

Summary
We investigated serotonin signaling in C. elegans as a paradigm for neural regulation of energy balance and found that serotonergic regulation of fat is molecularly distinct from feeding regulation. Serotonergic feeding regulation is mediated by receptors whose functions are not required for fat regulation. Serotonergic fat regulation is dependent on a neurally expressed channel and a GPCR that initiate signaling cascades which ultimately promote lipid breakdown at peripheral sites of fat storage. In turn, intermediates of lipid metabolism generated in the periphery modulate feeding behavior. These findings suggest that, as in mammals, C. elegans feeding behavior is regulated by extrinsic and intrinsic cues. Moreover, obesity and thinness are not solely determined by feeding behavior. Rather, feeding behavior and fat metabolism are coordinated but independent responses of the nervous system to the perception of nutrient availability.
Introduction
To survive, grow and reproduce, animals must maintain adequate energy reservoirs in environments where food availability is often scarce and dynamic. In mammals, the nervous system regulates energy balance through myriad behavioral, physiological, and metabolic responses elicited by extrinsic and intrinsic cues of food availability and energy demand (Morton et al., 2006). The molecular underpinnings of these responses and their coordination remain key challenges in understanding energy balance and fat regulation.
The neurotransmitter 5-hydroxytryptamine (5-HT, serotonin) regulates food-related behaviors and physiology in diverse vertebrate and invertebrate phyla (Tecott, 2007). A loss-of-function mutation in the mammalian 5-HT2c receptor leads to obesity (Tecott et al., 1995) and pharmacological manipulations that increase central nervous system 5-HT cause weight loss in rodents (Edwards and Stevens, 1991; Vickers and Dourish, 2004) and humans (Sugrue, 1987; Weintraub et al., 1984). Recent efforts have begun to delineate the molecular mechanisms through which 5-HT exerts its control on mammalian energy balance (Heisler et al., 2003; Heisler et al., 2002; Heisler et al., 2006). However, this is a challenging task given the complexity of mammalian energy regulatory pathways, compensatory mechanisms, and roles of both peripheral and central serotonergic pathways in energy balance (Fernstrom and Choi, 2008; Lam and Heisler, 2007; Tecott, 2007).
C. elegans provides a tractable system for defining the molecular genetics of fat regulation and food-related behaviors and for disentangling the complex homeostatic mechanisms that operate in multiple tissues. Evolutionarily conserved pathways regulate food-related behaviors as well as fat regulatory mechanisms in C. elegans (Ashrafi, 2006; de Bono and Maricq, 2005). For instance, neuropeptide Y, dopamine, and 5-HT signaling pathways modulate food-related behaviors in mammals (Morton et al., 2006) and C. elegans (de Bono and Bargmann, 1998; Hills et al., 2004; Sze et al., 2000). Moreover, C. elegans fat reservoirs, which are stored in intestinal and skin-like epidermal cells (Ashrafi, 2006; Kimura et al., 1997), are regulated through insulin and other neuroendocrine and transcriptional regulators of metabolic pathways that are also conserved in mammals (McKay et al., 2003; Ogg et al., 1997; Van Gilst et al., 2005).
We examined C. elegans serotonergic fat regulation as a paradigm for elucidating how the nervous system coordinates energy balance. In hermaphrodite C. elegans, 5-HT synthesis is limited to only a few neurons (Sze et al., 2000). Animals deficient in 5-HT biosynthesis are viable but display phenotypes mimicking responses seen in food-deprived wild-type animals such as reduced mating and retention of eggs (Loer and Kenyon, 1993; Sze et al., 2000; Waggoner et al., 1998). Conversely, exogenously-supplied 5-HT stimulates egg-laying and mating, responses associated with the re-exposure of food deprived animals to food (Horvitz et al., 1982; Loer and Kenyon, 1993). 5-HT signaling is also required for the dramatic reduction of locomotion when food-deprived animals re-encounter food ensuring that these animals spend more time a food-replete environment (Sawin et al., 2000). 5-HT signaling also underlies experience-dependent choices that C. elegans display in selecting growth-promoting bacteria and in avoiding pathogens (Shtonda and Avery, 2006; Zhang et al., 2005).
5-HT is a potent modulator of C. elegans feeding rate and fat content. C. elegans ingest bacteria through pharyngeal pumping. Defects in pharyngeal pumping reduce food intake and are associated with a starved appearance (Avery and Horvitz, 1990). C. elegans match their feeding rate to food availability: feeding rate increases with increasing concentrations of food and decreases as food concentration diminishes (Avery and Horvitz, 1990). However, the experience of starvation modulates feeding rate such that animals that have undergone a period of food deprivation temporarily feed faster than well-fed worms (Avery and Horvitz, 1990). Thus, C. elegans feeding behavior is modulated by both food availability and experience. 5-HT treatment elicits increased feeding rate even in the absence of food (Avery and Horvitz, 1990) while tph-1(−) mutants, which fail to synthesize 5-HT, display reduced feeding (Sze et al., 2000). Despite reduced feeding, tph-1(−) mutants accumulate excess fat (Sze et al., 2000). Furthermore, here we report that exogenous administration of 5-HT causes potent fat reduction in C. elegans despite increasing feeding rate. Thus, similar to mammals, the deficiency and excess of 5-HT lead to fat accumulation and loss, respectively, in C. elegans. Unlike mammals, the regulation of C. elegans fat levels by 5-HT is inversely correlated with 5-HT induced feeding rates.
To decipher the molecular relationships between various 5-HT-regulated responses, we identified molecular determinants of 5-HT-induced fat reduction. Our findings indicate that in C. elegans, serotonergic regulation of fat is molecularly dissociated from serotonergic regulation of feeding. We identified both neurally- and peripherally-expressed genes that mediate serotonergic fat reduction, as well as internal homeostatic signals that modulate feeding rate. Our findings suggest that feeding behavior and fat metabolism are coordinated but independent responses of the nervous system to intrinsic and extrinsic cues of nutrient availability. Since several lines of evidence suggest that 5-HT-induced changes in feeding alone cannot account for the observed effects on fat content in mammals (Fernstrom and Choi, 2008), we speculate that the regulation of fat content independent of feeding may be the principal, evolutionarily-conserved mechanism of serotonergic fat metabolism.
Results
Increased 5-HT signaling causes fat reduction in C. elegans
Chronic administration of exogenous 5-HT to wild-type animals decreased fat content (Figure 1) but increased feeding rate (Horvitz et al., 1982) (Figure 2A). Reduced fat content of 5-HT-treated animals was visualized by Nile Red staining (Figure 1A,B) and confirmed by thin-layer chromatography (TLC) quantitation of total triglycerides extracted from vehicle- and 5-HT-treated worms (Figure 1C) and by Sudan Black fat staining (Figure S1). 5-HT-induced fat decrease was dose-dependent and reversible (Figure 1 and Figure S1). The 5mM 5-HT dose has been typically used for locomotor, egg-laying and feeding-rate assays (Dempsey et al., 2005; Hobson et al., 2006; Horvitz et al., 1982) and it is estimated that the effective 5-HT concentration in worms is approximately 1000-fold below that added to culture plates. 5-HT treatment showed similar fat and feeding responses in both larval-stage animals and adults (not shown), indicating that the observed phenotypes were due to regulatory rather than developmental processes.
Figure 1. 5-HT reduces fat content.
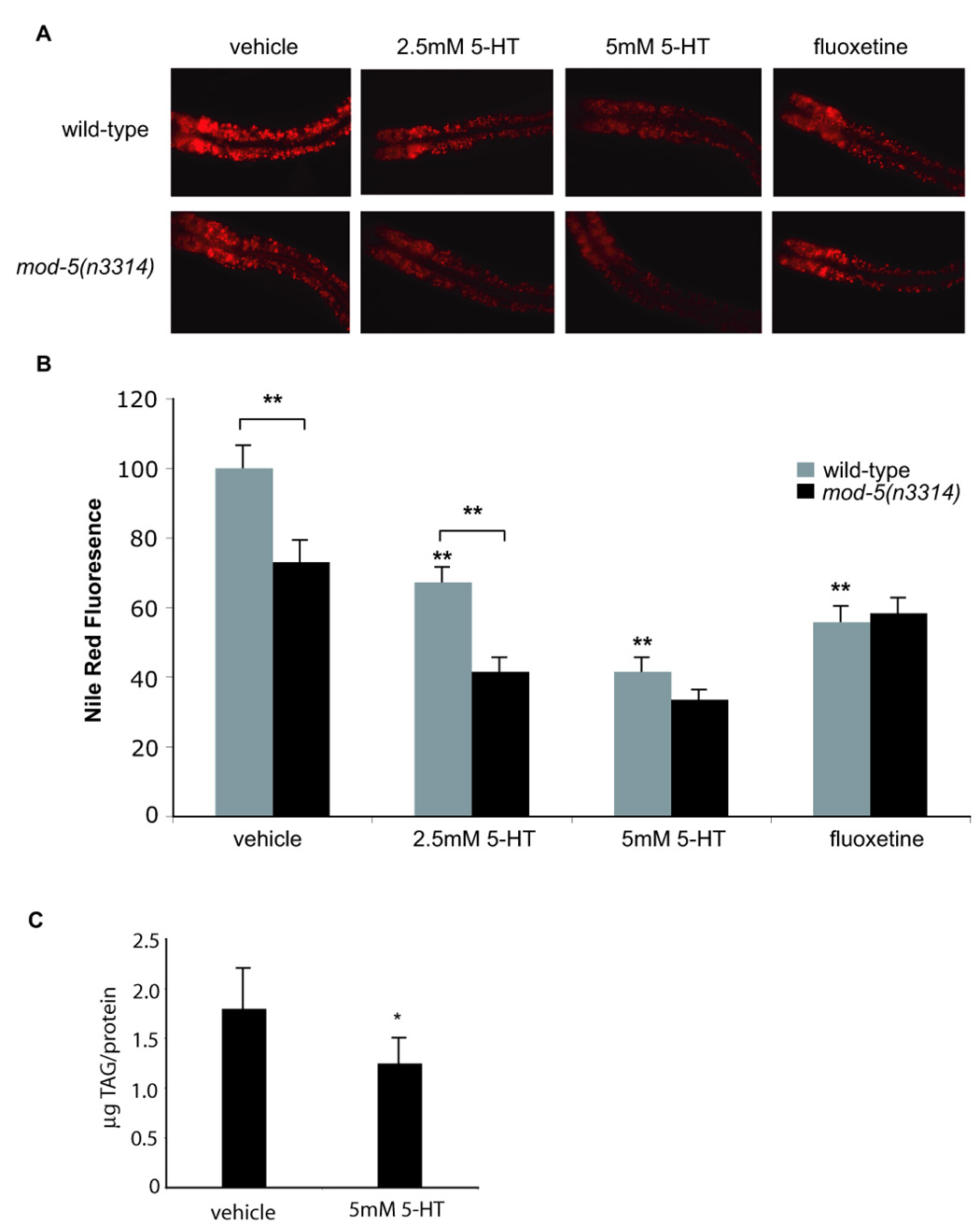
(A) Images of Nile Red-stained wild-type and mod-5(n3314) animals treated with 5-HT or fluoxetine. In all images the anterior end of the animals is oriented towards the left. (B) Quantification of Nile Red fluorescence (n=8 animals per condition). The data are expressed as a percentage of vehicle-treated animals on OP50 bacteria. (**, p<0.005 when compared to vehicle-treated controls or indicated comparisons). (C) TLC measurement of extracted triacylglycerides (TAG) show that 5-HT treatment leads to fat loss (*, p<0.05).
Figure 2. Serotonergic feeding increase requires ser-1 and ser-7 whereas serotonergic fat decrease requires mod-1 and ser-6.
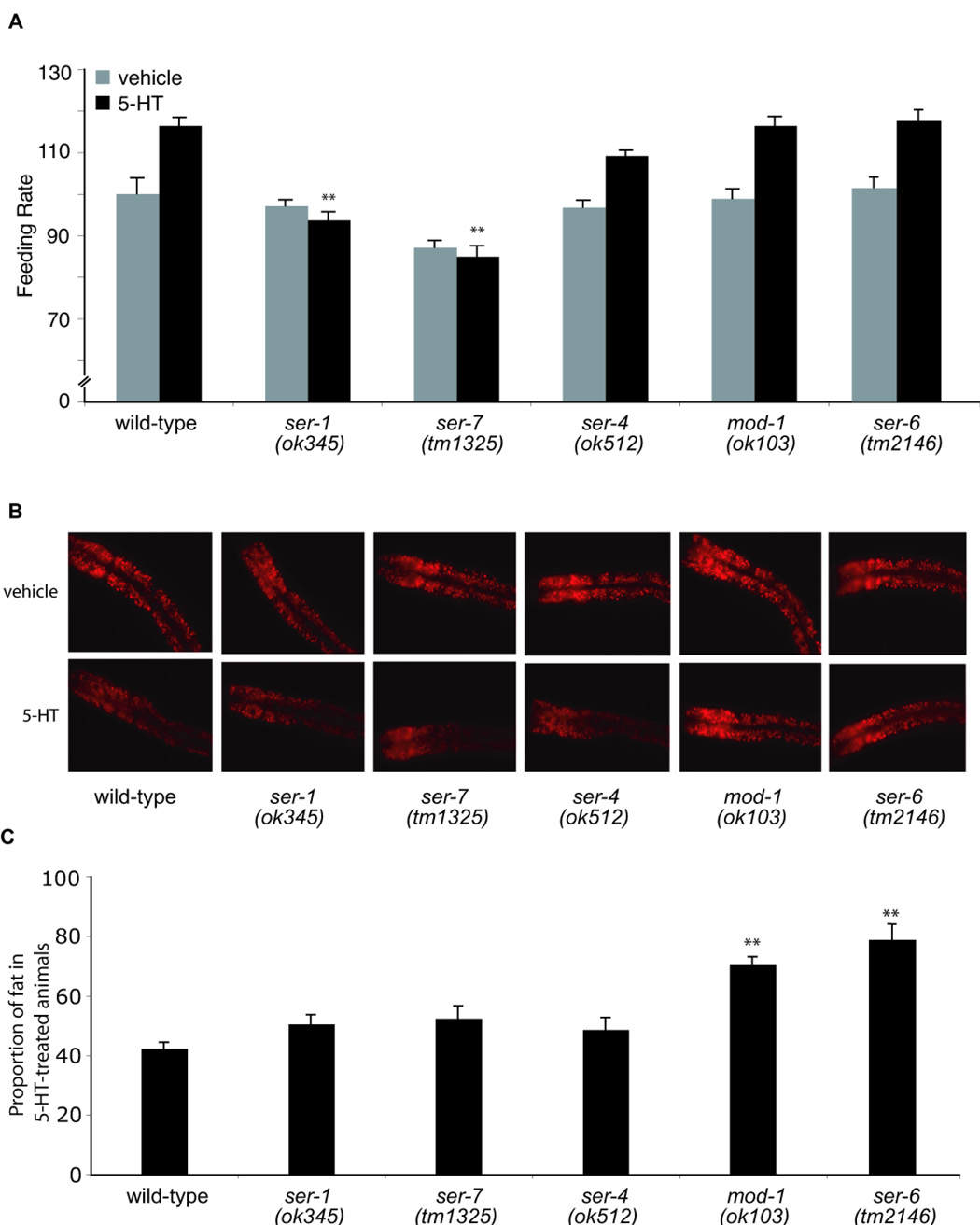
(A) Feeding rates were measured in wild-type controls and indicated mutants (n=10) treated with either vehicle (gray bars) or 5mM 5-HT (black bars). Data are expressed as a percentage of vehicle-treated wild-type animals (**, p<0.005, when compared to 5HT-treated wild-type animals). (B) Representative images of Nile Red-stained animals treated with vehicle (top row) or 5mM 5-HT (bottom row). The anterior end of the animals is oriented towards the left. (C) The proportion of fat remaining in 5-HT-treated relative to vehicle-treated animals for each genotype (n=8). Nile Red fluorescence intensities are reported in Table 1. mod-1(ok103) and ser-6(tm2146) animals retained a significantly greater proportion of their fat upon 5-HT treatment when compared to wild-type (**, p<0.005).
Other manipulations to increase endogenous concentrations of synaptic 5-HT also reduced fat content. Administration of fluoxetine, a 5-HT-specific re-uptake inhibitor (Messing et al., 1975), elicited fat reduction similar in magnitude to exogenous 5-HT treatment (Figure 1) while animals deficient in mod-5, a 5-HT re-uptake transporter (Ranganathan et al., 2001), displayed reduced fat levels relative to wild-type animals (Figure 1). While mod-5 mutants were further susceptible to the fat-reducing effects of exogenously administered 5-HT, consistent with previous identification of mod-5 as a target of fluoxetine (Ranganathan et al., 2001), they were resistant to further fat reduction by fluoxetine treatment (Figure 1A,B). Thus, despite increasing feeding rate, excess 5-HT signaling from exogenous and endogenous sources reduces fat content.
Distinct receptors mediate serotonergic regulation of fat, feeding, and egg-laying
To determine the relationships between various 5-HT-regulated processes, we examined the fat content of animals defective in various 5-HT-modulated behaviors. ser-1 and ser-7, which encode two serotonergic G protein-coupled receptors (GPCRs), are required for the feeding-rate- and egg-laying rate-increases of 5-HT-treated animals (Hobson et al., 2006; Xiao et al., 2006). ser-1(ok345) and ser-7(tm1325) mutants displayed wild-type fat content, were resistant to 5-HT-induced feeding increase but remained subject to fat reduction by exogenous 5-HT to an extent indistinguishable from wild-type animals (Figure 2). Thus, receptors that mediate the serotonergic increase in feeding rate were distinct from pathways mediating serotonergic fat reduction.
5-HT modifies of the physiological properties of pharyngeal muscles leading to faster contraction-relaxation cycles thereby affecting food intake (Niacaris and Avery, 2003). These cycles are regulated by excitatory acetylcholinergic (McKay et al., 2004) and inhibitory glutamatergic signals (Avery, 1993). eat-2 and eat-18, encoding a pharyngeal muscle-localized nicotinic acetylcholine receptor and a predicted transmembrane protein, and avr-15, encoding a glutamate-gated chloride channel, are required for nicotinic and glutamatergic regulations of pharyngeal muscle, respectively (Dent et al., 1997; McKay et al., 2004). Without 5-HT treatment, avr-15(ad1051) mutants had wild-type fat content while eat-2(ad1113) and eat-18(ad1110) animals showed dramatically diminished fat levels (Figure S2). The fat content of all three mutants was further diminished by 5-HT (Figure S2), demonstrating that 5-HT exacerbated the reduced fat of pumping-defective animals.
As with feeding, we found that the serotonergic modulation of egg-laying was molecularly independent of serotonergic fat regulation. In addition to ser-1 and ser-7, egl-30, egl-8, and egl-19 which encode Gαq, PLC-β and an L-type calcium channel α-subunit, respectively, are required for serotonergic stimulation of egg-laying (Schafer, 2006). Egg-laying-defective mutants egl-30(js126), egl-8(n488) and egl-19(ad1015) (Trent et al., 1983) had nearly wild-type fat and were fully susceptible to the fat-reducing effects of 5-HT (Figure S2). Similar results were obtained for the egg-laying-constitutive mutants unc-2(e55), and unc-36(e251), which encode subunits of an L-type voltage-gated calcium channel required for adaptation to 5-HT, wild-type feeding rate and movement (Schafer, 2006; Schafer and Kenyon, 1995) (Figure S2) and for animals mutant in ser-4, which encodes a serotonergic GPCR implicated in egg-laying (Dempsey et al., 2005) (Figure 2).
Based on sequence similarity to mammalian receptors, the C. elegans genome encodes at least sixteen putative biogenic amine GPCRs (Bargmann, 1998; Chase and Koelle, 2007) as well as MOD-1, a 5-HT-gated chloride channel required for the reduction in locomotor rate when food deprived animals re-encounter food (Ranganathan et al., 2000). In addition to serotonergic SER-1, SER-4, and SER-7 receptors, functional and pharmacological analyses have placed a subset of the other thirteen GPCRs in dopaminergic, octopaminergic, and tyraminergic signaling pathways (Chase and Koelle, 2007) (Table S1). We examined available loss-of-function mutants in twelve of the thirteen putative biogenic amine GPCRs (Table S1) as well as the serotonergic chloride channel mod-1 for their potential roles in 5-HT-induced fat reduction. Only loss-of-functions mutations in mod-1 and the GPCR encoded by Y54G2A.35/ser-6 partially abrogated 5-HT-induced fat reduction (Figure 2). By contrast, egg-laying and feeding rates of mod-1(ok103) and ser-6(tm2146) animals were indistinguishable from wild-type animals with or without 5-HT treatment (Dempsey et al., 2005) (Figure 2A and data not shown).
Without 5-HT treatment, mod-1(ok103) animals had excess fat while fat levels of ser-6(tm2146) mutants were indistinguishable from wild-type (Figure 2). We confirmed the excess fat content of mod-1(ok103) mutants by TLC analysis (Figure S3A). Relative to their basal levels, wild-type, mod-1(ok103), and ser-6(tm2146) mutants retained ~ 40%, ~70% and ~80% of their fat upon 5-HT treatment, respectively (Figure 2). Thus, mod-1 and ser-6 mutants were proportionally less susceptible to the fat-reducing effects of 5-HT. To evaluate whether the role of mod-1 simply reflected increased basal fat content, we examined the effects of 5-HT on ~100 RNAi induced gene-inactivations that increased fat (Ashrafi et al., 2003). Despite increasing overall fat content, none of these RNAi-treatments blocked the fat-reducing effects of 5-HT (not shown). Together, these findings suggested that the MOD-1 channel and the SER-6 GPCR play specific roles in 5-HT induced fat reduction.
We found that a GFP-reporter fused to putative promoter sequence of ser-6 exclusively expressed in a subset of head and tail neurons (Figure S3B). GFP-reporter expression of mod-1 was previously reported in a few head, ventral cord and tail neurons (Ranganathan et al., 2000). Together, these results indicated that serotonergic regulation of fat is mediated through neurally-expressed receptors that are distinct from serotonergic receptors that modulate feeding and egg-laying behaviors of animals.
5-HT-mediated fat loss is independent of insulin signaling
As in mammals, insulin signaling is a major neuroendocrine regulator of energy balance in C. elegans (Nelson and Padgett, 2003). Moreover, several lines of evidence suggest a cross talk between C. elegans 5-HT and insulin signaling pathways. For instance, active signaling through the DAF-2 insulin receptor inactivates the FOXO transcription factor DAF-16 by inhibiting its translocation from the cytoplasm to the nucleus (Liang et al., 2006). Food deprivation, inactivation of insulin signaling and 5-HT deficiency promote DAF-16 nuclear localization (Liang et al., 2006). In turn, exogenous 5-HT shifts DAF-16 to the cytoplasm in a DAF-2 dependent manner (Liang et al., 2006). Although daf-2 mutation causes excess fat accumulation (Kimura et al., 1997), we found that these mutants remained susceptible to 5-HT-induced fat reduction (Figure S4). Similarly, daf-16 loss-of-function did not alter fat reducing effects of 5-HT (Figure S4). Thus, insulin signaling is not required for the fat-reducing effects of 5-HT.
Peripheral lipid oxidation pathways are required for 5-HT-induced fat reduction
Our results indicated a dissociation between serotonergic fat and feeding mechanisms. Fat-reducing effects of 5-HT could not be attributed to enhanced locomotory activity as increased 5-HT signaling suppresses C. elegans locomotory rate (Horvitz et al., 1982). Therefore, we investigated the contribution of metabolic pathways by assessing the requirement of each of ~100 metabolic genes, encoding various components of fatty acid β-oxidation, ketogenesis, triacylglycerol synthesis, and fatty acid elongation and desaturation (Table S2) to 5-HT mediated fat reduction. 5-HT-treated animals on control or most test RNAi clones only retained ~35% or less of their basal fat levels (data not shown). By contrast, each of nine gene-inactivations partially blocked the fat-reducing effect of 5-HT allowing for 60%–83% fat retention (Figure 3 and Table 1).
Figure 3. β-oxidation genes are required for the fat-reducing effect of 5-HT.
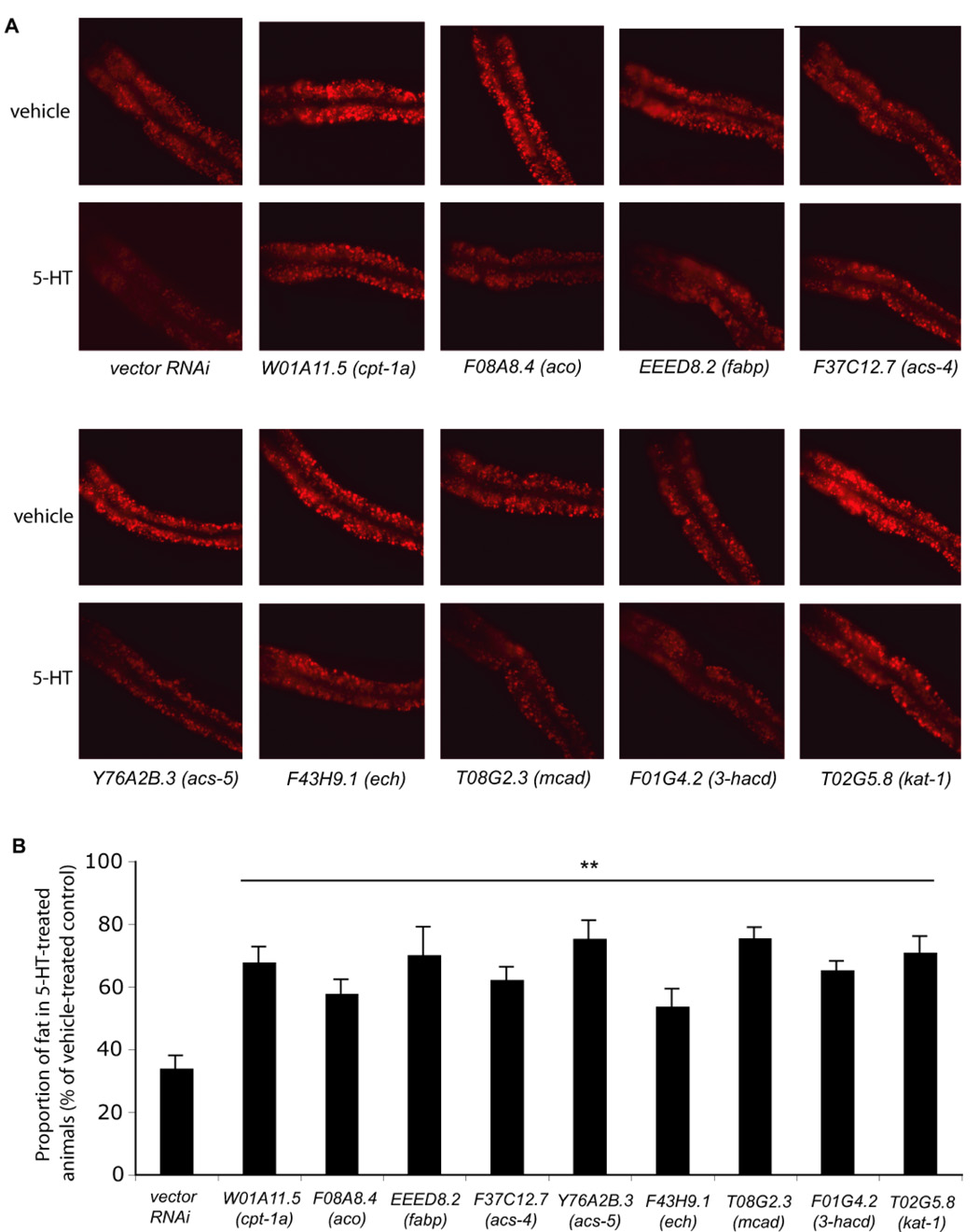
(A) Representative images of Nile Red-stained animals treated with the indicated RNAi clones exposed to either vehicle (top rows) or 5-HT (bottom rows). The anterior end of the animals is oriented towards the left. (B) The proportion of fat remaining in 5-HT-treated relative to vehicle-treated animals on HT115 bacteria exposed to each indicated RNAi (n=8). Nile Red fluorescence intensities are reported in Table 1. (**, p<0.005, when compared to wild type on vector control RNAi).
Table 1.
Effects of various gene inactivations on fat content as assessed by Nile Red fluorescence.
| Gene ID | Strain or Reciprocal Blast Match | Nile Red Fluorescencea | Subcellular | Tissue | |
|---|---|---|---|---|---|
| Vehicle | 5-HT | Localization | Localization | ||
| Wild-type (N2)* | 100.0 ± 6.0 | 42.3 ± 5.1 | |||
| F59C12.2 | ser-1(ok345)* | 95.2 ± 12.1 | 44.8 ± 5.0 | Plasma membrane | Pharyngeal Muscle, Vulvag |
| C09B7.1 | ser-7(tm1325) * | 103.3 ± 9.0 | 45.6 ± 9.3 | Plasma membrane | Interneuronsg |
| Y22D77AR.13 | ser-4(ok512) * | 110.0 ± 9.9 | 45.6 ± 8.9 | Plasma membrane | Pharyngeal Neurons, Vulvag |
| K06C4.6 | mod-1(ok103) * | 128.1 ± 11.4 | 86.8 ± 4.5 | Plasma membrane | Interneuronsh |
| Y54G2A.35 | ser-6(tm2146)* | 98.0 ± 6.9 | 70.0 ± 3.1 | Plasma membrane | Sensory Neurons, Interneuronsf |
| R13H8.1 | daf-16(mgDf47) * | 90.5 ± 4.1 | 30.6 ± 4.8 | Cytoplasmic/Nucleare | Ubiquitouse |
| vector RNAi# | 100.0 ± 7.6 | 34.0 ± 3.7 | |||
| W01A11.5 | Carnitine Palmitoyl Transferase-1a# | 138.3 ± 8.3 | 93.7 ± 7.8 | Mitochondrialb | epidermisf |
| F08A8.4 | Acyl CoA Oxidase# | 115.6 ± 7.5 | 68.5 ± 6.0 | Peroxisomalb | Intestinef |
| EEED8.2 | Fatty Acid Binding Protein# | 94.5 ± 9.2 | 66.3 ± 6.9 | Cytoplasmicb | epidermis, Intestinef |
| F37C12.7 | Acyl CoA Synthase-4# | 99.5 ± 5.5 | 82.5 ± 5.0 | - | Intestine, Pharyngeal Musclef |
| Y76A2B.3 | Acyl CoA Synthase-5# | 113.3 ± 8.1 | 85.4 ± 6.4 | Mitochondrialb | epidermis, Intestinef |
| T08G2.3 | Mitochondrial Acyl CoA Dehydrogenase# | 113.0 ± 5.6 | 81.7 ± 6.6 | Mitochondrialb | epidermis, Intestinef |
| F43H9.1 | Enoyl CoA Hydratase# | 103.2 ± 5.6 | 78.5 ± 4.4 | Peroxisomalb | Intestinef |
| F01G4.2 | 3'-Hydroxy Acyl CoA Dehydrogenase# | 90.2 ± 9.4 | 58.9 ± 2.9 | Mitochondrial | epidermis, Intestinef |
| T02G5.8 | Acetoacyl CoAThiolase (kat-1) # | 158.3 ± 7.8 | 112.2 ± 8.7 | Mitochondrialc | Intestine, Body Wall Musclec |
Wild-type and mutant strains were grown on OP50 bacteria.
Wild-type (N2) grown on indicated RNAi clones produced by HT115 bacteria.
5-HT was approximately 10% more efficacious on HT115 than OP50. All comparisons were made to appropriate control conditions. All values are means ± s.e.m. For each condition, n=8 animals.
Predicted using TargetP and PSORT as described (Van Gilst et al., 2005).
Mukhopadhyay et al., 2006.
This study.
Six of these RNAi clones targeted genes predicted to encode conserved components of mitochondrial and peroxisomal β-oxidation pathways. These were an acyl-CoA oxidase (F08A8.4/aco) and an enoyl-CoA hydratase (F43H9.1/ech), which catalyze the first two steps of peroxisomal β-oxidation (Salway, 1999); a carnitine palmitoyl transferase-1a (W01A11.5/cpt-1a), required for transport of fatty acyl-CoAs into mitochondria, a medium-chain acyl-CoA dehydrogenase (T08G2.3/mcad), a 3’-hydroxy-acyl-CoA dehydrogenase (F01G4.2/3’OH-acd), and an acetoacetyl-CoA thiolase (T02G5.8/kat-1), all of which are mitochondrial components of fatty acyl-CoA breakdown (Salway, 1999) (Figure S5A). Additionally, this screen identified a fatty-acid binding protein (EEED8.2/fabp) and two acyl-CoA synthases, F37C12.7/acs-4 and Y76A2B.3/acs-5, homologous to mammalian acs-4 and acs-5, respectively. ACS proteins catalyze the attachment of co-enzyme A to fatty acids activating them for breakdown or synthesis while FABPs facilitate intracellular transport of fatty acids (Salway, 1999). Among these nine genes, we selected Y76A2B.3/acs-5 and confirmed by Sudan Black staining that this gene inactivation suppressed 5-HT-induced fat reduction (Figure S6A–F). Together, these results suggested that the flux of stored fats through β-oxidation pathways partially accounted for the fat reduction in 5-HT-treated animals.
For each of the nine genes identified, the C. elegans genome encodes multiple family members, all of which were included in our RNAi screen (Table S2). While we do not know the efficacy of RNAi for all clones tested, our findings suggested that the fat-reducing effects of 5-HT were dependent on specific family members. In support of this assertion we found that RNAi inactivations/loss-of-function mutations in some of the other family members caused dramatic increases in the fat content without blocking the fat-reducing effects of 5-HT (not shown).
To determine sites of function of these metabolic genes, we generated reporter fusions utilizing 2–3 kb of each gene’s predicted promoter sequence fused to GFP. These transcriptional reporters primarily showed expression in the intestine and in the skin-like epidermis, two major sites of C. elegans fat storage (Figure 4A–H and Table 1). These data were consistent with a previous report of kat-1/T02G5.8 thiolase expression in intestinal and muscle tissues (Mak et al., 2006). The predicted peroxisomal F08A8.4/aco and F43H9.1/ech were most strongly expressed in intestinal cells while the predicted mitochondrial β-oxidation genes showed expression in both the epidermis and the intestine.
Figure 4. Expression patterns of β-oxidation genes.
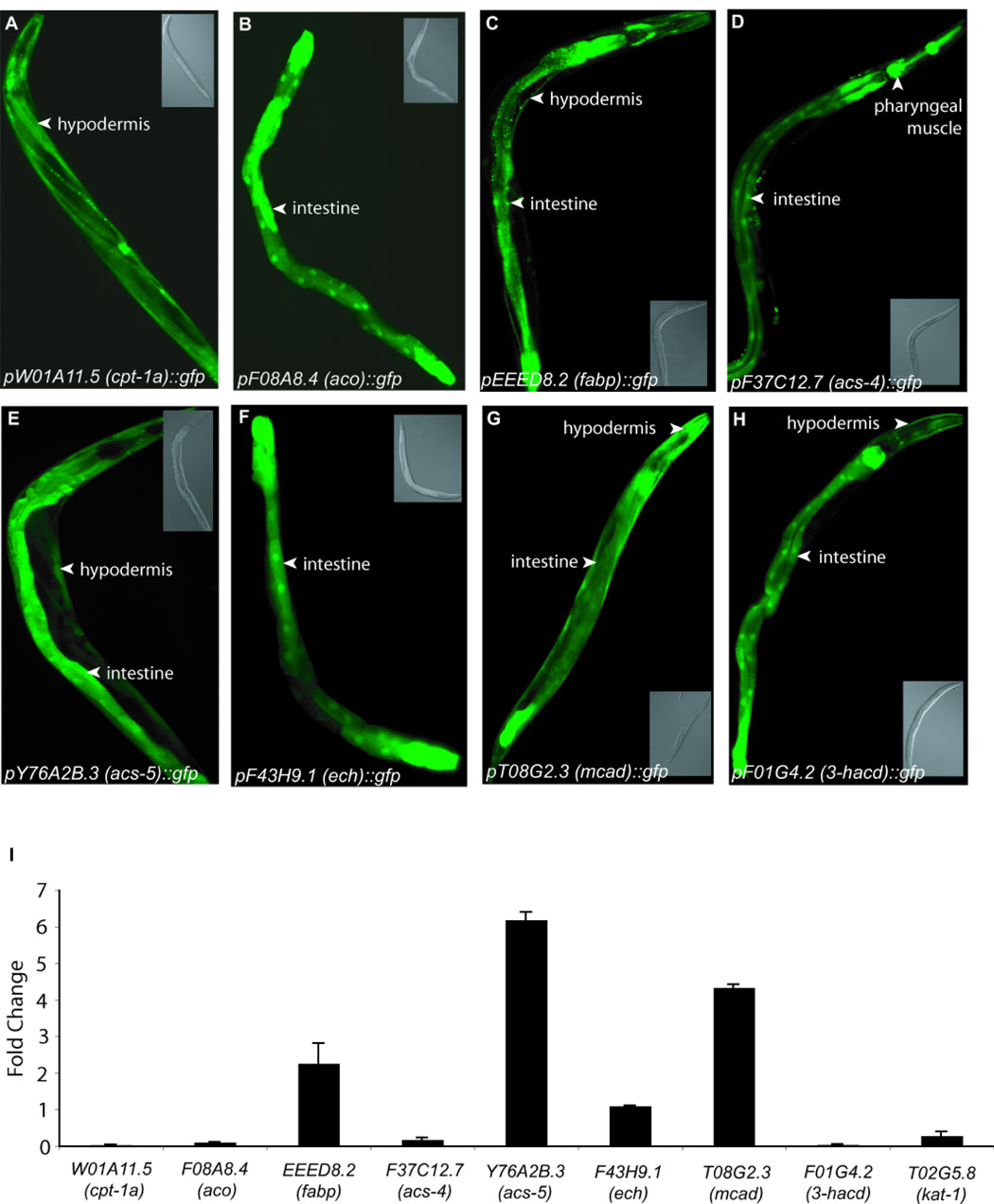
(A–H) Representative images of transgenic animals expressing GFP-reporter fusions for promoters of indicated genes. Inset panels show corresponding DIC images. (I) Change in transcript levels of indicated metabolic genes upon 5HT treatment in wild-type animals as determined by qRT-PCR. The data are reported as the average of two independent cDNA preparations ± s.e.m.
To further evaluate the roles of the nine metabolic genes, we assessed their transcriptional responses to 5-HT by quantitative real-time polymerase chain reaction (qRT-PCR). 5-HT treatment elicited transcriptional upregulation in several of the nine genes including 4–6 fold increases in Y76A2B.3/acs-5 and T08G2.3/mcad (Figure 4I). These findings indicated that neural 5-HT signaling initiates a signaling cascade to ultimately promote fat utilization via select lipid oxidation pathways in the periphery.
5-HT treatment increases energy expenditure fueled by fat oxidation
One explanation for increased activation of lipid oxidation pathways may be an increased demand for energy expenditure in 5-HT-treated animals. To assess this possibility, we designed and validated an assay for oxygen consumption as a measure of total body energy expenditure (Figure S5B–D). Oxygen consumption in 5-HT-treated animals was increased by ~15% relative to vehicle-treated animals while it was reduced by ~30% in 5-HT-deficient tph-1(mg280) mutants compared to wild-type animals (Figure 5A and Figure S5D). Without 5-HT, animals exposed to control or each of the nine metabolic RNAi clones displayed similar oxygen consumption rates as wild-type animals (Figure 5A). By contrast, inactivation of each of F37C12.7/acs-4, Y76A2B.3/acs-5, W01A11.5/cpt-1a and F08A8.4/aco fully suppressed the increased oxygen consumption of 5-HT-treated animals (Figure 5A). While inactivations of each of EEED8.2/fabp, T08G2.3/mcad, F43H9.1/ech, F01G4.2/acd, and T02G5.8/kat-1 partially blocked 5-HT induced fat reduction, they had no effects on increased oxygen consumption rates of 5-HT treated animals (Figure 3 and Figure 5A).
Figure 5. Effects of β-oxidation genes on oxygen consumption and feeding rate.
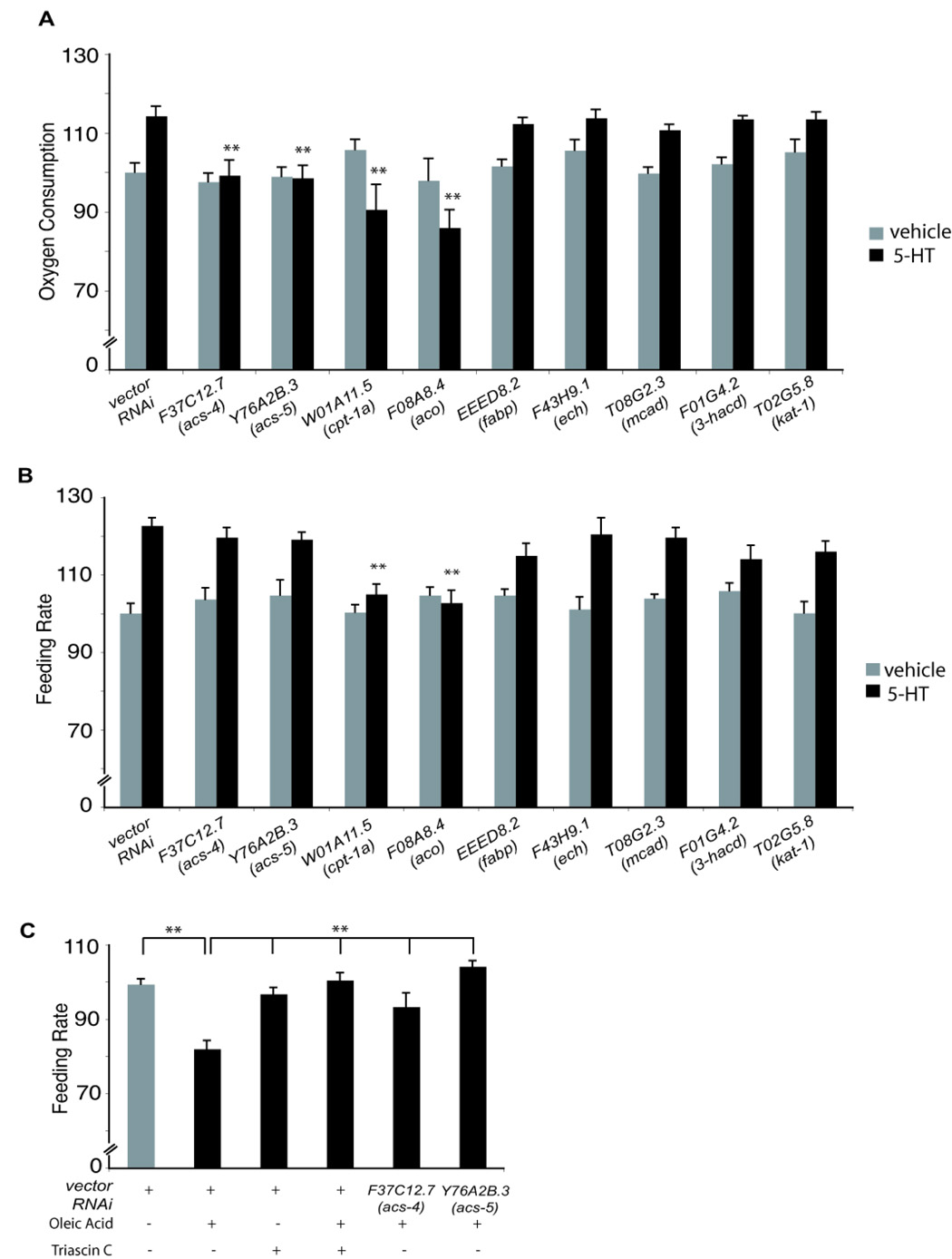
(A) Oxygen consumption measurements (n=800 animals per condition) with or without 5-HT for vector and RNAi-treated animals. The data are expressed as a percentage of wild-type vehicle-treated animals, (**, p<0.005 when compared to 5-HT-treated animals on vector control). (B) Effects of 5-HT on feeding rate (n=10 animals per condition) (**, p<0.005 when compared to 5-HT-treated animals on vector control RNAi). For panels A and B, gray bars represent vehicle-treated animals and black bars, 5-HT-treated animals. The data are expressed as a percentage of wild-type vehicle-treated animals. (C) Effects of dietary oleic acid, Triascin C and inactivations of acs genes on feeding rate (**, p<0.005 when comparing the indicated treatments).
Given that ACS proteins activate fatty acids into acyl-CoA moieties that are subsequently imported into peroxisomes and mitochondria by actions of ACO and CPT-1a proteins (see Figure S5A) (Salway, 1999), our findings suggested that flux of fats through β-oxidation pathways is a principal mechanism for meeting increased energy expenditure of 5-HT treated animals. However, once fatty acyl-CoAs gain entry into peroxisomal or mitochondrial oxidation pathways, blockage of their breakdown uncouples fat reduction and energy expenditure perhaps through activation of compensatory mechanisms of ATP generation through glycolysis or protein degradation. Indeed, pharmacological or genetic inactivation of mammalian mitochondrial fatty-acid β-oxidation pathways elicit such compensatory increases in glucose oxidation (Adhikari et al., 2007) and oxygen consumption (Hutter et al., 1985).
Metabolic intermediates regulate feeding rate
Inactivations of either F08A8.4/aco or W01A11.5/cpt-1a not only abrogated 5-HT induced increase in oxygen consumption but reduced oxygen consumption below the levels observed when animals were treated with RNAi alone (Figure 5A). In the mammalian hypothalamus, build-up of long chain fatty acyl-CoAs and inactivation of CPT-1a, which leads to acyl-CoA accumulation, inhibit feeding (Lam et al., 2005). Since accumulation of acyl-CoA moieties correlate with plentiful energy supplies, their effects on food intake have been interpreted as homeostatic satiety signals (Lam et al., 2005). Similarly, we found that inactivation of F08A8.4/aco or W01A11.5/cpt-1a but none of the other metabolic genes, abrogated the increased feeding rate of 5-HT-treated animals (Figure 5B).
Given the mammalian precedents, we hypothesized that the normalized feeding rates of 5-HT-treated animals in which either F08A8.4/aco or W01A11.5/cpt-1a had been inactivated reflected accumulation of acyl-CoA moieties. Direct biochemical measurements of acyl-CoA moieties from C. elegans extracts are challenging since these moieties are produced in small quantities and in only a subset of cells. To provide evidence for our hypothesis, we tested whether excess dietary fatty acids affected feeding rate of well-fed wild-type animals. Exposure to 1µM oleic acid (C18:1n-9) for 30 minutes resulted in ~20% reduction in feeding rate (Figure 5C) but no other obvious concomitant behavioral and morphological changes. This reduction in feeding was abrogated by a 30-minute pre-treatment with 1nM Triascin C, a pharmacological inhibitor of ACS enzymes, and also by RNAi-mediated inactivation of either F37C12.7/acs-4 or Y76A2B.3/acs-5 (Figure 5C). At the indicated concentration, Triascin C administration alone had no effects on feeding rate (Figure 5C) supporting the notion that conversion of dietary oleic acid to its CoA derivatives causes decreased feeding rate.
These results revealed that the simultaneous mobilization of stored fatty acids via serotonergic signals and the prevention of their breakdown via inactivation of genes that function as gatekeepers of β-oxidation, elicit a homeostatic reduction in feeding rate. Thus, as in mammals, acyl-CoA moieties or their metabolic derivates likely function as feeding regulatory signals in C. elegans.
mod-1 and ser-6 link neural serotonergic signals to peripheral β-oxidation pathways
To determine pathway relationships between neural and peripheral components required for serotonergic fat reduction, we performed pairwise epistasis analyses between several pathway components. Upon 5-HT treatment, mod-1(ok103);ser-6(tm2146) double mutants retained a greater proportion of their fat compared to either single mutant alone (Figure 6A, B). Moreover, RNAi inactivation of Y76A2B.3/acs-5 and F08A8.4/aco in ser-6(tm2146) did not alter basal fat levels or fat retention of these mutants when treated with 5-HT. By contrast, inactivations of either Y76A2B.3/acs-5 or F08A8.4/aco improved the proportion of fat retained in 5-HT-treated mod-1 mutants without altering the fat content of untreated animals (Figure 6C and Table S3). These findings suggested that mod-1 and peripherally-expressed metabolic gene define a pathway of serotonergic fat regulation that is mechanistically distinct from that defined by ser-6.
Figure 6. ser-6 and mod-1 mediate serotonergic fat regulation through distinct mechanisms.
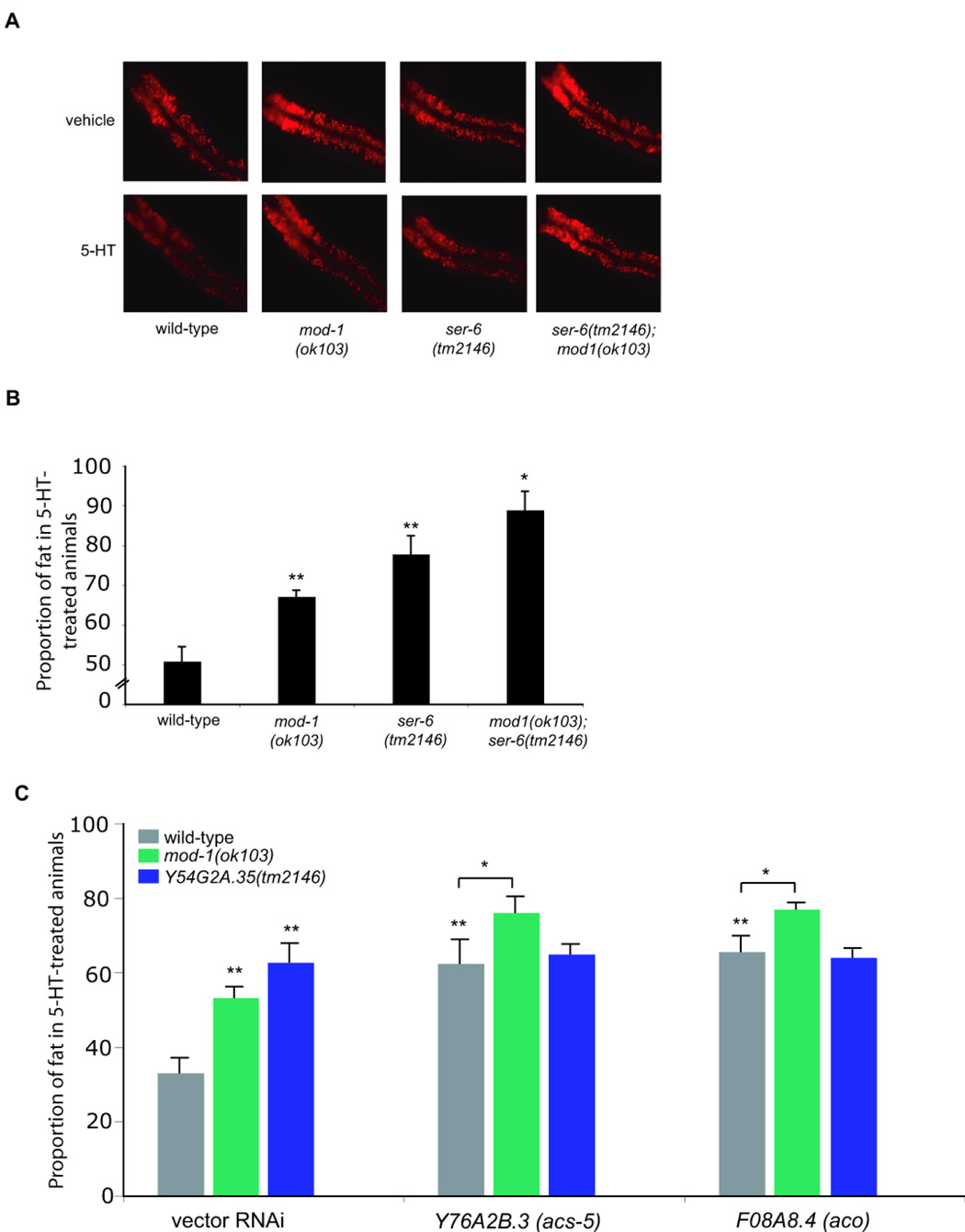
(A) Representative images of Nile Red-stained animals exposed to either vehicle (top row) or 5-HT (bottom row). The anterior end of the animals is oriented towards the left. (B) mod-1(ok103); ser-6(tm2146) double mutants block serotonergic fat reduction to a greater degree than either single mutant alone (n=8, and *, p<0.05). (C) RNAi inactivations of Y76A2B.3/acs-5 and F08A8.4/aco in mod-1(ok103) but not in ser-6(tm2146) animals cause a further block in serotonergic fat reduction (n=8, and *, p<0.05). Nile Red intensity measurements used to generate graphs (B) and (C) are reported in Table S3.
We examined the transcriptional profiles of the nine peripheral lipid oxidation genes by qRT-PCR in mod-1(ok103) and ser-6(tm2146) with or without exogenous 5-HT. Inactivation of mod-1 did not alter the expression levels of these metabolic genes in the absence of 5-HT treatment but abrogated increased transcriptional changes elicited by 5-HT treatment (Figure 4I and Figure S6G). In the absence of 5-HT, inactivation of ser-6 resulted in transcriptional downregulation of a subset of peripheral lipid oxidation genes (Figure S6G). Although 5-HT treatment of ser-6 mutants caused transcriptional upregulation of peripheral lipid oxidation genes, expression levels of these genes remained below that observed in 5-HT treated wild type animals, and in some cases, even below that of untreated animals (Figure S6G). These findings further supported the notion that ser-6 and mod-1 define distinct mechanisms mediating serotonergic fat regulation.
Together, these analyses suggested that peripheral lipid oxidation genes remain effectively inactivated in ser-6 mutants such that their inactivations by RNAi does not further improve fat retention in 5-HT treated ser-6 animals. By contrast, since these genes display wild-type basal expression levels in mod-1 animals, they contribute to 5-HT induced fat reduction because mod-1 is required for 5-HT induced upregulation of these genes.
Discussion
In this report, we delineate a genetic and molecular framework for C. elegans serotonergic fat and feeding regulation. Our findings suggest that serotonergic regulation of fat is not a consequence of other 5-HT-regulated behaviors but rather a distinct metabolic response initiated in the nervous system. We identified the 5-HT-gated chloride channel encoded by mod-1 and the GPCR encoded by ser-6, as components of signaling mechanisms linking neural 5-HT to peripheral mobilization of fat stores. This feeding-independent neural fat regulatory pathway ultimately causes fat reduction by promoting lipid oxidation via select β-oxidation genes in the periphery. In turn, we found evidence that C. elegans modulate their feeding rate in response to peripheral accumulation of acyl-CoAs or their derivates, which are postulated to be gauges of fuel availability in mammals (Lam et al., 2005). Thus, the C. elegans nervous system can regulate fat content independent of feeding behavior but this behavior is subject to homeostatic regulation by internal cues of energy availability.
In both C. elegans and mammals, excess 5-HT signaling is associated with fat loss while reduced signaling is associated with fat accumulation. Unlike mammals, serotonergic effects on C. elegans fat are inversely correlated to its effects on feeding. Interestingly, several lines of evidence from mammalian studies suggest that changes in feeding alone cannot account for the observed effects of 5-HT deficiency or excess on fat content. First, while 5-HT2c receptor mutant mice are chronically hyperphagic and hyperactive, they do not develop obesity until 5–6 months of age (Nonogaki et al., 2003). This obesity develops without a further increase in hyperphagia and despite increased hyperactivity. Second, in both rodents and humans, chronic administration of serotonergic compounds promotes fat reduction that can be maintained for extended periods of time as long as drug treatment is continued, however, the reported feeding reduction, while significant upon initiation of drug treatment, is only transient and quickly rebounds to pretreatment levels despite continued drug treatment and maintenance of reduced weight (Fernstrom and Choi, 2008; Vickers et al., 2000). Upon discontinuation of drug treatment, weight gain resumes without further increases in feeding (Choi et al., 2002). Third, the central melanocortin pathway, a principal regulator of fat and feeding (Ellacott and Cone, 2006), has been shown to mediate the anorectic effects of increased central 5-HT signaling (Heisler et al., 2003). Selective expression of the melanocortin-4 receptor (MC4R) in subpopulations of neurons in mc4r null mice revealed that the effect of MC4R on food intake is dissociable from its effect on energy expenditure (Balthasar et al., 2005). Finally, similar to C. elegans, changes in central serotonergic signaling also elicit changes in energy expenditure in both rodents and humans (Even and Nicolaidis, 1986; Lam and Heisler, 2007; Le Feuvre et al., 1991; Rothwell and Stock, 1987). Thus, in mammals as in C. elegans, 5-HT regulation of fat may be largely independent of feeding, highlighting the ancient origins of 5-HT as a regulator of energy balance and that the basic organization of central vertebrate serotonergic nervous systems was already established in their invertebrate ancestors (Hay-Schmidt, 2000).
Since C. elegans directly match their feeding rates to increasing and decreasing food concentrations, 5-HT-induced effects on fat and feeding in C. elegans are consistent with the role of this neuromodulator as a sensory gauge of nutrient availability. As in animals preparing for the hibernating dauer state which is characterized by build-up of fat reservoirs, the excess fat of 5-HT-deficient animals is a response consistent with the perception of depleting food availability and a shift in metabolism that favors energy conservation and directs nutrients into fat reservoirs. Accordingly, 5-HT-induced fat reduction is consistent with the re-activation of energy-utilizing pathways that accompany the resumption of growth and reproduction once food is available. Moreover, we found that 5-HT treatment caused increased feeding rate even when animals were already well-fed. Since C. elegans can distinguish food quality in a 5-HT-dependent manner (Shtonda and Avery, 2006) and their feeding rate can be modulated based on previous experience of starvation (Avery and Horvitz, 1990), we postulate that rather than a simple switch indicating food presence, 5-HT signaling functions as a sophisticated gauge whose relative abundance could reflect various food-related contexts and experiences. The full range of environmental and physiological signals that could operate through 5-HT signaling are not yet known.
Given the contributions of the serotonergic pathway to energy balance across phylogeny, we speculate that human counterparts of feeding-independent fat regulatory genes identified in our study may similarly regulate energy balance.
Experimental Procedures
C. elegans maintenance and strain constructions
Nematodes were cultured on OP50 bacterial lawns on nematode growth media (NGM) plates at 20°C. N2 Bristol was used as the wild-type reference strain. The following out-crossed strains were analyzed: eat-18(ad1110)I, egl-30(js126)I, daf-16(mgDf47)I, mod-5(n3314)I, eat-2(ad1113)II, tph-1(mg280)II, ser-4(ok512)III, unc-36(e251)III, daf-2(e1370)III, egl-19(ad1015)IV, ser-6(tm2146)IV, mod-1(ok103)V, avr-15(ad1051)V, egl-8(n488)V, ser-1(ok345)X, ser-7(tm1325)X and unc-2(e55)X. GPCR mutants are listed in Table S2. When generating double mutants, desired genotypes arising from a cross were determined by PCR analysis. For all experiments, strains were synchronized by hypochlorite treatment of gravid adults.
5-HT, fluoxetine, oleic acid and Triascin C treatment
5-HT hydrochloride (Sigma, St. Louis) powder was dissolved in 0.1M HCl to a concentration of 0.1M, and added to NGM plates seeded with either OP50 or vector RNAi (HT115) to a final plate concentration of 5mM, or 10mM (only when noted). Plates were allowed to equilibrate overnight and used the next day. For vehicle-treated controls 0.1M HCl was added to plates to a final concentration of 5mM. This HCl treatment did not affect growth, viability or fecundity. Synchronized L1-stage animals were plated on vehicle or 5-HT-treated plates and fat content, feeding rate and oxygen consumption were measured in young adults. We noted that 5-HT was more efficacious on HT115 bacteria (60–70% fat reduction) compared to OP50 bacteria (50–60% fat reduction). Fluoxetine (Sigma, St. Louis) was used at a final concentration of 20 µg/ml. Oleic acid (Sigma, St. Louis) was solubilized in 45% (w/v in dH2O) 2-hydroxypropyl)-β-cyclodextrin to 1M, and then added to NGM/OP50 plates with PBS to a final concentration of 1µM. Triascin C (Sigma, St. Louis) was solubilized in DMSO and used at 1nM on NGM/OP50 plates. In both cases, L4-stage animals were transferred to plates containing the appropriate vehicle or drug for the designated time period before counting feeding rate. Chronic treatment with Triascin C (1µM) resulted in larval arrest.
Nile Red Staining, Image Acquisition and Quantitation
Nile Red staining of nematodes was conducted as previously described (Ashrafi et al., 2003). For routine examination and screening, Nile Red fluorescence was visualized using a Zeiss SV11 M2-bio microscope equipped with a Texas Red filter (excitation, 596nm; emission, 613nm). For image analyses and quantitation, images were captured using a digital CCD camera (Hamamatsu C4742-95-12ER) attached to a Zeiss Axioplan II. All Nile Red images were acquired using identical settings and exposure times to allow direct comparisons. Images used for quantitation were captured such that pixel intensities were linear with exposure time and below saturation. Reported Nile Red intensities of the first two pairs of intestinal cells were quantified using the Image J software (NIH). This region accounts for most of the variation seen between all tested strains and conditions. For all Nile Red quantifications, 8–10 worms from each genotype were randomly selected under the dissecting microscope (without fluorescence) and imaged as described. Each experiment was repeated at least three times and all reported results were consistent in all repeats.
RNAi
HT115 bacteria containing each RNAi vector (see Table S1) were tested as described (Ashrafi et al., 2003) with the following modification: after overnight growth, bacteria were pelleted and resuspended to 5X concentration, 0.1 ml of which was used for seeding each well of a 12-well plate containing 4ml NGM agar, 6 mM IPTG and 25 µg/ml carbenicillin. Next, Nile Red and 5-HT (or HCl “vehicle”) were added on top of each well and the plates were allowed to equilibrate overnight. 20–30 synchronized wild-type or mutant L1 animals were placed per well and incubated at 20°C to adulthood. Nile Red assessment was performed on gravid adult animals. For each RNAi experiment, HT115 (vector alone) and OP50 control wells were also included. All 5-HT suppression phenotypes were confirmed by multiple (5–10) additional rounds of testing on the selected clones, whose identities were confirmed by DNA sequencing. At the time of scoring Nile Red phenotypes, the identities of the target RNAi clones were unknown.
Sudan Black B staining
Staining was performed as previously described (Kimura et al., 1997) with the following modification: 5-HT-treated animals were marked with FITC and mixed in with vehicle-treated animals and stained in the same tube to minimize staining variation. Fat decrease in 5-HT-treated animals was scored blind and then matched to genotype and conditions using FITC staining.
Measurement of triacylglycerides
Synchronized L4-stage animals were washed off 6 cm plates using 0.9% NaCl and centrifuged gently to pellet worms, and washed twice in 0.9% NaCl. After incubation for 20 minutes at room temperature, nematodes were pelleted by centrifugation and resuspended in 500 µl H2O. 100 µl were removed for protein determination and total cellular lipids were extracted from 400 µl nematode suspension as described (Bligh and Dyer, 1959). For the quantification of triacylglycerols, extracts were applied to Silica Gel 60 plates (Merck) with the aid of a sample applicator (Linomat IV; CAMAG, Muttenz, Switzerland), and chromatograms were developed using the solvent system hexane/diethyl ether/acetic acid (70:30:1, v/v). Triacylglycerols were visualized by post-chromatographic staining by dipping plates for 6s into a developing reagent consisting of 0.63 g of MnCl2 · 4H2O, 60 ml of water, 60 ml of methanol, and 4 ml of concentrated sulfuric acid, briefly dried, and heated at 100°C for 30 min. Quantification of triacylglycerols was carried out by densitometric scanning at 400 nm using a Typhoon Trio Variable Mode Scanner (Amersham Biosciences) with triolein (Sigma, St. Louis, USA) as a standard. The worm suspension removed for protein determination (100 µl) was adjusted to a final volume of 400 µl with H2O, and sonicated twice for 30 s (Branson, intensity level 20). Tris-HCl and SDS were added to the extracts to a final concentration of 40 mM and 0.1%, respectively, and incubated for one hour at 4°C. The protein concentration was determined using the Qubit assay system according to the manufacturer (Invitrogen, Carlsbad, Califonia, USA). All measurements were done in triplicate per strain and each strain was subject to at least two independent rounds of triglyceride measurement.
Feeding Rate Assay
Feeding rates were measured in gravid adults raised at 20°C on OP50 or RNAi bacteria. All measurements were done at room temperature (22°C). Feeding rate was measured for each animal by counting the rhythmic contractions of the pharyngeal bulb over a 10-second period under the M2Bio microscope (Zeiss). For each genotype, 10 animals were counted per condition and the experiment repeated at least three times.
Oxygen Consumption Assay
Oxygen consumption was measured in gravid adult animals washed off culture plates with S-basal and then washed once more with S-basal. For each genotype, 200 animals were deposited in a single well of a 96-well plate containing a biosensor film whose fluorescence intensity increases in proportion to the amount of oxygen consumed (BD Biosciences). Oxygen consumption was measured over a three-hour period, which we determined empirically to be required for the biosensor film to reach equilibrium. The data reported are end-point measurements since they accurately reflect changes in oxygen concentration of the samples rather than changes due to equilibration of the biosensor film. Within a single experiment, each genotype was measured in quadruplicate, and each experiment was repeated 35 times. Fluorescence (excitation 485nm, emission 590nm with cutoff at 550nm) was measured using a Molecular Devices FlexStation™. The mitochondrial uncoupler Rotenone (Salway, 1999) (Sigma) decreased oxygen consumption in a dose-dependent manner and was used to validate the assay.
Quantitative RT-PCR
Total RNA extraction, cDNA preparation and qRT-PCR were performed as described (Van Gilst et al., 2005). Primer sequences used for the genes tested are available upon request. The data were standardized to GAPDH, which did not vary by more than one cT value in all conditions tested. For all conditions tested, two independent rounds of nematode growths followed by multiple cDNA preparations from each growth were performed to ensure the accuracy and consistency of the observed changes.
Generation of GFP-fusion reporter constructs and transgenic animals
GFP reporter constructs were generated using Gateway Technology (Invitrogen) reagents. The promoters for W01A11.5, F37C12.7, F43H9.1, F01G4.2, T02G5.8 and EEED8.2 were obtained from Open Biosystems. These promoters included an approximately 2 kb region upstream of the start site for each corresponding gene. For Y54G2A.35, Y76A2B.3 and T25G12.5, we PCR amplified approximately 2–3 kb upstream of each gene’s start site, typically where the next-upstream coding sequence was located. We tested two promoters for F08A8.4. A short 0.5 kb promoter and a longer 2 kb promoter beginning within the pen-ultimate exon of the upstream gene were both tested giving identical results. Promoters were cloned into the Gateway™ vector pDONR-P4-P1R (a gift from Marc Vidal) and sequenced to confirm their identity. In all cases, promoters were placed upstream of a GFP reporter with an unc-54 3’ UTR.
Purified plasmids were injected into the gonads of wild-type N2 animals to obtain stable transgenic lines. At least 10 independent transgenic lines were generated for each construct, and 20–30 animals from 3–5 lines were examined in detail to confirm the consistency of expression patterns. Images were captured using a digital CCD camera (Hamamatsu C4742-95-12ER) attached to a Zeiss Axioplan II microscope equipped with a FITC/GFP filter (emission 500–515 nm).
Statistical analyses
Student’s t-test was used to determine the significance of the data for feeding rate, fat content, and oxygen consumption assays.
Supplementary Material
Acknowledgments
S.S. was supported by the Ellison/AFAR Senior Postdoctoral Fellowship and by the K99/R00 grant from the NIH/NIDDK (DK-077427). This work was supported by research grants from the Sandler Opportunity Fund and the Richard and Susan Smith Family Foundation Pinnacle Program Project Award. Additional support was received from the Searle Scholar’s Award and a Burroughs Welcome Career Award to K.A. We are grateful to the Japanese Knockout Consortium (Tokyo Women's Medical University, Tokyo, Japan) and the Caenorhabditis Genetics Center (University of Minnesota, Twin Cities, USA) for strains, Dr. Bruce R. Conklin (The Gladstone Institutes, San Francisco) for use of the Flexstation™ and members of the Ashrafi laboratory for discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adhikari S, Erol E, Binas B. Increased glucose oxidation in H-FABP null soleus muscle is associated with defective triacylglycerol accumulation and mobilization, but not with the defect of exogenous fatty acid oxidation. Mol Cell Biochem. 2007;296:59–67. doi: 10.1007/s11010-006-9298-0. [DOI] [PubMed] [Google Scholar]
- Ashrafi K. Obesity and the Regulation of Fat Metabolism. In: A. V. a. M. Maricq SL, editor. Wormbook. 2006. http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268–272. doi: 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- Avery L. Motor neuron M3 controls pharyngeal muscle relaxation timing in Caenorhabditis elegans. J Exp Biol. 1993;175:283–297. doi: 10.1242/jeb.175.1.283. [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans. J Exp Zool. 1990;253:263–270. doi: 10.1002/jez.1402530305. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Chase DL, Koelle MR. Biogenic amine neurotransmitters in C. elegans. WormBook. 2007:1–15. doi: 10.1895/wormbook.1.132.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Jonak EM, Simpson L, Patil V, Fernstrom JD. Intermittent, chronic fenfluramine administration to rats repeatedly suppresses food intake despite substantial brain serotonin reductions. Brain Res. 2002;928:30–39. doi: 10.1016/s0006-8993(01)03330-3. [DOI] [PubMed] [Google Scholar]
- de Bono M, Bargmann CI. Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell. 1998;94:679–689. doi: 10.1016/s0092-8674(00)81609-8. [DOI] [PubMed] [Google Scholar]
- de Bono M, Maricq AV. Neuronal substrates of complex behaviors in C. elegans. Annu Rev Neurosci. 2005;28:451–501. doi: 10.1146/annurev.neuro.27.070203.144259. [DOI] [PubMed] [Google Scholar]
- Dempsey CM, Mackenzie SM, Gargus A, Blanco G, Sze JY. Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics. 2005;169:1425–1436. doi: 10.1534/genetics.104.032540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent JA, Davis MW, Avery L. avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. Embo J. 1997;16:5867–5879. doi: 10.1093/emboj/16.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Stevens R. Peripherally administered 5-hydroxytryptamine elicits the full behavioural sequence of satiety. Physiol Behav. 1991;50:1075–1077. doi: 10.1016/0031-9384(91)90441-p. [DOI] [PubMed] [Google Scholar]
- Ellacott KL, Cone RD. The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond B Biol Sci. 2006;361:1265–1274. doi: 10.1098/rstb.2006.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even P, Nicolaidis S. Metabolic mechanism of the anorectic and leptogenic effects of the serotonin agonist fenfluramine. Appetite. 1986 7 Suppl:141–163. doi: 10.1016/s0195-6663(86)80059-9. [DOI] [PubMed] [Google Scholar]
- Fernstrom JD, Choi S. The development of tolerance to drugs that suppress food intake. Pharmacol Ther. 2008;117:105–122. doi: 10.1016/j.pharmthera.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Hay-Schmidt A. The evolution of the serotonergic nervous system. Proc Biol Sci. 2000;267:1071–1079. doi: 10.1098/rspb.2000.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler LK, Cowley MA, Kishi T, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Zigman JM, et al. Central serotonin and melanocortin pathways regulating energy homeostasis. Ann N Y Acad Sci. 2003;994:169–174. doi: 10.1111/j.1749-6632.2003.tb03177.x. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, et al. Activation of central melanocortin pathways by fenfluramine. Science. 2002;297:609–611. doi: 10.1126/science.1072327. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Jobst EE, Sutton GM, Zhou L, Borok E, Thornton-Jones Z, Liu HY, Zigman JM, Balthasar N, Kishi T, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51:239–249. doi: 10.1016/j.neuron.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Hills T, Brockie PJ, Maricq AV. Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J Neurosci. 2004;24:1217–1225. doi: 10.1523/JNEUROSCI.1569-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson RJ, Hapiak VM, Xiao H, Buehrer KL, Komuniecki PR, Komuniecki RW. SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics. 2006;172:159–169. doi: 10.1534/genetics.105.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD. Serotonin and octopamine in the nematode Caenorhabditis elegans. Science. 1982;216:1012–1014. doi: 10.1126/science.6805073. [DOI] [PubMed] [Google Scholar]
- Hutter JF, Piper HM, Spieckerman PG. Effect of fatty acid oxidation on efficiency of energy production in rat heart. Am J Physiol. 1985;249:H723–H728. doi: 10.1152/ajpheart.1985.249.4.H723. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lam DD, Heisler LK. Serotonin and energy balance: molecular mechanisms and implications for type 2 diabetes. Expert Rev Mol Med. 2007;9:1–24. doi: 10.1017/S1462399407000245. [DOI] [PubMed] [Google Scholar]
- Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci. 2005;8:579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- Le Feuvre RA, Aisenthal L, Rothwell NJ. Involvement of Involvement of corticotrophin releasing factor (CRF) in the thermogenic and anorexic actions of serotonin (5-HT) and related compounds. Brain Res. 1991;555:245–250. doi: 10.1016/0006-8993(91)90348-y. [DOI] [PubMed] [Google Scholar]
- Liang B, Moussaif M, Kuan CJ, Gargus JJ, Sze JY. Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 2006;4:429–440. doi: 10.1016/j.cmet.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Loer CM, Kenyon CJ. Serotonin-deficient mutants and male mating behavior in the nematode Caenorhabditis elegans. J Neurosci. 1993;13:5407–5417. doi: 10.1523/JNEUROSCI.13-12-05407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak HY, Nelson LS, Basson M, Johnson CD, Ruvkun G. Polygenic control of Caenorhabditis elegans fat storage. Nat Genet. 2006;38:363–368. doi: 10.1038/ng1739. [DOI] [PubMed] [Google Scholar]
- McKay JP, Raizen DM, Gottschalk A, Schafer WR, Avery L. eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics. 2004;166:161–169. doi: 10.1534/genetics.166.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay RM, McKay JP, Avery L, Graff JM. C elegans: a model for exploring the genetics of fat storage. Dev Cell. 2003;4:131–142. doi: 10.1016/s1534-5807(02)00411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing RB, Phebus L, Fisher LA, Lytle LD. Analgesic effect of fluoxetine hydrochloride (Lilly 110140), a specific inhibitor of serotonin uptake. Psychopharmacol Commun. 1975;1:511–521. [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Nelson DW, Padgett RW. Insulin worms its way into the spotlight. Genes Dev. 2003;17:813–818. doi: 10.1101/gad.1090203. [DOI] [PubMed] [Google Scholar]
- Niacaris T, Avery L. Serotonin regulates repolarization of the C. elegans pharyngeal muscle. J Exp Biol. 2003;206:223–231. doi: 10.1242/jeb.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonogaki K, Abdallah L, Goulding EH, Bonasera SJ, Tecott LH. Hyperactivity and reduced energy cost of physical activity in serotonin 5-HT(2C) receptor mutant mice. Diabetes. 2003;52:315–320. doi: 10.2337/diabetes.52.2.315. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Cannon SC, Horvitz HR. MOD-1 is a serotonin-gated chloride channel that modulates locomotory behaviour in C. elegans. Nature. 2000;408:470–475. doi: 10.1038/35044083. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Sawin ER, Trent C, Horvitz HR. Mutations in the Caenorhabditis elegans serotonin reuptake transporter MOD-5 reveal serotonin-dependent and -independent activities of fluoxetine. J Neurosci. 2001;21:5871–5884. doi: 10.1523/JNEUROSCI.21-16-05871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. Effect of diet and fenfluramine on thermogenesis in the rat: possible involvement of serotonergic mechanisms. Int J Obes. 1987;11:319–324. [PubMed] [Google Scholar]
- Salway JG. Metabolism at a glance. 2nd edition edn. Blackwell Science; 1999. [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Schafer WF. Genetics of egg-laying in worms. Annu Rev Genet. 2006;40:487–509. doi: 10.1146/annurev.genet.40.110405.090527. [DOI] [PubMed] [Google Scholar]
- Schafer WR, Kenyon CJ. A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature. 1995;375:73–78. doi: 10.1038/375073a0. [DOI] [PubMed] [Google Scholar]
- Shtonda BB, Avery L. Dietary choice behavior in Caenorhabditis elegans. J Exp Biol. 2006;209:89–102. doi: 10.1242/jeb.01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue MF. Neuropharmacology of drugs affecting food intake. Pharmacol Ther. 1987;32:145–182. doi: 10.1016/0163-7258(87)90057-x. [DOI] [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- Tecott LH. Serotonin and the orchestration of energy balance. Cell Metab. 2007;6:352–361. doi: 10.1016/j.cmet.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 2005;3:e53. doi: 10.1371/journal.pbio.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers SP, Benwell KR, Porter RH, Bickerdike MJ, Kennett GA, Dourish CT. Comparative effects of continuous infusion of mCPP, Ro 60-0175 and d-fenfluramine on food intake, water intake, body weight and locomotor activity in rats. Br J Pharmacol. 2000;130:1305–1314. doi: 10.1038/sj.bjp.0703443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers SP, Dourish CT. Serotonin receptor ligands and the treatment of obesity. Curr Opin Investig Drugs. 2004;5:377–388. [PubMed] [Google Scholar]
- Waggoner LE, Zhou GT, Schafer RW, Schafer WR. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 1998;21:203–214. doi: 10.1016/s0896-6273(00)80527-9. [DOI] [PubMed] [Google Scholar]
- Weintraub M, Hasday JD, Mushlin AI, Lockwood DH. A double-blind clinical trial in weight control. Use of fenfluramine and phentermine alone and in combination. Arch Intern Med. 1984;144:1143–1148. [PubMed] [Google Scholar]
- Xiao H, Hapiak VM, Smith KA, Lin L, Hobson RJ, Plenefisch J, Komuniecki R. SER-1, a Caenorhabditis elegans 5-HT2-like receptor, and a multi-PDZ domain containing protein (MPZ-1) interact in vulval muscle to facilitate serotonin-stimulated egg-laying. Dev Biol. 2006;298:379–391. doi: 10.1016/j.ydbio.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lu H, Bargmann CI. Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature. 2005;438:179–184. doi: 10.1038/nature04216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.