes. These protein spots were then identified through peptide mass fingerprinting and peptide sequencing using mass spectrometry. One of the identified proteins exhibiting more than twofold increased phosphorylation in ocular hypertensive samples was 14-3-3, which is the focus of this study. As shown in Figure 1, the identified 14-3-3 isoforms included alpha/beta (α/β, Ywhab), delta/zeta (δ/ζ, Ywhaz), eta (η, Ywhah), gamma (γ, Ywhag), and theta (τ, tau, Ywhaq). Similar to the findings of the 2-D PAGE-based proteomic analysis, a gel-free proteomic technique using LC-MS/MS analysis of enriched phosphoproteins also identified different isoforms of 14-3-3 proteins with significant matches (Fig. 1B).
FIGURE 1.

Proteomic analysis. A 2-D PAGE-based proteomic approach was initially used for phosphoprotein detection and identification. (A) 2-D gels obtained using ocular hypertensive and control samples after protein gel stain or phosphoprotein staining. Proteins revealed by phosphoprotein staining (Pro-Q Diamond; Invitrogen/Molecular Probes) were then identified through peptide mass fingerprinting and peptide sequencing. The five spots shown in the box correspond to different isoforms of 14-3-3. Comparison of phosphoprotein-stained 2-D gels using ocular hypertensive and control samples indicates many other proteins in addition to 14-3-3 (black arrows) that exhibit new or increased phosphorylation in ocular hypertensive eyes. Consistent with the results of 2-D gel analysis, experiments through a gel-free proteomic technique identified the same 14-3-3 proteins. (B) Results of gel-free analysis using tandem mass spectrometry.
Quantitative analysis of protein gel-stained 2-D gels detected a more than twofold increase (2.6 ± 0.5) in the expression level of 14-3-3 protein in ocular hypertensive eyes compared with controls (Fig. 2A). Immunodetection on 2-D Western blots using a specific antibody matched with the position of the 14-3-3 spot identified by mass spectrometric analysis of 2-D gel pieces. Figure 2B shows Western blots of 2-D gels obtained using a narrower immobilized pH gradient to zoom in on the 14-3-3 spot for better resolution. These Western blots further support phosphorylation-related acidic shift of the 14-3-3 protein spot in ocular hypertensive samples from its initial position on control 2-D gels.
FIGURE 2.
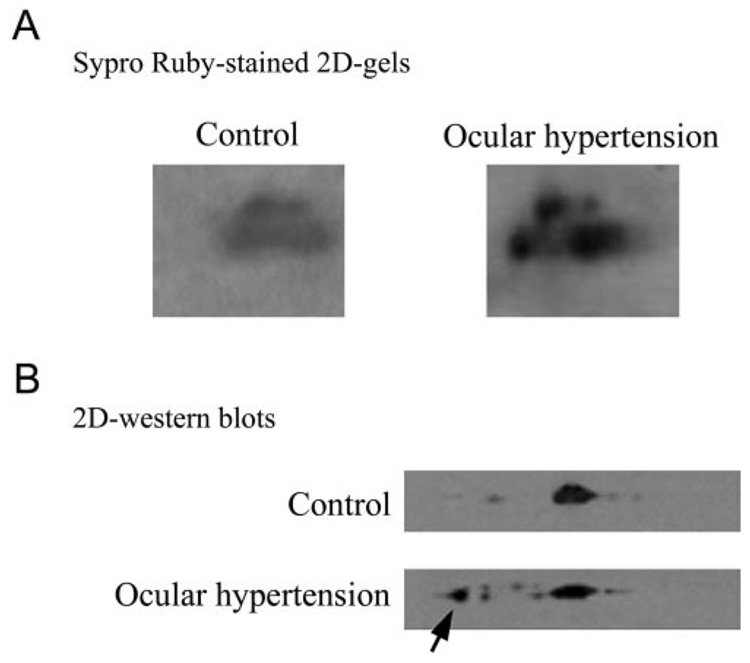
Comparative analysis of 2-D gels. (A) 14-3-3 protein spots on protein gel-stained 2-D gel images obtained using control and ocular hypertensive samples. Quantitative image analysis detected a more than twofold increase in the expression level of 14-3-3 protein in ocular hypertensive eyes compared with controls. To provide additional confirmation for the mass spectrometric protein identification from 2-D gel pieces, Western blot analysis was performed using a specific 14-3-3 antibody. (B) Western blots of 2-D gels obtained using a narrower pH gradient (4.5–5.5). An acidic shift of the 14-3-3 protein spot detected on these Western blots (arrow) further supports the phosphorylation of this protein in ocular hypertensive eyes.
To determine the extent and cellular localization of 14-3-3 proteins, histologic sections of the retina and optic nerve head were immunolabeled with specific antibodies to different 14-3-3 isoforms (Fig. 3). Retina and optic nerve head sections exhibited prominent immunolabeling for the 14-3-3 isoforms identified by proteomic analysis, including alpha/beta, delta/zeta, eta, and gamma. The extent of 14-3-3 immunolabeling was found to be relatively greater in ocular hypertensive eyes than in normotensive control eyes. Using digital image analysis, the extent of 14-3-3 immunolabeling (mean ± SD) was 36% ± 5% in ocular hypertensive eyes but was less than 10% in controls. Based on the assessment of morphologic characteristics of cell types and double immunolabeling, different 14-3-3 isoforms were widely detectable in different cell types, which prominently included RGCs and their axons.
FIGURE 3.
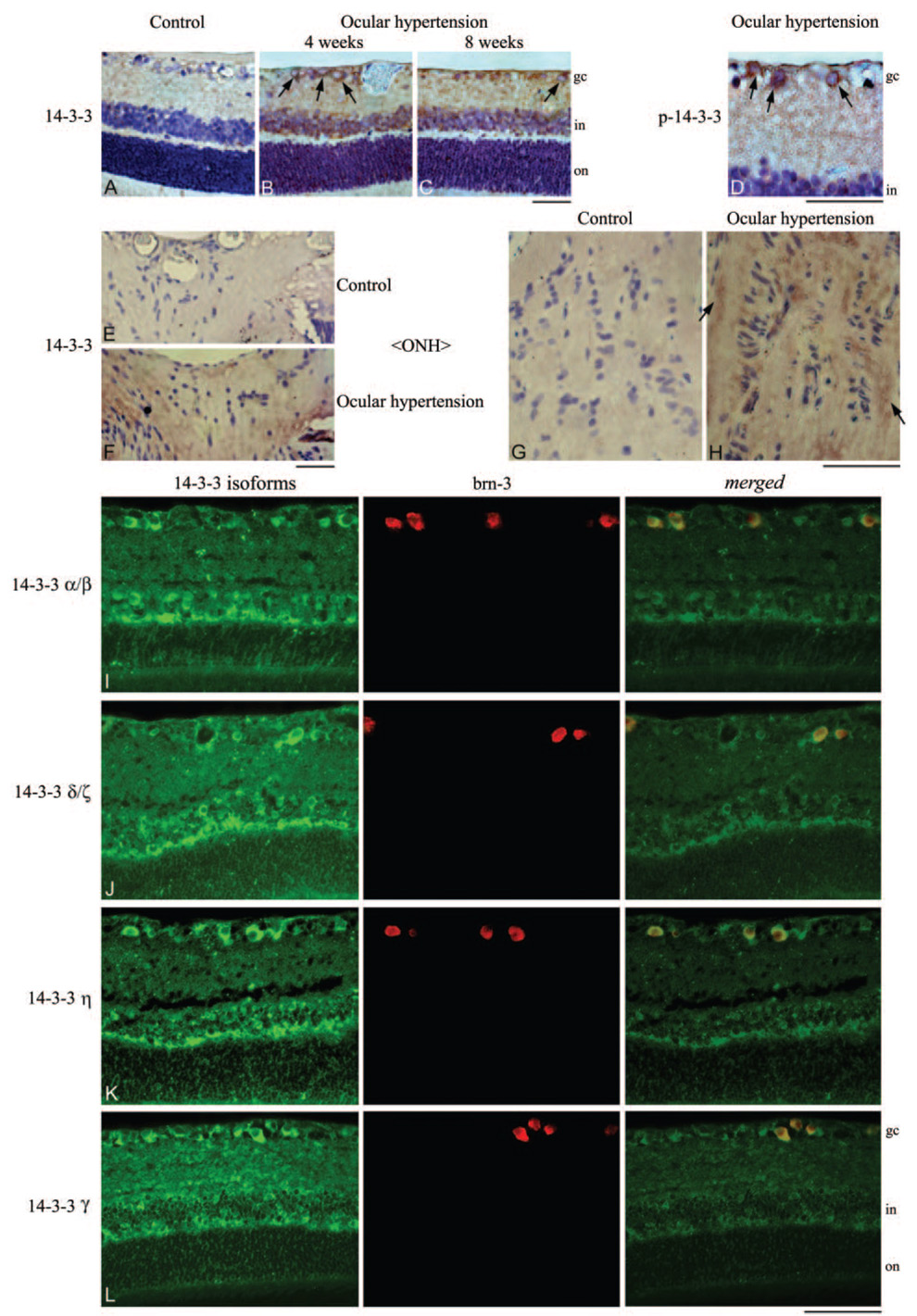
Immunohistochemical analysis of 14-3-3 proteins. To determine the cellular distribution of 14-3-3, retina and optic nerve head sections obtained from control and ocular hypertensive rat eyes were immunolabeled using specific 14-3-3 antibodies. (A–C) Immunoperoxidase labeling of retinal sections using a common antibody for 14-3-3 isoforms in control and ocular hypertensive eyes at 4 or 8 weeks after IOP elevation. An increase was detectable in the intensity of the retinal 14-3-3 immunolabeling in ocular hypertensive eyes compared with controls. Based on morphologic characteristics of different cell types, widespread cytoplasmic immunolabeling for 14-3-3 included RGCs (arrows). (D) Prominent immunoperoxidase labeling of RGCs (arrows) using a phosphorylation site-specific antibody to 14-3-3. (E, F) Immunoperoxidase labeling of optic nerve head sections for 14-3-3. (G, H) 14-3-3 immunolabeling of optic nerve heads under higher magnification. Increased 14-3-3 immunolabeling of the glaucomatous optic nerve head was prominent in glial cells and nerve bundles (arrows). To determine the cellular localization of 14-3-3, double-immunofluorescence labeling was performed using antibodies to different 14-3-3 isoforms (green) and brn-3 (red) as a marker for RGCs. Merged images indicate prominent localization of 14-3-3 isoforms to RGCs (yellow). gc, ganglion cell; in, inner nuclear; on, outer nuclear. Scale bar, 100 µm.
In addition, immunolabeling of ocular hypertensive tissues with a phospho-14-3-3 antibody was mainly detectable in RGCs. In addition to the phosphoprotein staining of 2-D gels followed by LC-MS/MS analysis and the acidic shift of specific protein spots on 2-D gels, findings of tissue immunolabeling using a phosphorylation site-specific antibody were consistent with the phosphorylation of 14-3-3 in ocular hypertensive eyes (Fig. 3). These findings support the presence of 14-3-3 isoforms in RGCs and their upregulation and phosphorylation during glaucomatous neurodegeneration.
Interaction of 14-3-3 with Bad Constitutes a Regulatory Pathway of RGC Death Signaling in a Phosphorylation-Dependent Manner
To detect proteins interacting with 14-3-3, 14-3-3–containing protein complexes were eluted through coimmunoprecipitation using a monoclonal antibody against 14-3-3. As a complementary approach, recombinant protein-based affinity pull-down was performed using GST-tagged 14-3-3 protein. As shown in Figure 4, SDS-PAGE confirmed the purified protein complexes before their further analysis for mass spectrometric protein identification.
FIGURE 4.
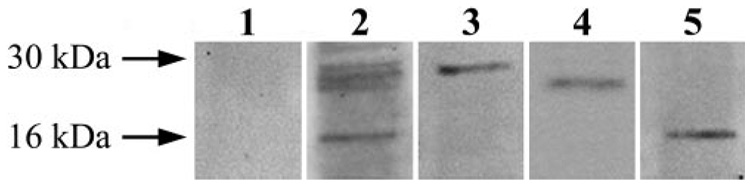
Isolation of 14-3-3– containing protein complexes. To detect proteins interacting with 14-3-3, 14-3-3– containing protein complexes were eluted through coimmunoprecipitation using antibodies against different 14-3-3 isoforms. As a complementary approach to elute 14-3-3– containing protein complexes, recombinant protein-based affinity pull-down was also performed using GST-tagged 14-3-3 protein isoforms. Eluted 14-3-3– containing protein complexes were then subjected to gel-free mass spectrometric analysis to identify proteins interacting with 14-3-3. SDS-PAGE was performed to confirm the purified protein complexes before their mass spectrometric analysis. Lanes 1 and 2 indicate protein staining of gels obtained using a control immunoprecipitate with IgG or the immunoprecipitate with 14-3-3 antibody, respectively. Lanes 3, 4, and 5 are immunoblots of the 14-3-3 immunoprecipitate shown in lane 2 using 14-3-3, Bad, or calmodulin antibodies, respectively. Because no band was detected in the negative control, these data verify that the proteins subsequently identified through mass spectrometric analysis are molecular constituents of the 14-3-3 protein complex. Western blot analysis using specific antibodies also confirm the results of mass spectrometric protein identification that the proteins interacting with 14-3-3 include Bad and calmodulin.
To identify proteins in eluted 14-3-3–containing protein complexes, gel-free LC-MS/MS analysis and Western blot analysis were performed. The proteins identified through consecutive experiments included calmodulin and proapoptotic members of the Bcl-2 family and many other proteins such as zinc-finger protein, protein kinase C, ubiquitin, and HSP70. Regarding interactions between 14-3-3 and proapoptotic Bcl-2 proteins, though our findings also support 14-3-3/Bax interaction, the experiments presented herein focus on 14-3-3/Bad interaction.
As presented in Figure 4, Western blots using specific antibodies confirmed the presence of Bad and calmodulin in the 14-3-3– containing protein complex. As shown in Figure 5A, Western blot analysis using a phosphorylation site-specific antibody demonstrated that the Bad in the 14-3-3–containing protein complex is phosphorylated and that phospho-Bad expression exhibits a more than threefold decrease in ocular hypertensive samples compared with controls. In addition, Bad expression was found to increase in the mitochondrial fraction of ocular hypertensive RGC proteins while decreasing in the cytoplasmic fraction of the same samples (Fig. 5A). This is supported by another observation from quantitative Western blot analysis that the 14-3-3–containing protein complexes repeatedly eluted from ocular hypertensive samples contained much lower levels of Bad than did controls (a more than threefold decrease in relative protein quantity). In some samples, Bad was not even detectable in the eluted 14-3-3 protein complex.
FIGURE 5.
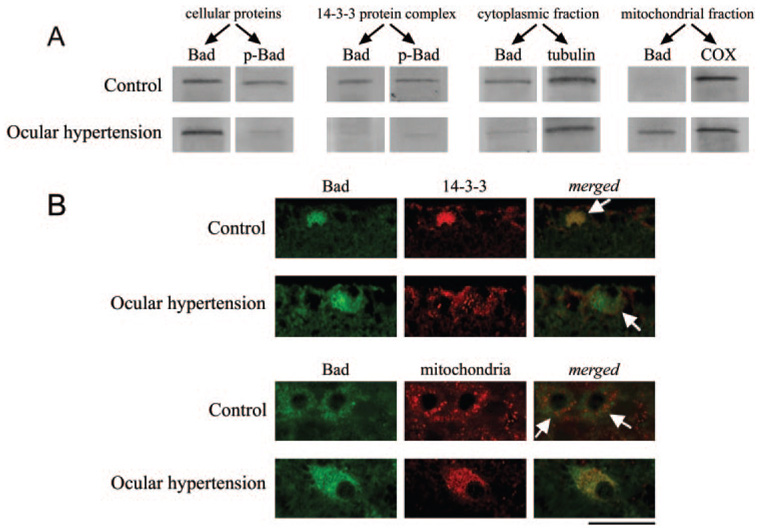
Analysis of 14-3-3/Bad interaction. (A) Western blot analysis using a phosphorylation site-specific antibody demonstrated a prominent decrease in phospho-Bad in ocular hypertensive eyes compared with controls, whereas Bad expression exhibited an increase, thereby supporting the dephosphorylation of Bad in ocular hypertensive eyes. Phospho-Bad was found to be associated with the 14-3-3– containing protein complex. However, this association exhibited a significant decrease in ocular hypertensive eyes. In addition, Bad expression was found to increase in the mitochondrial fraction of ocular hypertensive protein samples while it decreased in the cytoplasmic fraction. In accordance with the findings of Western blot analysis, immunolabeling of retina sections with specific antibodies demonstrated differential subcellular localization of Bad in ocular hypertensive eyes compared with control RGCs. (B, bottom) To determine the subcellular localization of Bad, immunolabeling was also performed using an antibody against prohibitin, a mitochondrial marker. (B, top) Double-immunofluorescence labeling of the control and ocular hypertensive retinas for Bad (green) and 14-3-3 (red). Merged images show colocalization of these proteins in the control retina (yellow). However, in the ocular hypertensive retina, colocalization of 14-3-3 and Bad was no more prominent. Arrows: RGCs based on morphologic assessment. (B, bottom) Double-immunofluorescence labeling with Bad (green) and mitochondrial marker antibodies (red). Yellow in merged image supports mitochondrial translocation of Bad in ocular hypertensive RGCs (arrows). Scale bar, 50 µm.
In accordance with the findings of Western blot analysis, immunolabeling of retina sections with specific antibodies demonstrated differential subcellular localization of Bad in ocular hypertensive eyes compared with controls (Fig. 5B). Double-immunofluorescence labeling with Bad and 14-3-3 antibodies supported their strong interaction in control retinas. However, double immunolabeling with Bad and mitochondrial marker antibodies confirmed the mitochondrial translocation of Bad in many RGCs in ocular hypertensive eyes.
These findings support that the interaction of Bad with 14-3-3 is important for the subcellular localization and functional activity of this proapoptotic protein in a phosphorylation-dependent manner. In addition, 14-3-3 binding to phospho-Bad keeps this proapoptotic protein sequestered in the cytoplasm. However, this association is disrupted in ocular hypertensive eyes in correlation with Bad dephosphorylation. When dissociated from the 14-3-3 protein complex, Bad translocates to mitochondria, where its binding to Bcl-2/Bcl-x(L) is known to lead to the initiation of apoptotic cell death signaling.
Inhibition of 14-3-3 Phosphorylation and Bad Dephosphorylation Complementarily Secure 14-3-3 Scaffold of Bad in the Cytoplasm, Thereby Preserving Optic Nerve Axons in Experimental Glaucoma
To determine the functional importance of the phosphorylation-dependent interaction between 14-3-3 and Bad, in vivo treatment experiments were performed using known modulators of the phosphorylation status of 14-3-3 and Bad. DJNKI and FK506 treatments were therefore applied to inhibit JNK activity and protein phosphatase activity, respectively. These treatments were also applied in combination to determine their additive effect. After a treatment period of 4 weeks, we detected that the inhibition of JNK and protein phosphatase activities prominently affects the interaction between 14-3-3 and Bad. Treatment with FK506, a protein phosphatase inhibitor, resulted in sustained phosphorylation of Bad and continued interaction of this proapoptotic protein with 14-3-3, thereby inhibiting its mitochondrial transport in ocular hypertensive eyes (Fig. 6).
FIGURE 6.
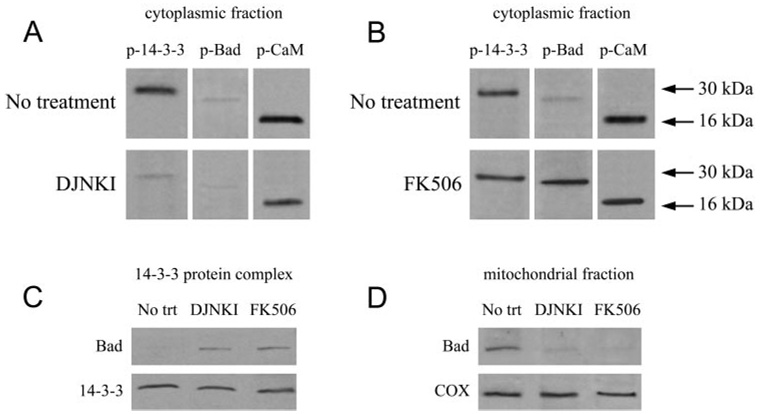
Phosphorylation dependence of 14-3-3 protein interactions. To determine phosphorylation-dependent protein interactions, ocular hypertensive rats were treated with DJNKI or FK506. (A) After a treatment period of 4 weeks, inhibition of JNK using DJNKI resulted in a prominent decrease in 14-3-3 phosphorylation, although JNK inhibition had no prominent effect on Bad phosphorylation. (B) However, treatment with FK506, a protein phosphatase inhibitor, resulted in sustained phosphorylation of Bad. Inhibition of 14-3-3 phosphorylation by DJNKI treatment and maintenance of Bad phosphorylation by FK506 treatment both caused inhibition in the mitochondrial transport of Bad. (C) Bad was undetectable in the 14-3-3 protein complex obtained using untreated ocular hypertensive samples. However, this proapoptotic protein was prominently detectable in the 14-3-3 protein complex eluted using ocular hypertensive samples after DJNKI or FK506 treatment. These findings support a treatment effect on reestablishment of the 14-3-3/Bad interaction in the cytoplasm. Consistent with these findings, (D) mitochondrial fractions exhibit a prominent decrease in Bad after DJNKI and FK506 treatment of ocular hypertensive eyes.
Another important observation was that JNK-inhibiting treatment similarly inhibited Bad translocation to mitochondria, though JNK inhibition had no prominent effect on Bad phosphorylation. However, as shown in Figure 6A, JNK-inhibiting treatment with DJNKI was found to result in inhibited phosphorylation of 14-3-3. This finding supports that the inhibition of mitochondrial translocation of Bad after anti-JNK treatment is dependent on the phosphorylation status of 14-3-3.
As also presented in Figure 6, Bad was undetectable in the 14-3-3 protein complex eluted using untreated ocular hypertensive samples. However, this proapoptotic protein was prominently detectable in the 14-3-3 protein complex eluted using ocular hypertensive samples after DJNKI or FK506 treatment, thereby supporting a treatment effect on reestablishment of the Bad/14-3-3 interaction in the cytoplasm.
Consistent with these observations, mitochondrial fractions exhibited a prominent decrease in Bad after DJNKI and FK506 treatments of ocular hypertensive animals. Thus, the phosphorylation of 14-3-3 and the dephosphorylation of Bad in ocular hypertensive eyes affected the subcellular redistribution of Bad from 14-3-3 scaffold in the cytoplasm to mitochondria. Although the phosphorylation of Bad is critical for its dissociation from 14-3-3 and its translocation to mitochondria, 14-3-3 phosphorylation by JNK is also important to reduce the affinity of this cytoplasmic anchor protein for Bad.
A treatment effect on neuronal survival was detectable by a prominent decrease in retinal TUNEL labeling in treated ocular hypertensive animals (Figs. 7F, 7G). Parallel to this beneficial effect on decreasing RGC apoptosis, treatments also had a protective effect on optic nerve damage. When optic nerve cross-sections were examined, the overall structure of the optic nerve was well preserved in treated ocular hypertensive rats, whereas optic nerve cross-sections obtained from untreated controls exhibited widespread degenerative changes characterized by axon loss, disorganized axon morphology, and myelin debris (Figs. 7A–E).
FIGURE 7.
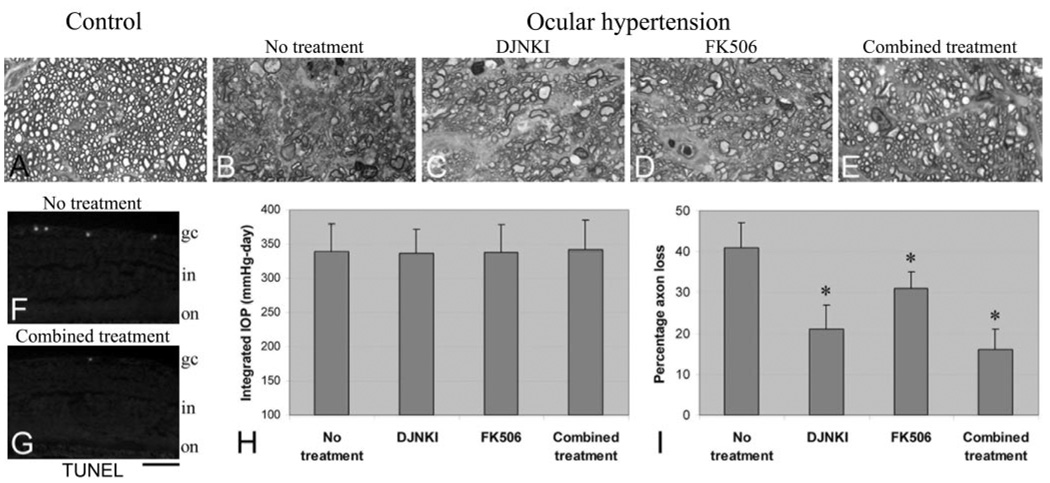
Optic nerve injury in ocular hypertensive eyes and protection by DJNKI and FK506 treatments. (A–E) Optic nerve cross-sections. Compared with those from normotensive control eyes (A), optic nerve cross-sections obtained from untreated ocular hypertensive eyes exhibited widespread degenerative changes characterized by axon loss, disorganized axon morphology, and myelin debris (B). However, the overall structure of the optic nerve was well preserved in treated ocular hypertensive rats (C–E). (F, G) Parallel to optic nerve findings, a decrease was also detectable in TUNEL-positive RGCs in the retinas of treated ocular hypertensive animals. Optic nerve axons were counted to quantitatively determine the treatment effect on neuronal damage. Axon counts revealed that although the treatments did not affect IOP in ocular hypertensive animals (H), they did result in a significant decrease in axon loss (I). Combined treatment with DJNKI and FK506 resulted in the greatest neuroprotective effect (combined treatment vs. no treatment; P = 0.0001). The neuroprotective effect of the combined treatment was found to be significantly greater than FK506 treatment applied alone (P = 0.001); however, the greater neuroprotective effect of the combined treatment was not statistically significant compared with DJNKI treatment applied alone (P = 0.1). gc, ganglion cell; in, inner nuclear; on, outer nuclear. *P < 0.05; statistically significant. Scale bar, 200 µm.
Optic nerve axons were counted to assess for the degree of any treatment effect on neuronal damage. Axon counts revealed that although the used treatments did not affect IOP in ocular hypertensive animals (Fig. 7H; Mann-Whitney U test; P > 0.05), they did result in a significant decrease in axon loss. Despite prominent axon loss in the untreated group of ocular hypertensive rats, in rats receiving DJNKI or FK506, significantly less optic nerve damage was observed (Fig. 7I; P = 0.001 and P = 0.02, respectively). When a combined treatment with DJNKI and FK506 was applied, the neuroprotective effect was greatest (combined treatment vs. no treatment, P = 0.0001). The neuroprotective effect of the combined treatment was found to be significantly greater than FK506 treatment applied alone (P = 0.001). However, though the neuroprotective effect of the combined treatment was also greater than DJNKI treatment applied alone, this difference was not statistically significant (P = 0.1). These findings further support an important association of 14-3-3 phosphorylation and Bad dephosphorylation with neuronal damage during glaucomatous neurodegeneration in ocular hypertensive eyes.
DISCUSSION
Findings of this in vivo study identified that an important protein family associated with checkpoint control pathways, 14-3-3, is involved in cellular signaling during glaucomatous neurodegeneration in a phosphorylation-dependent manner. Using an experimental rat model of chronic pressure-induced glaucoma and controls, phosphorylated Bad was found to be sequestered in the cytoplasm by 14-3-3 scaffold, thereby preventing its mitochondrial translocation to induce apoptosis. Neuronal damage in ocular hypertensive rat eyes was found to be associated with the phosphorylation of 14-3-3 and the dephosphorylation of Bad. Inhibition of 14-3-3 phosphorylation by D-JNKI treatment and inhibition of Bad dephosphorylation by FK506 treatment resulted in an additive neuroprotective effect in ocular hypertensive eyes. Relatively greater protection by anti-JNK treatment is supportive of additional roles of JNK in cellular signaling during the neurodegenerative process.12,13,24,26
Named for their characteristic migration pattern on electrophoretic gels, 14-3-3 proteins were originally identified in 1967 as abundant brain proteins with preferential localization to neurons. However, almost 25 years passed before it became clear that these proteins are involved in the regulation of many cellular processes, including metabolic pathways, redox-regulation, transcription, protein synthesis, protein folding and degradation, cell cycle, cytoskeletal organization, and cellular trafficking. These adaptor proteins form dimers that provide two binding sites for phosphorylated ligand proteins, thereby bringing two proteins into close proximity. Through phosphorylation-dependent protein interactions, 14-3-3 proteins regulate other proteins by cytoplasmic sequestration, occupation of interaction domains, export or import sequences, prevention of degradation, and activation or inactivation of protein activity and protein modifications. More than 300 binding partners of 14-3-3 proteins that have been identified until now include the proteins identified in this study, Bad and calmodulin. Additionally, 14-3-3 proteins are implicated in antagonizing apoptotic signals, mainly through the cytoplasmic sequestration of the proapoptotic proteins Bad and Bax.27–30 For example, a substantial proportion of Bad is bound to 14-3-3 proteins in the cytosol of healthy cells, regulated by phosphorylation through the PI3-kinase-Akt pathway.31,32
We demonstrated that the 14-3-3 protein similarly constitutes an important mechanism of cell death regulation in RGCs and that the activity of Bad in RGCs is regulated by reversible phosphorylation in such a way that phosphorylated Bad remains sequestered in the cytoplasm by 14-3-3 scaffold. This association is disrupted in ocular hypertensive eyes in correlation with Bad translocation to mitochondria. Although phosphorylation prevents Bad from promoting cell death, its dephosphorylated form dissociates from 14-3-3 and redistributes to mitochondria, where cell death can be induced through binding of Bcl-2/Bcl-x(L). These findings outline an important role of 14-3-3 in proapoptotic protein trafficking during RGC apoptosis in ocular hypertensive eyes.
The phosphorylation dependence of Bad location between the cytoplasm and the mitochondria is consistent with previous evidence of calcineurin-mediated Bad dephosphorylation in ocular hypertensive eyes, which has been associated with the mitochondrial pathway of RGC apoptosis.11 Given that the binding of calmodulin with 14-3-3 inhibits calcium/calmodulin-dependent activity,33 which is critical for calcineurin activity, it may be speculated that the 14-3-3/calmodulin interaction we detected in RGCs may be an intrinsic counterbalancing effort to prevent the proapoptotic function of Bad. It would be interesting to test the validity of such an association.
In addition to Bad dephosphorylation, increased phosphorylation of 14-3-3 was observed to be similarly important for the proapoptotic signaling in ocular hypertensive eyes. In accordance with our findings, previous studies have demonstrated that because dimerization of 14-3-3 proteins is required for phosphorylation-dependent binding to protein ligands, phosphorylation at the dimerization interface of 14-3-3 prevents dimerization, thereby inhibiting its adaptor function.34–36 Our treatment experiments using a JNK inhibitor revealed that by mediating the phosphorylation of 14-3-3, JNK interferes with the 14-3-3/Bad interaction and promotes the dissociation of Bad from 14-3-3 and translocation to the mitochondria. This is consistent with previous observations supporting that JNK is required for stress-induced mitochondrial dysfunction and apoptosis in association with Bcl-2 family proteins. For example, the phosphorylation of 14-3-3 by JNK and the release of Bad from 14-3-3 has been found to antagonize the effects of Akt signaling, thereby regulating cell death.37 Because Bad shares the 14-3-3-binding motif with other proapoptotic proteins, JNK-mediated phosphorylation of 14-3-3 also regulates other proapoptotic proteins and makes cells more susceptible to apoptotic signals. Indeed, JNK activity has been shown to promote Bax translocation to mitochondria through 14-3-3 phosphorylation and Bax dissociation,38 and inhibition of JNK has provided neuroprotection through the inhibition of the Bax-mediated mitochondrial apoptosis pathway.39 This decisive role of JNK in neurodegenerative injury is also consistent for glaucomatous injury because the findings of previous in vitro and in vivo studies support that JNK activity is critical in switching the life balance toward RGC death.12,13,24,26
There are many other interesting aspects of the phosphorylation-dependent 14-3-3/Bad interaction relevant to neurode-generation. For example, considerable evidence supports that the release of Bad from 14-3-3 after JNK-mediated 14-3-3 phosphorylation may also be necessary for Bad dephosphorylation. It has been shown that 14-3-3 and protein phosphatases compete for Bad. Although 14-3-3 binding maintains the phosphorylated state of Bad, JNK-mediated release from 14-3-3 makes Bad accessible to phosphatases, thereby facilitating its dephosphorylation.40 What is also interesting is that the phosphorylation of Bad at another site in proximity to the 14-3-3 binding site may negatively modulate its interaction with 14-3-3, thereby activating cell death signaling.41,42 Such complexity in the nature of phosphorylation-dependent protein interactions is consistent with the additive neuroprotective effect of treatments inhibiting the JNK and protein phosphatase activity we detected.
In summary, cellular signal transduction pathways involve protein kinases, protein phosphatases, and phosphoprotein-interacting domain–containing cellular proteins, such as 14-3-3, to provide multidimensional, dynamic, and reversible regulation of many biological activities.43
Multiple intrinsic adaptive/protective mechanisms are known to be activated in response to multiple pathogenic mechanisms associated with glaucomatous neurodegeneration, and a critical balance of diverse signals determines whether an RGC will live or die. Present evidence supports that 14-3-3 serves as a key integration point of stress and survival signals in RGCs, thereby contributing to life-and-death decisions (Fig. 8). In the context of 14-3-3 protein interactions detected in this study, there appears to be some threshold at which apoptosis signaling is triggered but cannot lead to cell death, and 14-3-3 can function to raise this hypothetical threshold for the gain of RGC survival by acting as an anchor for several proapoptotic proteins on phosphorylation, including Bad. Given that regulation of the diverse functions of Bcl-2 family proteins is critical for the initiation of mitochondrial dysfunction, a critical component of RGC death signaling, targeting the upstream 14-3-3 scaffolding may be a promising strategy for RGC protection. This warrants further studies to better determine the functional importance of 14-3-3 protein interactions as a possible treatment target for neuroprotection in glaucoma.
FIGURE 8.

Flow diagram illustrating 14-3-3/Bad interaction. By controlling the subcellular localization and function of Bad in a phosphorylation-dependent manner, 14-3-3 proteins constitute an important regulatory pathway of RGC death signaling during glaucomatous neurodegeneration. Although phosphorylated Bad normally remains sequestered in the cytoplasm by 14-3-3 scaffold, phosphorylation of 14-3-3 and dephosphorylation of Bad in ocular hypertensive eyes lead to translocation of this proapoptotic protein to mitochondria. Findings of this study support that the maintenance of the 14-3-3/Bad interaction in the cytoplasm by treatments inhibiting 14-3-3 phosphorylation and Bad dephosphorylation may increase RGC survival in ocular hypertensive eyes.
Acknowledgments
Supported in part by National Eye Institute Grants 2R01 EY013813, 1R01 EY017131, and R24 EY015636; an unrestricted grant to University of Louisville Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness Inc.; and the University of Louisville School of Medicine.
Footnotes
Disclosure: X. Yang, None; C. Luo, None; J. Cai, None; W.M. Pierce, None; G. Tezel, None
References
- 1.Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–8700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tezel G, Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci. 2004;45:4049–4059. doi: 10.1167/iovs.04-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tezel G, Yang X, Cai J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest Ophthalmol Vis Sci. 2005;46:3177–3187. doi: 10.1167/iovs.05-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tezel G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 2006;25:490–513. doi: 10.1016/j.preteyeres.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickells RW. Apoptosis of retinal ganglion cells in glaucoma: an update of the molecular pathways involved in cell death. Surv Ophthalmol. 1999;43 suppl 1:S151–S161. doi: 10.1016/s0039-6257(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–213. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- 7.Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- 8.Libby RT, Li Y, Savinova OV, et al. Susceptibility to neurodegeneration in a glaucoma is modified by bax gene dosage. PLoS Genet. 2005;1 doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji J, Chang P, Pennesi ME, et al. Effects of elevated intraocular pressure on mouse retinal ganglion cells. Vision Res. 2005;45:169–179. doi: 10.1016/j.visres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Kim HS, Park CK. Retinal ganglion cell death is delayed by activation of retinal intrinsic cell survival program. Brain Res. 2005;1057:17–28. doi: 10.1016/j.brainres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Fileta JB, Dobberfuhl A, et al. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci U S A. 2005;102:12242–12247. doi: 10.1073/pnas.0505138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003;44:3025–3033. doi: 10.1167/iovs.02-1136. [DOI] [PubMed] [Google Scholar]
- 13.Tezel G, Yang X. Comparative gene array analysis of TNF-α-induced MAPK and NF-κB signaling pathways between retinal ganglion cells and glial cells. Exp Eye Res. 2005;81:207–217. doi: 10.1016/j.exer.2005.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanov D, Dvoriantchikova G, Nathanson L, McKinnon SJ, Shestopalov VI. Microarray analysis of gene expression in adult retinal ganglion cells. FEBS Lett. 2006;580:331–335. doi: 10.1016/j.febslet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 15.Skoulakis EM, Davis RL. 14-3-3 proteins in neuronal development and function. Mol Neurobiol. 1998;16:269–284. doi: 10.1007/BF02741386. [DOI] [PubMed] [Google Scholar]
- 16.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 17.Yaffe MB, Rittinger K, Volinia S, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 18.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 2000;19:349–358. doi: 10.1093/emboj/19.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 20.Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997;64:85–96. doi: 10.1006/exer.1996.0184. [DOI] [PubMed] [Google Scholar]
- 21.Schulenberg B, Aggeler R, Beechem JM, Capaldi RA, Patton WF. Analysis of steady-state protein phosphorylation in mitochondria using a novel fluorescent phosphosensor dye. J Biol Chem. 2003;278:27251–27255. doi: 10.1074/jbc.C300189200. [DOI] [PubMed] [Google Scholar]
- 22.Schulenberg B, Goodman TN, Aggeler R, Capaldi RA, Patton WF. Characterization of dynamic and steady-state protein phosphorylation using a fluorescent phosphoprotein gel stain and mass spectrometry. Electrophoresis. 2004;25:2526–2532. doi: 10.1002/elps.200406007. [DOI] [PubMed] [Google Scholar]
- 23.Tezel G, Wax MB. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci. 2000;20:3552–3562. doi: 10.1523/JNEUROSCI.20-10-03552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996:202–212. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Tezel G, Wax MB. Hypoxia-inducible factor 1α in the glaucomatous retina and optic nerve head. Arch Ophthalmol. 2004;122:1348–1356. doi: 10.1001/archopht.122.9.1348. [DOI] [PubMed] [Google Scholar]
- 26.Levkovitch-Verbin H, Quigley HA, Martin KR, et al. The transcription factor c-jun is activated in retinal ganglion cells in experimental rat glaucoma. Exp Eye Res. 2005;80:663–670. doi: 10.1016/j.exer.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 27.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 28.Franke TF, Cantley LC. Apoptosis: a Bad kinase makes good. Nature. 1997;390:116–117. doi: 10.1038/36442. [DOI] [PubMed] [Google Scholar]
- 29.Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 32.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 33.Davare MA, Saneyoshi T, Guire ES, Nygaard SC, Soderling TR. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J Biol Chem. 2004;279:52191–52199. doi: 10.1074/jbc.M409873200. [DOI] [PubMed] [Google Scholar]
- 34.Shen YH, Godlewski J, Bronisz A, et al. Significance of 14-3-3 self-dimerization for phosphorylation-dependent target binding. Mol Biol Cell. 2003;14:4721–4733. doi: 10.1091/mbc.E02-12-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J Biol Chem. 2003;278:36323–36327. doi: 10.1074/jbc.M304689200. [DOI] [PubMed] [Google Scholar]
- 36.Powell DW, Rane MJ, Joughin BA, et al. Proteomic identification of 14-3-3zeta as a mitogen-activated protein kinase-activated protein kinase 2 substrate: role in dimer formation and ligand binding. Mol Cell Biol. 2003;23:5376–5387. doi: 10.1128/MCB.23.15.5376-5387.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuruta F, Sunayama J, Mori Y, et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004;23:1889–1899. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao Y, Signore AP, Yin W, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 40.Chiang CW, Kanies C, Kim KW, et al. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol Cell Biol. 2003;23:6350–6362. doi: 10.1128/MCB.23.18.6350-6362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- 42.Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- 43.Sawyer TK, Shakespeare WC, Wang Y, et al. Protein phosphorylation and signal transduction modulation: chemistry perspectives for small-molecule drug discovery. Med Chem. 2005;1:293–319. doi: 10.2174/1573406053765486. [DOI] [PubMed] [Google Scholar]