

Abstract
Autosomal-dominant sensorineural hearing loss is genetically heterogeneous, with a phenotype closely resembling presbycusis, the most common sensory defect associated with aging in humans. We have identified SLC17A8, which encodes the vesicular glutamate transporter-3 (VGLUT3), as the gene responsible for DFNA25, an autosomal-dominant form of progressive, high-frequency nonsyndromic deafness. In two unrelated families, a heterozygous missense mutation, c.632C→T (p.A211V), was found to segregate with DFNA25 deafness and was not present in 267 controls. Linkage-disequilibrium analysis suggested that the families have a distant common ancestor. The A211 residue is conserved in VGLUT3 across species and in all human VGLUT subtypes (VGLUT1-3), suggesting an important functional role. In the cochlea, VGLUT3 accumulates glutamate in the synaptic vesicles of the sensory inner hair cells (IHCs) before releasing it onto receptors of auditory-nerve terminals. Null mice with a targeted deletion of Slc17a8 exon 2 lacked auditory-nerve responses to acoustic stimuli, although auditory brainstem responses could be elicited by electrical stimuli, and robust otoacoustic emissions were recorded. Ca2+-triggered synaptic-vesicle turnover was normal in IHCs of Slc17a8 null mice when probed by membrane capacitance measurements at 2 weeks of age. Later, the number of afferent synapses, spiral ganglion neurons, and lateral efferent endings below sensory IHCs declined. Ribbon synapses remaining by 3 months of age had a normal ultrastructural appearance. We conclude that deafness in Slc17a8-deficient mice is due to a specific defect of vesicular glutamate uptake and release and that VGLUT3 is essential for auditory coding at the IHC synapse.
Introduction
Age-related hearing impairment, or presbycusis, is the most common sensory disorder in older individuals, with prevalence increasing with age.1 Both genetic factors and environmental factors such as noise exposure influence the development of presbycusis.2,3 Although a remarkable number of genes responsible for hereditary hearing impairment have been identified in recent years, a significant number of genes associated with mapped deafness loci still await discovery.4 The dominant (DFNA) loci represent interesting candidates for presbycusis, because the hearing loss associated with these loci is typically delayed-onset and progressive, and with few exceptions, predominantly affects high frequencies. We previously mapped one such locus, DFNA25 (MIM 605583), to chromosome 12q21-q24.5
Molecular genetic studies of mutant mice and of humans with hereditary hearing loss have contributed to our understanding of the unique properties of the afferent synapse of cochlear inner hair cells.6,7 Bassoon (MIM 604020) is a presynaptic scaffolding protein that is required to anchor the ribbon at the presynaptic active zone of the sensory inner hair cell (IHC) synapse and for synchronous signaling to the auditory nerve.8 Bassoon mutant mice show reduction of the presynaptic readily releasable pool of vesicles and impaired synchronous and sound-evoked activation of spiral ganglion neurons. Otoferlin (MIM 603681), a protein encoded by the OTOF gene, is essential for the late step of synaptic-vesicle exocytosis and may act as the major Ca2+ sensor that triggers membrane fusion at the IHC ribbon synapse.9 Mutations in OTOF are responsible for DFNB9 (MIM 601071) deafness, which is characterized by profound hearing loss with documentation of preserved outer hair cell (OHC) function as measured by otoacoustic emission (OAE) in some patients.10,11
Very recently, Seal et al.12 identified another key component of the IHC synapse, the vesicular glutamate transporter-3 (VGLUT3 [MIM 607557]), which is encoded by the SLC17A8 gene and transports the neurotransmitter into synaptic vesicles before it is released into the synaptic cleft. The genetic deletion of Slc17a8 in mice results in profound deafness because of the lack of glutamate release. Because SLC17A8 maps within the DFNA25 interval, we evaluated SLC17A8 as a candidate gene and studied a mouse with a targeted deletion of Slc17a8 exon 2.13 Here, we provide the first evidence that a glutamate-transmission deficit is directly responsible for human sensorineural deafness, DFNA25, because of a mutation of SLC17A8, segregating in two families. Although Slc17a8−/− mice exhibit no response to sound, electrical stimulation applied to the round-window membrane elicits clear auditory brainstem responses. In addition, we demonstrate that IHC presynaptic function and morphology are intact up to the stage of transmitter release after vesicle fusion, making Slc17a8−/− mutant mice an excellent model to test therapeutic interventions in the inner ear.
Subjects and Methods
Human Subjects
These studies were approved by the Institutional Review Boards of the University of Michigan (IRBMED) and the University of Iowa.
Phenotype and Mapping of DFNA25
The DFNA25 locus was mapped in an American family (family 1) of Czech descent segregating autosomal-dominant, progressive, high-frequency sensorineural hearing loss.5 The DFNA25 interval as defined by key recombination events spanned 20 cM and 14.3 Mb on chromosome 12q21-q245 and includes more than 60 genes, including SLC17A8 (UCSC Genome Browser). Penetrance was shown to be age dependent and possibly also dependent on the sex of the parent of origin.14
Iowa Family 1490, an American family of German descent, segregated a similar type of high-frequency, autosomal-dominant progressive hearing loss. A genome-wide linkage analysis was performed as previously described15 and identified a locus on 12q21.2-q24.21 defined by markers D12S326 and D12S79, spanning 38.9 cM, with a maximum LOD score of 3.91 at θ = 0 for D12S78. In both families, hearing loss appeared to be nonsyndromic, without a history of seizure disorders or other neurologic dysfunction in affected family members.
Sequencing SLC17A8 from Human Genomic DNA Samples
DNA sequencing of all 12 coding exons and intron-exon junctions was performed in four DNA samples from individuals in family 1 (three affected and one unaffected) and one affected individual from family 1490. In addition, five DNA samples from individuals (three affected and two unaffected) in an unrelated family (family 63) with nonsyndromic, dominant, progressive, high-frequency hereditary hearing impairment were assayed. Primers for polymerase chain reaction (PCR) were designed with the Primer 3.0 program (Table S1 available online). PCR was done with Failsafe (Epicenter Biotechnologies, Madison, WI, USA) or Bioline reagents (Bioline, Taunton, MA, USA) according to the manufacturer's instructions. Standard thermocycling conditions were used with 50°C–70°C annealing temperatures and 30 s extension for 35 cycles. PCR products were purified with the QiaQuick DNA extraction kit (QIAGEN, Valencia, CA, USA) and sequenced by dye-terminator cycle sequencing on a 3730xl DNA analyzer (Applied Biosystems, Weiterstadt, Germany) at the University of Michigan DNA Sequencing Core or a 3130xl Genetic Analyzer (Applied Biosystems) at the Molecular Otolaryngology Research Laboratory with the manufacturer's recommended reagents and software. Sequence data were compared to reference sequences NM_139319 (mRNA) and NP_647480 (protein) with Lasergene (DNAStar, Madison, WI, USA).
Assaying the Mutation for Segregation in the Families and Presence in Controls
The presence of the c.632T→C mutation was assayed by sequencing exon 5 in remaining available DNA samples (17 affected, 19 unaffected, 9 unknown status, and 8 married-in spouses) in family 1 and in controls. A total of 267 unrelated controls were sequenced of which 90 were from the DNA Polymorphism Discovery Resource Panel, 99 were from the Caucasian North American Panel, and 78 were from Human Variation Panels, consisting of Northern European (ten samples), Czech (ten samples), African American, Mexican, Southeast Asian, South American, Middle Eastern, South African, Ashkenazi Jewish, Chinese, and North African panels (Coriell Cell Repositories, Camden, NJ, USA).
For family 1490, the presence of the c.632T→C mutation was determined by KpnI (New England Biolabs, Ipswich, MA, USA) restriction digest of exon 5 in remaining available DNA samples (seven affected, five unaffected, and five married-in spouses).
Conservation Analysis
Amino acid sequences representing exons 5 and 6 of select proteins from the VGLUT protein family in H. sapiens (VGLUT1 [MIM 605208], VGLUT2 [MIM 607563], and VGLUT3), R. norvegicus, and M. musculus and homologous proteins in C. elegans and D. melanogaster were obtained from the NCBI database and aligned with ClustalX2 software.
Identification of Previously Unreported STRs and SNPs
We identified short tandem repeats (STRs) and single-nucleotide polymorphisms (SNPs) not previously reported by searching sequences from bacterial artificial chromosomes mapping to the region of the SLC17A8 gene with Tandem Repeats Finder software. Primer design and PCR for screening newly identified STRs was done as described above for SLC17A8 exon primers. PCR product was separated by polyacrylamide gel electrophoresis and detected with silver staining. Primer design and PCR for SNP screening was also done as described above for SLC17A8 exon primers, and PCR products were analyzed by restriction digests. Restriction enzymes for screening SNPs were used according to the manufacturer's instructions (New England Biolabs) and are as follows: rs79703820 with BsrI; rs11110349 with AseI; rs7485480 with DdeI; rs11568544 with NdeI; rs11110390 with NheI; and rs35735 with BstEII.
Haplotype Analysis
Haplotype analysis was done with STRs and single nucleotide SNPs (Table S2). Primers to amplify previously reported STRs were designed on the basis of reports (NCBI) and fluorescently labeled. PCR was done with AmpliTaq Gold polymerase and buffers (Applied Biosystems) according to the manufacturer's instructions, and products were evaluated on a 3130xl Genetic Analyzer (Applied Biosystems). Data analysis was performed with GeneMarker software (Softgenetics, State College, PA, USA), and haplotype reconstruction was done with custom-made GeneScreen software (Itty Bitty Computers, San Jose, CA, USA).
Animals
The care and use of animals followed the animal-welfare guidelines of the Institut National de la Santé et de la Recherche Médicale (Inserm), and was approved by the Ministère Français de l'Agriculture et de la Pêche (authorization number A 3417231). All efforts were made to minimize the number and suffering of the animals used.
Generation of the Slc17a8−/−Mice
The entire procedure to generate the Slc17a8 knockout mice was performed by the Institut Clinique de la souris (Illkirch, France). In brief, the null mutation in the Slc17a8 mouse gene, located on chromosome 10, was created by homologous recombination.13 Removal of exon 2, after insertion of the targeting construct, introduced a stop codon into exon 3 in frame with exon 1, resulting in a complete loss of allele function. Consequently, mutant mice no longer expressed Slc17a8 mRNA. All experiments were performed with F2 wild-type and homozygous littermates derived from crossing F1 heterozygous Slc17a8+/− mice (129/Sv × C57BL/6). Mice were identified by PCR analysis of tail DNA with three primers located in intron 2–3 of the Slc17a8 gene or in the neomycin cassette.
Animal Surgery
Animals were anaesthetized by an intraperitoneal injection of a mixture of Rompun 2% (3 mg/kg) and Zoletyl 50 (40 mg/kg). Both pinna were severed to facilitate the insertion and placement of the distortion product otoacoustic emissions (DPOAEs) probe. A retroauricular incision of the skin was performed on both sides, and the bulla tympani were opened to expose the round windows.
Distortion Product Otoacoustic Emission Recordings
DPOAEs were recorded in the external auditory canal with an ER-10C S/N 2525 probe (Etymotic Research, Elk Grove Village, IL, USA) consisting of two emitters and one microphone. The two primary tones were generated, and the distortion was processed by the Cubdis system HID 40133DP (Mimosa Acoustics, Champaign, IL, USA). The probe was self-calibrated for the two stimulating tones before each recording. The two tones were presented simultaneously, sweeping f2 from 0.5 kHz to 20 kHz by quarter octave steps, and maintaining the f2/f1 ratio constant at 1.2. The primary intensities of f2 and f1 were set at 60 dB and 55 dB SPL (ref. 2 × 10−5 Pa), respectively. For each frequency, the cubic distortion product (2f1-f2) and the neighboring noise magnitudes were measured and expressed as a function of f2. Data are expressed as means ± standard error of the mean (SEM).
Recording of Cochlear- and Auditory-Nerve Potentials
The acoustical stimuli were generated by an arbitrary function generator (type 9100R; LeCroy Corporation, Chestnut Ridge, NY, USA), consisting of 9 ms tone bursts with a 1 ms rise and fall time delivered at a rate of 10/s. Tone bursts were passed through a programmable attenuator and delivered to the animal by a JBL 075 loudspeaker (James B. Lansing Sound, Los Angeles, CA, USA) in a calibrated free field. Cochlear potentials were obtained via a silver electrode placed on the round window and then amplified (×2000) by a Tektronix (TM 503) differential amplifier and digitalized (50 kHz sampling rate, with a 12-bit dynamic range and 1024 samples per burst), averaged 256 times, and stored on a computer (Dell Dimensions, Austin, TX, USA). This signal was then digitally filtered either with a low-pass filter (cutoff frequency 3.5 kHz) to display the compound action potential (CAP) and the summating potential (SP) or with a band-pass filter centered on the frequency of stimulation with a 4 kHz span to display the cochlear microphonic (CM). We obtained intensity-amplitude functions of these potentials at each frequency tested (2, 4, 6, 8, 10, 12, 16, 20, 26, 32, and 64 kHz) by varying the intensities of the tone bursts from 0 to 100 dB SPL, in 5 dB steps. The CAP, SP, and CM thresholds were defined as the minimum sound intensity necessary to elicit a clearly distinguishable response.
Electrical Stimulation of the Auditory Nerve
Electrical stimulation consisted in 25 μs rectangular pulses of alternate polarity delivered at a rate of 20/s by a custom-made DC stimulator. The intensity of the pulses was graded from 100 μA to 2 mA by 100 μA steps. Stimuli were delivered through a pair of electrodes placed respectively on the round window and in the neighboring muscles. To insure that the electrical pulses directly activated cochlear spiral ganglion neurons, we applied a droplet of artificial perilymph containing 10 μM of Na2+ channel blocker tetrodotoxin (TTX) into the round-window niche. Auditory brainstem responses (ABRs) were recorded before and 30 min after TTX application. The artificial perilymph solution had the following composition: 137 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM NaHCO3, 11 mM glucose, and 10 mM HEPES. The pH was 7.4 and the osmolarity was maintained at 300 mOsm/kg H2O.
Brainstem-Response Recordings
Brainstem-evoked responses to either acoustical stimulation of the ear or to electrical stimulation of the cochlear nerve were differentially recorded by needle electrodes placed subcutaneously respectively at the vertex and at base of the pinna, whereas the ground electrode was placed in the muscles of the posterior leg. The physiological signal was amplified 100,000 times by a Grass P5 series amplifier (Grass Instruments, Quincy, MA, USA), digitalized (50 kHz sampling rate, with a 12-bit dynamic range and 1024 samples per burst), and averaged 1000 times (Dell Dimensions).
Patch-Clamp Recordings of Ca2+ Currents and Membrane Capacitance
The animals were maintained according to the animal-welfare guidelines of the University of Goettingen and the State of Lower Saxony. After cervical dislocation of mice (postnatal day 12 [P12] to 18 [P18]), IHCs of the apical coil of freshly dissected organs of Corti were patch-clamped at their basolateral face at room temperature in perforated-patch configuration as described previously.16,17 The pipette solution for perforated-patch recordings contained the following: 130 mM Cs-gluconate, 10 mM tetraethylammonium-Cl (TEA-Cl), 10 mM HEPES, 1 mM MgCl2, and 10 mM 4-Aminopyridine (4-AP) as well as 250 μg/ml amphotericin B. The extracellular solution contained the following: 105 mM NaCl, 35 mM TEA-Cl, 2.8 mM KCl, 1 mM MgCl2, 10 mM HEPES, 2 mM CaCl2, 1 mM CsCl, and 10 mM D-glucose. Solutions were adjusted to pH 7.2 and had osmolarities between 290 and 310 mOsm/kg H2O. All chemicals were obtained from Sigma (St. Louis, MO, USA), with the exception of CsOH (Aldrich, Milwaukee, WI, USA) and amphotericin B (Calbiochem, La Jolla, CA, USA). Ca2+ current integrals were calculated from the total depolarization-evoked inward current, including Ca2+ tail currents after P/10 leak subtraction (i.e., from the start of the depolarization step to 1.5 ms after the end of the depolarization step). Cells that displayed a membrane current exceeding −30 pA at −87 mV were discarded from the analysis. No series resistance (Rs) compensation was applied; however, recordings with Rs > 30 MΩ were discarded from the analysis. Current was low-pass filtered at 4 kHz and sampled at 10 kHz for capacitance measurements and at 40 kHz for recordings of Ca2+ current-voltage relationships. All voltages were corrected for the liquid junction potential between pipette and bath (calculated at 16.9 mV).
Capacitance Measurements
Membrane capacitance (Cm) was measured with the Lindau-Neher technique18 implemented in the software-lockin module of Pulse (HEKA Elektronik, Lambrecht, Germany) combined with compensation of pipette and resting cell capacitances by the EPC-9 (HEKA Elektronik) compensation circuitries. A 1 kHz, 70 mV peak-to-peak sinusoid was applied about the holding potential of −87 mV. ΔCm was estimated as the difference of the mean Cm over 400 ms after the end of the depolarization (the initial 250 ms was skipped) and the mean prepulse capacitance (400 ms). Mean ΔCm and Ca2+ current estimates present grand averages calculated from the mean estimates of individual IHCs. Data analysis was performed with Igor Pro software (WaveMetrics, Lake Oswego, OR, USA).
All the functional data (cochlear potentials, brainstem responses, distortion product otoacoustic emissions, and patch-clamp recordings) are expressed as means ± SEM.
Light-Microscopic Evaluation of Slc17a8 Deletion in the Cochlea
The immunocytochemistry was performed on whole-mount preparations of cochleas from knockout (n = 18) and wild-type (n = 13) mice. The mice were decapitated under deep anesthesia (pentobarbital 50 mg/kg), and their cochleas were removed from the temporal bone and fixed 1 hr in 4% paraformaldehyde diluted in PBS (4°C); afterward, they were dissected in PBS and immunocytochemically processed as whole mounts.
The cochleas were rinsed 3 × 8 min in PBS, preincubated 1 hr in 30% normal goat or donkey serum (depending on the species of the secondary antibodies) and 0.3% Triton X-100, and incubated overnight at 4°C in primary antibodies diluted in PBS containing 1% normal serum. They were then rinsed in PBS (3 × 8 min) and incubated for 2 hr in fluorescently labeled secondary antibodies. They were finally rinsed in PBS (3 × 8 min), mounted in Mowiol, and examined with the Zeiss LSM510 Meta or the BioRad MRC1024 laser-scanning confocal microscope of the Montpellier RIO Imaging IURC facility (Montpellier, France). The fluorescence emissions were collected sequentially in order to minimize bleed-through between the emitted fluorescences.
The three following pairs of primary antibodies were used: RIBEYE (CtBP2)/GluR2 or GluR2/3 and synaptophysin/parvalbumin. We used a mouse monoclonal anti-CtBP2 IgG1 antibody that recognizes both the B-domain of RIBEYE, a component of presynaptic ribbons, and the transcriptional corepressor CtBP2 at a dilution of 1:150-1:500 (BD Biosciences, San Diego, CA, USA). RIBEYE and CtBP2 are alternate protein products of the CTBP2 (MIM 602619) gene locus, and their sequences differ by only the first 20 N-terminal amino acids.19 This antibody is now routinely used to label IHC presynaptic ribbons in the cochlea.8,20,21
AMPA (alpha-amino-3hydroxy-5-methyl-4-isoxazole proprionic acid) receptors were located with a mouse monoclonal IgG2a antibody raised against its GluR2 (ionotropic glutamate receptor-2) subunit (clone 6C4, 1:200, Millipore, Temecula, CA, USA) and a rabbit polyclonal antibody raised against the C terminus of the GluR2/3 subunits (1:200, Millipore). This latter antibody has been fully characterized22 and is widely used in the cochlea to localize these subunits.8,23,24 The specificity of the monoclonal antibody has been previously assayed with radioimmunoassay, western blots, and immunocytochemistry for identification of GluR1- to GluR7-transfected human embryonic kidney (HEK293) cells.25 In addition, this antibody has been used in the cochlea to detect GluR2 in the maturing organ of Corti.26,27 It is an optimal antibody to follow the lateral mobility of GluR2-containing AMPA receptors to and from synaptic sites in rat-cultured hippocampal neurons.28
As in earlier experiments,26,29,30 IHCs were immunolabeled for the calcium-binding protein parvalbumin, and lateral olivocochlear (efferent) fibers were labeled for the synaptic-vesicle transmembrane protein synaptophysin. The mouse anti-synaptophysin (clone SVP-38, 1:500, Sigma) and the goat anti-parvalbumin (1:500, Swants, Bellizona, Switzerland) primary antibodies were revealed with donkey secondary antibodies against mouse IgGs and goat IgGs, respectively conjugated to Alexa 488 and Alexa 647 (1:500, Invitrogen, Eugene, OR, USA). Anti-RIBEYE (CtBP2) and anti-GluR2 mouse primaries were detected with goat secondary antibodies against mouse IgG1 and IgG2a, respectively conjugated to Alexa 488 and Alexa 568 (Invitrogen), at a dilution of 1:500. Anti-GluR2/3 rabbit and anti-RIBEYE (CtBP2) mouse primaries were detected with goat secondary antibodies against rabbit and mouse IgGs, respectively conjugated to Alexa-488- and Alexa-568-labeled secondary antibodies (Invitrogen, 1:200) for 2-week-old animals.
Negative controls were carried out by omitting the primary antibodies in the first incubation step or by substituting them with IgGs. In addition, we further assayed the species or isotype specificity of the secondary antibodies. The sections were incubated with only one primary antibody, then secondary antibodies. In all the cases, only one specific fluorescence was observed.
Whole-mount preparations of wild-type (n = 4) and Slc17a8−/− (n = 4) cochleas were immunostained for parvalbumin and synaptophysin. The entire image of the inner spiral bundle from the basal and the medial turns were acquired at the confocal microscope. The whole stacks of images were then processed with ImageJ (Wayne Rasband), median-filtered, and 3D reconstructed. The quantification was done with the particle analysis plug-in on the optical sections in which the synaptophysin-immunoreactive endings did not overlap. The quantitative data were expressed as the percentage of synaptophysin-positive endings per μm length of the inner spiral bundle. The Student's t test was used for the statistical analysis of the data.
Ultrastructural Evaluation of the Slc17a8 Gene Deletion in the Mouse Cochlea
The mouse cochleas were dissected as described above and fixed in 2.5% glutaraldehyde in 0.1 M (pH 7.3) phosphate buffer. They were then processed for either scanning (SEM) or transmission (TEM) electron microscopy. For SEM, two cochleas from two Slc17a8−/− mice were used. The otic capsule was dissected out, and the stria vascularis, tectorial, and Reissner's membranes were removed. After rinsing in phosphate buffer (0.2 M, pH 7.3), the samples were dehydrated in a graded series of ethanol (30%–100%), critical-point-dried in CO2, coated with gold palladium, and observed with a Hitachi S4000 microscope.
For TEM, both wild-type (n = 3) and Slc17a8−/− (n = 6) cochleas were used. The cochleas were postfixed in a 2% aqueous solution of osmium tetroxide for 1 hr, rinsed in buffer, dehydrated in a graded series of ethanol (30%–100%), and embedded in Epon resin. Thin (8 μm) transverse sections were cut from the basal turn and observed with light microscopy to have a gross morphological survey of the specimens. Then, ultra-thin sections (100 nm) were performed, mounted on formvar-coated or mesh grids, stained with uranyl acetate and lead citrate, and observed with a Hitachi 7100 microscope.
The synaptic ribbons present at the presynapse between the afferent dendrites of type I ganglion neurons and the IHCs were counted with TEM on transverse sections of the organ of Corti in both wild-type (n = 3 cochleas) and Slc17a8-deficient (n = 6 cochleas) mice. A total of 18 IHCs were investigated in the wild-type animals, and 30 IHCs were investigated in the Slc17a8-deficient ones. The synaptic ribbons were then classified into two groups corresponding to normal and atypical structures, and the proportion of synaptic ribbons in each group was calculated for the wild-type and Slc17a8-deficient mice. A single organelle attached to the presynaptic membrane and showing a circular or oval outline with a central electron-dense body ≤ 500 nm and surrounded by synaptic vesicles was defined as normal. All other types were considered atypical.
Measurements of vesicle outer diameter were performed in 16 normal ribbons in the wild-type and in 19 normal ribbons in the Slc17a8-deficient mice with the camera (AMT XR611) software on digitized images of synaptic ribbons taken at a magnification × 150,000. The number of vesicles surrounding the central dense body was compared between the wild-type and Slc17a8-deficient mice from seven normal ribbons in each strain. Results are presented as the number of vesicles present on both sides of the dense body reported to a 100 nm length. Results were statistically compared with the Student's t test and presented as mean ± SEM.
Results
Human Studies
A Mutation in SLC17A8 Is Responsible for Human Deafness DFNA25
DNA sequencing in family 1 and family 1490 revealed a heterozygous nonsynonymous missense mutation, nucleotide change c.632C→T in exon 5, leading to amino acid change p.A211V, segregating with DFNA25 deafness in both families (Figure 1). Moreover, the p.A211V mutation was not found in 267 controls (534 chromosomes) or in the dB SNP database. No other variations from the reference sequence were detected other than SNPs (Table 1 and Table S2).
Figure 1.
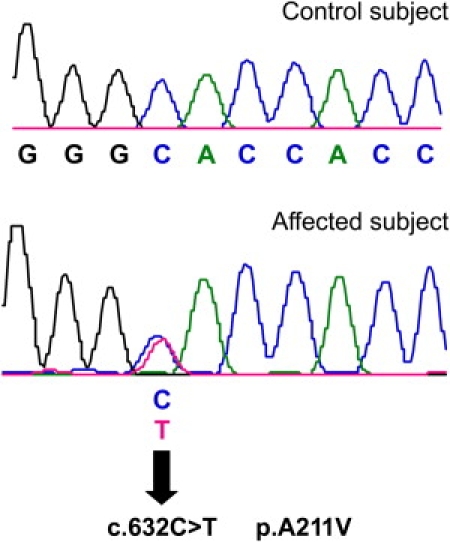
c.632C→T Mutation in SLC17A8
Sequencing chromatogram from normal-hearing control (top) shows the wild-type nucleotide C at position 632. The wild-type codon is GCA and codes for alanine. Sequencing chromatogram from affected individual from family 1 (bottom) shows heterozygous c.632C→T mutation (indicated by an arrow), leading to p.A211V amino acid change. The codon containing the mutation is GTA and the altered amino acid is valine.
Table 1.
Linkage Disequilibrium Analyses comparing STR and SNP Haplotypes for Michigan Family 1 and Iowa Family 1490
| Frequency of D Allele | Family 1 Genotypes | STR/SNP | Location in SLC17A8 | Mb | Family 1490 Genotypes | Frequency of D Allele | ||
|---|---|---|---|---|---|---|---|---|
| D | W | D | W | |||||
| 3 | 6 | D12S1063 | 97.223 | 5 | 4 | |||
| 6 | 6 | D12S1706 | 97.392 | 8 | 6 | |||
| 4 | 4 | D12S306 | 99.117 | 3 | 4 | |||
| 0.800 | T | T | rs7970382 | 99.128 | T | T | 0.8 | |
| 2 | 3 | AC026110.1 | 99.176 | 5 | 2 | |||
| 3 | 2 | AC026110.2 | 99.238 | 3 | 2 | |||
| 0.217 | T | C | rs11110349 | Intron 1 | 99.278 | T | C | 0.217 |
| 2 | 1 | AC126308.1 | Intron 1 | 99.298 | 2 | 1 | ||
| 0.737 | G | A | rs7485480 | Intron 2 | 99.305 | G | A | 0.737 |
| 2 | 1 | AC126308.2 | Intron 2 | 99.308 | 2 | 1 | ||
| T | C | c.632C→T (p.A211V) | Exon 5 | 99.314 | T | C | ||
| 0.672 | A | A | rs11568544 | Intron 6 | 99.319 | G | G | 0.328 |
| T | T | ss104807044a | Exon 8 | 99.321 | C | C | ||
| 3 | 3 | AC126308.3 | Intron 9 | 99.327 | 2 | 2 | ||
| 1 | 1 | AC126308.4 | 99.359 | 1 | 1 | |||
| 0.317 | T | C | rs11110390 | 99.399 | T | T | 0.317 | |
| 1 | 2 | AC010200.1 | 99.431 | 5 | 7 | |||
| 1 | 1 | AC010200.2 | 99.440 | 1 | 1 | |||
| 0.397 | G | T | rs35735 | 99.475 | T | G | 0.603 | |
| 1 | 2 | D12S1727 | 100.225 | 3 | 4 | |||
| 5 | 7 | D12S1607 | 100.782 | - | - | |||
| 2 | 2 | D12S1030 | 101.448 | 4 | 1 | |||
“D” indicates haplotype segregating with disease allele; “W” indicates haplotype segregating with wild-type allele in representative affected individuals. Bold indicates the portion of D haplotype shared between families 1 and 1490. Dashes indicate genotype data not available.
c.951T→C, p.Y317Y. T allele also found in family 63 and reported in AK_128319, NW_001838061.2, and NT_019546.15; C allele reported in NM_139319.
Sequence comparison of VGLUT3 to related proteins and across species demonstrates conservation of the A211 residue in the three human vesicular glutamate transporters (VGLUT1-3), vglut3 in mouse and rat, C. elegans eat-4, and Drosophila vglut (Figure 2). Molecular models suggest that residue A211 is in the cytoplasmic short linker region between transmembrane domains 4 and 5.31 Thus, this residue may be facing the pore and therefore could affect the entry of the glutamate ligand from the cytoplasm to the pore of the transporter and into the vesicle. While both alanine and valine have hydrophobic R groups, the addition of a second methyl group secondary to the substitution of valine for an alanine may increase the steric hindrance.
Figure 2.
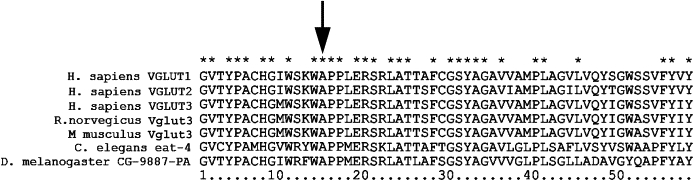
Conservation of Amino Acids of VGLUT3 Encoded by Exons 5 and 6
Amino acid sequence representing exons 5 and 6 of select proteins from the VGLUT protein family in H. sapiens, R. norvegicus, and M. musculus, and homologous proteins in C. elegans and D. melanogaster were aligned in ClustalX2. Asterisks show identical residues between the aligned sequences. An arrow indicates residue mutated in DFNA25 deafness.
Age-Dependent Penetrance of DFNA25 Deafness
Sequence results assaying for the p.A211V mutation in DNA samples from all related individuals in the DFNA25 pedigree were consistent with the family 1 haplotype results previously described.5 In other words, individuals who carried the portion of the haplotype common to the affected encompassing SLC17A8 (between D12S1063 and D12S1607) were found to carry the p.A211V mutation.
The phenotype of hearing loss varied in terms of severity, audiometric pattern, and age of onset among affected individuals within the family, as previously described.14 To further characterize the age of onset, we carried out a penetrance analysis to estimate the likelihood of a mutation-positive individual manifesting hearing loss by a certain age, given that this analysis was not possible on the basis of linkage analysis alone. There were 24 individuals with the p.A211V mutation for whom audiological data were available, of whom two were younger than age 20 at time of diagnosis. Penetrance was 77% (17/22) for those aged 20 and older; 80% (16/20) for those aged 30 and older; 83% (10/12) for those 40 and older; 86% (6/7) for those aged 50 and older; and 75% (3/4) for those 60 and older (Table S3). We did not screen for the mutation in individuals younger than 20 years of age without hearing loss.
SLC17A8 Mutation Screening in Other Subjects
Notably, there were three individuals with hearing loss who were not worse than expected for their age and sex and who were considered unaffected a priori to linkage analysis by criteria previously described.5,14 These subjects, whose hearing loss was consistent with presbycusis and who did not carry the DFNA25 haplotype, tested negative for the p.A211V mutation. The p.A211V mutation was not found in married-in spouses (with or without age-related hearing loss) in family 1 or in subjects from family 63.
Linkage-Disequilibrium Analysis
Linkage-disequilibrium analysis was performed by genotyping SNPs and STRs in the SLC17A8 region in family 1 and family 1490 (Table 1). There was a region of linkage disequilibrium spanning approximately 36 kb, for which the SNP genotypes were the same in both families. Interestingly, the LD is confined to SNPs within the gene but is not conserved for the entire gene; the two families have two different genotypes for rs11568544, 6 kb telomeric to the mutation.
Mouse Studies
Slc17a8 Deletion Does Not Induce Major Structural Changes at IHC Afferent Synapses
We first examined the overall morphology of the organ of Corti in Slc17a8−/− mice. Examination of the surface of the organ of Corti with SEM revealed that Slc17a8−/− mice display a full contingent of hair cells, except for a few scattered missing IHCs (less than 1% of the total number of IHCs) detected along the cochlear partition from base to apex. The shape of the stereocilia of both types of hair cells had a normal appearance (Figure 3A). Transverse light-microscopic sections of the organ of Corti showed a normal aspect with well-opened tunnel of Corti (Figure 3B). Transmission electron microscopy (Figures 3C–3J) confirmed that both OHCs (Figure 3C) and IHCs (Figure 3D) had a healthy appearance with a normal cytoplasmic content, well-defined organelles, and normal distribution of chromatin within the nucleus. The basal pole of the OHCs was innervated by large vesiculated efferent endings (axons of medial efferent neurons) and small afferent (dendrites from type II ganglion neurons, Figure 3E). The midbasal part of the IHCs was contacted by afferent dendrites (Figure 3F). Vesiculated axonal endings probably originating from the lateral efferent neurons were seen in the inner spiral bundle making classical en-passant axodendritic synapses with afferent fibers (Figure 3F). Occasional efferent contacts were also observed on IHCs (not shown).
Figure 3.
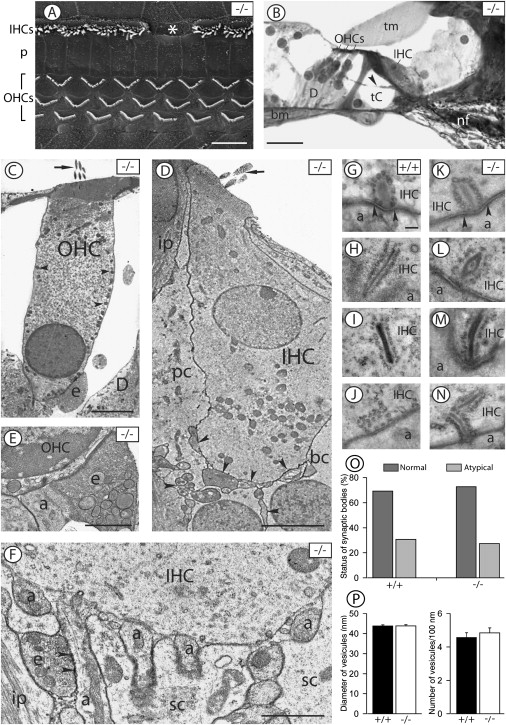
Slc17a8 Deletion Does Not Induce Major Change at IHC-Afferent Synapses
Electron (A and C–J) and light microscopy (B) of the organ of Corti in the basal turn of Slc17a8−/− (A–F and H–J) and wild-type (+/+, [G]) mice are shown.
(A) Surface of the organ of Corti observed with SEM. The stereociliary bundles are well organized in both the three rows of OHCs and the single row of IHCs. However, note the missing stereociliary bundle (asterisk) in the IHC row. “p” stands for pillar cells.
(B) Gross morphology of the organ of Corti seen in transverse section. The organ of Corti shows a normal appearance with well opened tunnel of Corti (tC), one IHC, and three OHCs. Note the presence of medial efferent fibers (indicated by an arrowhead) crossing the tunnel of Corti. The following abbreviations are used: D, Deiters cells; tm, tectorial membrane; and bm, basilar membrane. (C)–(M) show TEM.
(C) A normal appearing OHC showing erected stereocilia (indicated by an arrow), clear cytoplasm, a basal nucleus with peripherally located chromatin, typical distribution of mitochondria (indicated by arrowheads) along the lateral wall of the cell and below the nucleus. Note the efferent nerve ending (e) onto the basal pole of the OHC.
(D) Normal appearing IHC with erected stereocilia (indicated by an arrow), a clear cytoplasm, and a nucleus centrally positioned with peripheral chromatin. Note the nerve fibers underneath the IHC and in contact with its basal pole (indicated by arrowheads). The following abbreviations are used: pc, phalangeal cell; bc, border cell; and ip, inner pillar cell.
(E) Typical innervation pattern at the basal pole of an OHC showing a large efferent ending and a smaller afferent bouton (a).
(F) Basal pole of an IHC contacted by several afferent dendrites (a). Note the presence of a vesiculated efferent fiber (e) synapsing (indicated by arrowheads) with an afferent dendrite. “sc” stands for supporting cell.
(G–N) Structural variability of synaptic ribbons in IHCs from wild-type (G–J) and Slc17a8-deleted (K–N) mice. (G), (K), and (J) show normal synaptic ribbons. In (G) and (K), note the postsynaptic density (indicated by arrowheads) on the afferent bouton. (H)–(N) show atypical synaptic organelles including very long (H and M) and ectopic (I and N) ribbons and accumulation of synaptic vesicles without central electron-dense body (J).
(O) Proportion of normal versus atypical ribbons in wild-type (n = 3 cochleas) and Slc17a8−/− (n = 6 cochleas) mice. Quantification was done from 18 and 30 IHCs, respectively. Percents were calculated from 19 synaptic bodies in the wild-type and 23 in the Slc17a8−/− mice.
(P) The left panel shows quantitative analysis of the outer diameter of vesicles surrounding normal synaptic ribbons seen in IHCs from three wild-type mice (+/+) and six Slc17a8-deleted mice (−/−). A total of 16 synaptic ribbons were investigated in the wild-type mice against 19 synaptic ribbons in the Slc17a8−/− mice. A total number of 140 and 129 synaptic vesicles were measured in the wild-type and the deleted mice, respectively. The right panel shows quantification of vesicles per 100 nm of central dense body in normal synaptic ribbons from wild-type mice (n = 7 ribbons) and Slc17a8-deleted mice (n = 7 ribbons). No significant difference was found. Scale bars in (A) and (B) represent 10 μm, those in (C) and (D) represent 5 μm, those in (E) and (F) represent 1 μm, and those in (G)–(N) represent 100 nm.
The presynaptic site of the IHC afferent synapse displayed ribbons of various shapes facing afferent boutons (Figures 3G and 3H). Some ribbons showed classical structural organization (Figures 3G, 3K, and 3L), whereas others had an atypical appearance (Figures 3H–3J, 3M, and 3N). The normal synaptic bodies had round or oval outlines with a centrally positioned dense body, surrounded by vesicles and anchored to the presynaptic membrane. The atypical organelles included very long (ranging from 500 to 850 nm; Figures 3H and 3M) or double synaptic bodies (Figure 3N). A few synaptic ribbons were not located at the presynaptic membrane but were found floating within the IHC cytoplasm (Figures 3I and 3N). Occasional accumulations of synaptic vesicles without detectable central dense bodies were also seen either near the presynaptic membrane (Figure 3J) or in ectopic positions. The typical postsynaptic density was seen on the afferent boutons facing the presynaptic active zone (Figures 3G and 3K).
We performed a quantitative analysis to investigate the proportion of normal versus atypical synaptic ribbons in wild-type (n = 19 ribbons) and Slc17a8-deficient (n = 23 ribbons) mice (Table S4). Formations lacking a central dense body were not included in this analysis. Our results show that the proportion of normal versus atypical synaptic ribbons was similar, 69.23% versus 30.77%, as compared to 72.73% versus 27.27% in wild-type and Slc17a8−/− mice, respectively (Figure 3O). Measurements of the outer diameter of synaptic vesicles surrounding normal ribbons revealed no significant difference in the size of vesicles between the wild-type (140 vesicles from 16 ribbons) and the Slc17a8−/− mice (129 vesicles from 19 ribbons). The mean diameter of vesicles was respectively 45.84 nm ± 0.86 and 47.82 nm ± 1.53 (Figure 3P, left). Similarly, quantification of vesicles surrounding normal ribbons in wild-type (n = 7 normal ribbons) and Slc17a8−/− mice (n = 7 normal ribbons) shows no significant difference between wild-type and mutant mice with a mean of 4.56 vesicles ± 0.29 and 4.84 vesicles ± 0.30 per 100 nm of dense body length, respectively (Figure 3P, right).
Slc17a8 Deletion Causes a Slowly Progressing Loss of IHC Synapses and Spiral-Ganglion Neurons
Studies in mice demonstrated that, of the three vesicular glutamate transporters, only Vglut3 is selectively expressed in the IHCs (Figure S1A and Seal et al.12). The deletion of the Slc17a8 gene in mice abolished the Vglut3 expression in the IHCs (Figure S1B) but did not affect the expression of the presynaptic proteins such as cysteine-string protein and synaptogyrin (Figures S1C and S1D).
We used immunoreactivities of RIBEYE/CtBP2 and GluR2 to visualize the presynaptic ribbons and postsynaptic AMPA receptors at IHC synapses in 3-month-old mice. No fluorescence was observed in the negative controls. Both immunoreactivities were juxtaposed in Slc17a8−/− cochleas (Figure 4A). However, a lower density of ribbons was seen as a consequence of the gene deletion. We thus quantified their number in the wild-type and Slc17a8−/− mice. Counting RIBEYE/CtBP2-positive dots per IHC revealed that wild-type IHCs possessed 12.55 ± 0.63 presynaptic ribbons (Table S5A). The gene deletion induced a significant reduction (43.90%) in the number of immunostained dots, leading to 7.04 ± 0.53 ribbons per IHC (Figure 4B). This is consistent with the significant reduction (42%) observed in the number of spiral-ganglion neurons in Slc17a8−/− mice (Figures S2A–S2C and Table S5B). No sign of degeneration was seen in the remaining neurons. In fact, Slc17a8−/− mice showed a low density of spiral-ganglion neurons with a regular shape, clear cytoplasm, and round nucleus, surrounded by several rows of myelin (Figures S2D–S2F).
Figure 4.
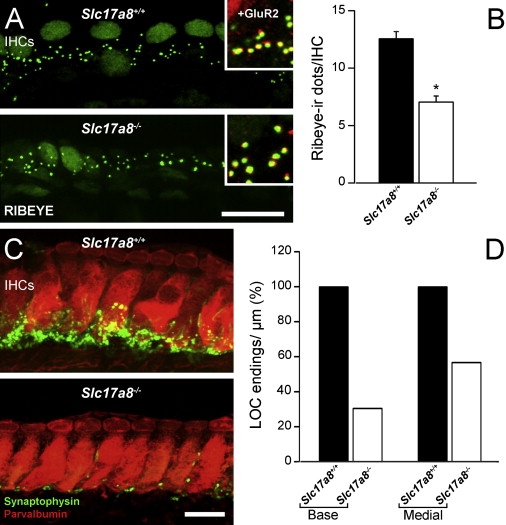
Slc17a8 Deletion Reduced the Number of IHC Synapses and Lateral Efferent Endings
(A) Confocal microscopy of RIBEYE immunoreactivity through basal turn inner hair cells of Slc17a8+/+ and −/− mice. The hair cell nuclei display the immunoreactivity to the CtBP2 transcription factor. The RIBEYE-immunoreactive dots are seen at the hair cell (IHC) bases in the Slc17a8−/− mouse, although they are less densely distributed than in the wild-type. The inserts show that in both mice, RIBEYE-immunoreactive dots face GluR2 immunoreactive spots. The scale bar represents 15 μm.
(B) RIBEYE dots count in +/+ and −/− cochleas (n = 3 and 4 respectively) shows a strong (43.90%) and significant (p = 9.17 × 10−8) decrease of the number of immunoreactive dots at the IHC bases of SLC17A8−/− mice.
(C) Confocal microscopy through the basal turn of Slc17a8+/+ and −/− mice. IHCs are labeled with parvalbumin (red), and lateral efferent endings are labeled with synaptophysin (green). Note the strong reduction in the number of immunoreactive endings in the Slc17a8−/− mouse. The scale bar represents 10 μm.
(D) Quantitative analysis of the number of synaptophysin-immunoreactive endings beneath IHCs from the basal and medial cochlear turns of wild-type (n = 4 cochleas) and Slc17a8−/− (n = 4 cochleas) mice. The reduction in the number of synaptophysin-immunoreactive endings is more pronounced in the basal turn (69.51% reduction) than in the medial turn (43.26%).
Slc17a8 Deletion Reduced the Number of Lateral Efferent Endings
We used the synaptophysin immunoreactivity to identify presynaptic endings of lateral olivocochlear efferents forming axodendritic synapses with the dendrites of spiral-ganglion neurons. A striking consequence of the Slc17a8 gene deletion was a strong decrease of the synaptophysin immunoreactivity in the inner spiral bundle in the basal turn of the cochleas (Figure 4C). Counting synaptophysin-immunoreactive endings along the inner spiral bundle of the basal and medial turns (Table S5C) showed that the number of the lateral efferent endings was reduced by 69.5% in the basal turn and 43.3% in the medial turn (Figure 4D).
Slc17a8 Deletion Does Not Affect the Transduction Mechanism
We then measured cochlear responses from an electrode apposed onto the round-window membrane, and DPOAEs were recorded in the external ear canal (see Figure 5). Wild-type Slc17a8+/+ (n = 10) and Slc17a8+/− heterozygous mice (n = 10) showed the classical sound-evoked CAP waveform in responses to all sound stimulation from 2 to 64 kHz with thresholds between 20 and 45 dB sound pressure level (SPL). In contrast, the Slc17a8−/− (n = 12) littermates showed no visible CAP responses, even for stimulus intensities of 100 dB SPL (Figures 5A–5C). Assessment of the functioning of the transduction mechanisms of sensory hair cells showed no significant difference in CM and SP amplitude-intensity functions in responses to 12 kHz tone-bursts among in Slc17a8+/+, Slc17a8+/−, or Slc17a8−/− mice (Figures 5D and 5E). Furthermore, DPOAEs were not significantly different in the wild-type and in the mutant mice, reflecting normal outer hair cell function at all stimulus frequencies and intensities studied (Figure 5F). Mechanoelectrical transduction of hair cells remained fully functional in the absence of Slc17a8 as indicated by the receptor potentials (SP and CM) of both IHCs and OHCs.
Figure 5.
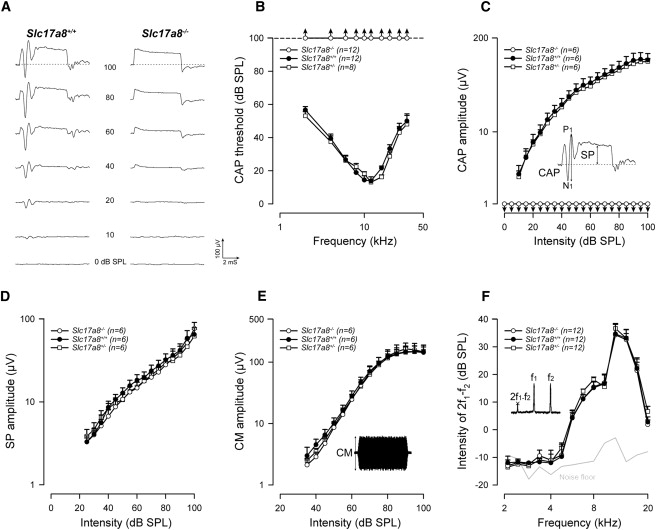
Slc17a8 Deletion Leads to a Profound Deafness, but Does Not Affect the Functioning of Cochlear Mechanotransduction
(A) Typical examples of averaged cochlear action potentials (CAPs) recorded from an electrode apposed onto the round-window membrane for wild-type and Slc17a8−/− mice. The potentials were recorded in response to 12 kHz tone bursts presented from 0 to 100 dB SPL. Slc17a8+/+ (n = 12) and Slc17a8+/− heterozygous mice (n = 8, not shown) showed the classical sound-evoked N1-P1 CAP waveform in responses to all sound stimulation. In contrast, the Slc17a8−/− (n = 12) littermates showed no visible CAP response, even for high stimulus intensities. Note that the summating potential (SP), which reflect the functional state of the inner hair cells, remains intact in Slc17a8−/− mice.
(B) CAP audiograms for Slc17a8−/−, Slc17a8+/+ and Slc17a8+/− mice were obtained by plotting CAP thresholds as a function of the stimulation frequency (2–64 kHz tone-bursts). Compared to Slc17a8+/+ (black circles) and Slc17a8+/− (open squares) mice, Slc17a8−/− mice (open circles) show no sound-evoked response at any stimulating frequency and intensity.
(C–E) Input-output functions of the CAP, SP, and cochlear microphonic (CM) evoked by a 12 kHz tone-bursts from 0 to 100 dB SPL in Slc17a8−/−, Slc17a8+/+ and Slc17a8+/− mice. As shown in (C), Slc17a8−/− mice displayed a complete abolition of CAP. The inset in (C) shows a typical trace of averaged cochlear potentials evoked by 12 kHz tone bursts presented at 80 dB sound pressure level (SPL). By contrast, normal SP (D) and CM (E) potentials were recorded in all animals tested.
(F) Recordings of distortion product otoacoustic emissions (2f1-f2) reflect the cochlear nonlinearity as a result of outer hair cell function. No significant change was measured between Slc17a8−/−, Slc17a8+/+, and Slc17a8+/− mice. The thick gray line indicates the background noise level of the recording system in the absence of sound. Values are mean ± SEM, and n indicates the number of animal tested.
Slc17a8 Deletion Does Not Affect Synaptic-Membrane Turnover
To determine whether the absence of Vglut3 impairs synaptic-vesicle cycling, we probed the Ca2+-triggered fusion of IHC synaptic vesicles with patch-clamp measurements of exocytic capacitance changes in 2-week-old mice (Slc17a8−/− and Slc17a8+/+). The Ca2+ currents of the two genotypes were similar in terms of kinetics and voltage dependence, but Slc17a8−/− IHCs showed larger Ca2+ current (Figure 6A). Fast exocytosis, mediated by the readily releasable pool of vesicles and sustained secretion (reviewed in Nouvian et al.7) were normal despite the absence of Vglut3 (Figures 6B and 6C). This finding indicates that IHCs maintain synaptic-vesicle cycling even though glutamate cannot be loaded and released. In line with these results, immunostaining of afferent IHC synapses (Figures 6D and 6E) for presynaptic ribbons (RIBEYE, green) and postsynaptic glutamate receptors (GluR2/3, red) revealed comparable numbers of ribbon-containing afferent synapses from 2-week-old mice (12.3 ± 0.3 for Slc17a8+/+, n = 76 IHCs of 5 mice, and 11.8 ± 0.3 for Slc17a8−/−, n = 112 IHCs of 4 mice; P13-P17).
Figure 6.
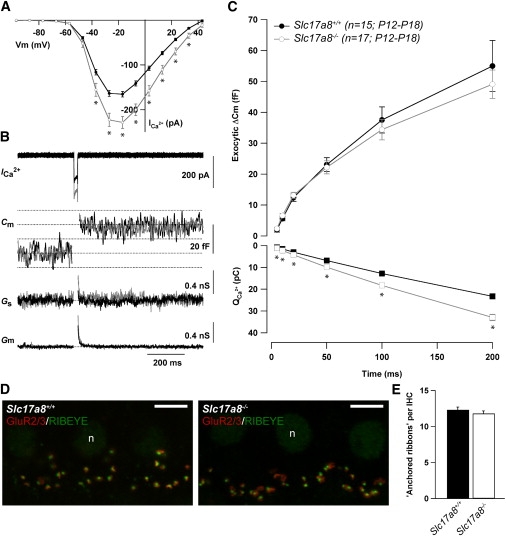
Ca2+-Triggered IHC Exocytosis Is Not Impaired by the Lack of Vglut3
(A) Ca2+ current steady-state I/V relationships of Slc17a8+/+ (black) and Slc17a8−/− (gray) IHCs from P12-18 mice. Steady-state amplitude was measured as the average over the last 5 ms of the 10 ms test pulse. Statistical significant difference is indicated by asterisks (p < 0.05, unpaired t test).
(B) Ca2+ current (ICa2+), membrane capacitance (Cm) low-pass filtered at 100 Hz, series conductance (Gs), and membrane conductance (Gm) traces (from top to bottom) of representative Slc17a8+/+ (black, P12) and Slc17a8−/− (gray, P13) IHCs elicited by 20 ms depolarization to the peak Ca2+ current potential.
(C) Kinetics of exocytosis (ΔCm, top) and corresponding Ca2+ current integrals (QCa2+, bottom) of Slc17a8+/+ (black) and Slc17a8−/− (gray) IHCs. ΔCm were obtained by multiple depolarizations of different durations to peak calcium current potential in each IHC and represent grand averages calculated from the means of the individual IHC. For Slc17a8+/+ and Slc17a8−/−, respectively, Rs = 19.6 ± 0.7 MΩ and 18.7 ± 0.9 MΩ; resting membrane capacitance = 8.9 ± 0.2 pF and 8.9 ± 1.4 pF, and resting current at holding potential (−87 mV) = −17.7 ± 1.1 pA and −17.9 ± 1.1 pA. Statistically significant difference is indicated by asterisks (p < 0.05, unpaired t test).
(D) Representative projections of confocal sections from P14 Slc17a8+/+ and P17 Slc17a8−/− apical organs of Corti stained for GluR2/3 (red) and RIBEYE/CtBP2 (green). “n” stands for nucleus; scale bars represent 5 μm.
(E) Quantitative analysis of synapse-anchored ribbons per IHC identified as small RIBEYE-positive spots underneath the IHC nuclei juxtaposing GluR2/3 immunofluorescence spots from P13-P17 mice.
Preserved Auditory-Nerve Function in Slc17a8−/−Mice
Next, we explored the functionality of the nerve by recording ABRs evoked by electrical stimulation applied to the round-window membrane. Whereas sound-evoked ABRs were elicited only in wild-type and in Slc17a8+/− mice, electrical stimulation evoked ABRs in Slc17a8−/− mice as well (Figures 7A and 7B). A slight reduction of ABR amplitude in Slc17a8−/− was noted in comparison with Slc17a8+/+ and Slc17a8+/− mice (Figures 7C and 7D), probably because of the 40% loss of spiral-ganglion neurons. We inhibited the generation of the action potentials of the spiral-ganglion neurons by applying 10 μM TTX into the round-window niche. The lack of electrically evoked ABRs in the presence of TTX demonstrated that electrical stimulation selectively activated the spiral-ganglion neurons. Thus, the preservation of functional transduction pathways in the IHCs, together with the response of spiral-ganglion neurons to electrical stimulation, demonstrates that the profound hearing impairment observed in Slc17a8−/− mice results from an IHC synaptic-transmission defect.
Figure 7.
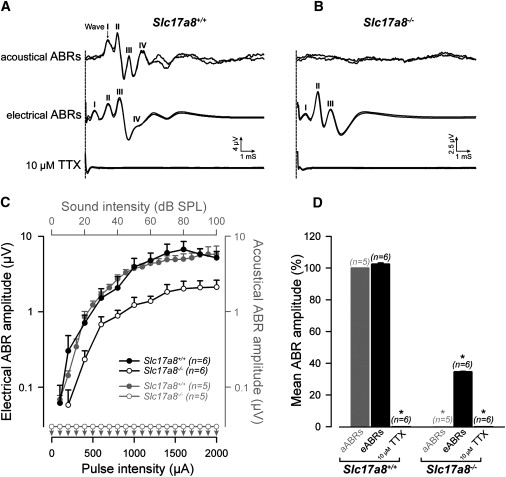
Slc17a8 Deletion Preserves the Integrity of the Auditory Nerve
(A) Typical auditory brainstem responses (ABRs) evoked by sound stimulation or by electrical stimulation applied via an electrode apposed onto the round-window membrane recorded in Slc17a8+/+ mice. The upper part of the figure shows superposed ABR traces evoked by a click presented at 50 dB SPL. Characteristic waves (I to IV) are clearly seen. The middle part of the figure shows ABR traces elicited by round-window electrical stimulation of 1 mA in the same animal. Note the similar pattern of ABR traces in both condition of stimulation. No electrophysiological response to sound or electrical pulses was recorded after round-window application of 10 μM Na+ channel blocker, TTX.
(B) No acoustical ABR response was seen in Slc17a8−/− mice whatever the intensity of sound stimulation (top trace). In contrast, this knockout mice shows clear electrically evoked-ABRs (middle trace) that could be abolished after injection of 10 μM TTX into the cochlea (bottom trace).
(C) Shown are input-output functions of ABRs wave I evoked by acoustical (gray symbols) or electrical (black symbols) stimulation in Slc17a8−/− and Slc17a8+/+ mice. Electrical ABRs amplitude is clearly reduced (black open circles) in Slc17a8−/− mice. Consistent with CAP data, no ABR could be evoked by sound stimulation in Slc17a8−/− mice (gray open circles).
(D) The histogram represents the mean amplitude from 0 to 100 dB SPL sound stimulation and 0 to 2 mA electrical pulses for acoustically evoked ABRs (aABRs; gray bar) and electrically evoked ABRs (eABRs; black bar) in Slc17a8+/+ and in Slc17a8−/− mice, respectively. No response was recorded after cochlear injection of 10 μM TTX. Data presented are mean ± SEM, and n indicates the number of animal tested.
Discussion
Here, we identify the gene responsible for human autosomal-dominant nonsyndromic DFNA25 deafness at the 12q21-q24 locus5 as SLC17A8, an 11 exon gene encoding the vesicular glutamate transporter 3. To our knowledge, this finding represents the first mutation in a VGLUT associated with human disease. The SLC17A subfamily of solute carrier membrane transport proteins includes VGLUTs and other proteins such as sialin (SLC17A5) involved in transport of organic anions.32
Expressivity and Penetrance
The expression of DFNA25 deafness is variable in terms of onset and rate of progression, with an age-dependent penetrance resembling an early-onset presbycusis.14 In general, for family 1, penetrance increases with age; the apparent decrease in penetrance at age 60 probably reflects the small number of subjects first ascertained at that age. Because penetrance in family 1 was incomplete even in the seventh decade of life,5 the DFNA25 candidate gene interval was based on recombination events in affected individuals only. Haplotype analysis of a second linked family (family 1490) did not reduce the size, and consequently, the DFNA25 interval was quite large. Several candidate genes in the interval have been sequenced without evidence of pathologic mutations (S.E., T.A.S., and M.M.L., unpublished data; J.M.V.R. and R.J.H.S., unpublished data).
Interestingly, we previously found that several “carriers” (those who inherited the haplotype segregating with deafness but with normal hearing for age and sex) had inherited the haplotype from their fathers rather than their mothers.14 We have previously excluded several deafness-associated mitochondrial mutations14 as modifiers, but other autosomal or mitochondrial modifier genes cannot be excluded. In addition, the phenotype may be influenced by environmental factors such as noise exposure and smoking.33
Linkage Disequilibrium
The HapMap project has identified large blocks of linkage disequilibrium across the human genome (International HapMap Consortium 2007). In European populations, haplotype blocks ranging from <1 to 173 kb have been reported, with an average minimum span of 18 kb.34 The 36 kb block of LD surrounding the p.A211V mutation in the two families is therefore well within the range of LD seen in the general population. Finding the same haplotype surrounding the mutation argues for a common, although distant, ancestor because the alternative hypothesis, that the identical mutation evolved separately, appears to be much less likely. Given the implied age of this mutation, it is likely that it will be found in additional individuals with hearing loss, but this mutation may be difficult to recognize because of the variability of the age of detection and rate of progression.
Selective Inner Hair Cell Dysfunction
Because Slc17a8−/− null mice clearly demonstrate robust OAEs, indicative of preserved OHC function, the question arises as to whether OAEs are preserved in human participants with heterozygous missense SLC17A8 mutations as well. Data were available for two affected individuals in family 1. One 68-year-old affected individual was found to have absent transient-evoked OAEs and no repeatable waveforms by ABR, consistent with moderate-severe to severe sensorineural hearing loss.5 A 38-year-old affected individual had absent DPOAEs and elevated pure tone thresholds between 4 and 8 kHz (M.M.L., unpublished data). The possibility that OAEs are preserved in hearing-impaired participants at younger ages and lost secondarily, as has been demonstrated for AUNA1 (MIM 609129) deafness35,36 and DFNB9 deafness,37 cannot be excluded.
Slc17a8 Deficiency Causes Profound Hearing Loss in Mutant Mice
Glutamate is the major excitatory neurotransmitter at the IHC afferent synapse.38 Before its exocytotic release, it must be loaded into synaptic vesicles by proton-driven membrane transporters.39,40 In contrast to the brain, in which it is sparsely distributed and often associated with other neurotransmitters,39–42 Vglut3 is strongly and selectively expressed in cochlear IHCs (our present study and Seal et al.12).
Direct patch-clamp recordings of hair cells from single postsynaptic boutons demonstrated the absence of synaptic transmitter release from the IHC at the first synapse of the auditory pathway.12 Our presynaptic capacitance measurements indicated that Ca2+-triggered synaptic-vesicle turnover occurs independently of glutamate loading, and such a finding is consistent with the normal cycling of synaptic vesicles observed in Slc17a7 (which encodes Vglut1) knockout mice.43 The observation of normal kinetics and amounts of hair cell exocytosis in Slc17a8 knockout mice demonstrates that even detailed synaptic properties are unchanged in the absence of functional transmission. The slight increase of the Ca2+ current in Slc17a8-deficient IHCs is currently unexplained but may reflect some adaptation. Hair cell function was also intact upstream from Ca2+-induced vesicle turnover as were presynaptic-transduction mechanisms.
The normal afferent synaptogenesis of IHCs in Slc17a8 knockout mice is consistent with findings in CaV1.3 (α subunit 1.3 forming L-type voltage-gated Ca2+ channel) and Otof knockout mice that also bear severe hair cell synaptopathies.9,30,44 Given the selective sensory deficit, it was possible to test the stability of the “silent” glutamatergic synapses. Strikingly, synaptic morphology in IHCs was astonishingly well maintained up into adulthood of the animals. We found mostly normal active zones and only a small number of atypical ribbons at age 3 months in mutant and wild-type IHCs, in contrast to the predominantly thin and elongated ribbons described in 3-week-old Slc17a8−/− mice by Seal and collaborators.12 The apparent discrepancy in these findings, however, may be explained by the differences in the genetic background. The mice in the present study were obtained by crossing heterozygous 129/Sv and C57BL/6 mice, whereas the Slc17a8−/− mice studied by Seal et al.12 were derived solely from C57BL/6 mice, a strain prone to age-related hearing loss.45 We were also unable to show any statistical difference in the number of vesicles per ribbon, in contrast to Obholzer et al.,46 who showed a reduction in vesicle number in Slc17a8 mutant zebrafish. However, the larger size and spherical shape of the zebrafish synaptic ribbon and the higher number of tethered synaptic vesicles may have permitted a more precise quantification in this model as compared to the mouse.
Because glutamate has been shown to provide trophic support for spiral-ganglion neurons,47 the deficiency of afferent synapses we observed is probably due to the lack of neurotransmitter release, which in turn leads to secondary degeneration of postsynaptic spiral-ganglion neurons. The neuronal loss in the spiral ganglion was milder than that of the CaV1.3 and Otof mutants, in which stimulus-secretion coupling is blocked at the level of Ca2+ influx or vesicle fusion. These findings raise the fascinating possibility that synapses and spiral-ganglion neurons of Slc17a8 knockout mice are better maintained because of functional secretion of trophic factors such as neurotrophin 3 (MIM 162660).48 This hypothesis and the possibility of release of other neuroactive substances from Slc17a8-deficient IHCs remain to be tested in future studies.
The significant reduction of lateral efferent endings below IHCs is difficult to interpret because their fate has not been investigated in other mutant mice lacking neurotransmitter release (e.g., Bassoon and Otoferlin). During development, Vglut3 is heavily expressed in GABA/glycinergic cell bodies and terminals of brainstem auditory nuclei, particularly the lateral superior olive, which contains the lateral efferent cell bodies.49–51 It may be possible that the absence of Slc17a8 expression during this period has resulted in a disruption of the auditory brainstem circuitry, leading to a secondary loss of lateral efferent neurons. Alternatively, the lack of input activity to the lateral superior olive may have caused this efferent neuron loss.
Impairment of VGLUT3 Causes Nonsyndromic Deafness DFNA25
Age-related hearing impairment is the most common sensory disorder in older individuals, with an incidence of up to 80% by the seventh decade of life. Both genetic factors and environmental factors such as noise exposure influence the development of presbycusis.2,33 Although many genes responsible for hereditary hearing impairment have been identified,4 SLC17A8 in particular may be a good candidate gene or locus for presbycusis. Like presbycusis, the DFNA25 phenotype is one of autosomal-dominant hearing loss with penetrance that increases with age and preferentially affects high frequencies. Because DFNA25 mutation carriers develop deafness despite the presence of the normal protein, and Slc17a8+/− mice have normal hearing responses and normal anatomy, haploinsufficiency does not appear to be a likely explanation of deafness due to the p.A211V mutation.
The demonstration of normal electrically evoked ABRs in Slc17a8−/− knockout mice suggests that cochlear implantation may be of benefit for patients with a deficiency of SLC17A8 deafness, especially if any with homozygous inactivating mutations are found. To our knowledge, no hearing-impaired member of either family 1 or family 1490 has undergone cochlear implantation. There is a critical period in which electrical stimulation by a cochlear implant will maintain the viability of spiral ganglion neurons despite a defective organ of Corti (reviewed in Roehm and Hansen52). Consequently, further investigations of the role of SLC17A8 may be helpful to understand the pathogenesis of and develop therapies for nonsyndromic deafness DFNA25, as well as to further elucidate the molecular mechanisms of the IHC afferent synapse.
The absence of the p.A211V mutation in controls and conservation of the A211 residue across species does not exclude the possibility that it is a benign polymorphism. The most convincing evidence that a mutation is pathologic rather than a benign polymorphism is identifying more than one mutation in the same gene in unrelated individuals of similar phenotype.53 Although one could argue that family 1490 is the same family as family 1, given that the exact same mutation was found, the small block of linkage disequilibrium suggests that the two families are no more closely related than any two in the general population. Because there is no other linked family available to narrow the large DFNA25 interval, we cannot rule out the possibility that a mutation in a different gene or genes explains the deafness in these families. Further characterization of the effects of the p.A211V mutation in experimental animals and assaying additional families for mutations in SLC17A8 may provide further evidence.
Supplemental Data
Supplemental Data include Supplemental Subjects and Methods, two figures, and five tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dB SNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/
Institut Clinique de la souris (Illkirch, France), http://www.mci.u.strasbg.fr
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer 3.0, http://fokker.wi.mit.edu/primer3/input.htm
Tandem Repeats Finder software, http://tandem.bu.edu/trf/trf.license.html
UCSC Genome Browser, http://genome.ucsc.edu/
Wayne Rasband, http://rsb.info.nih.gov/ij/index.html
Accession Numbers
The dB SNP accession number for the exon 8 synonomous SNP, c.951T→C, p.7317Y, is ss104807044.
Acknowledgments
Human genetic analyses were carried out by S.E. and J.M.V.R.; in vivo physiology studies were carried out by J.R. and G.R.; in vitro hair cell physiology studies were carried out by R.N.; T.B. and M.E. performed immunocytochemistry studies, and M.L. performed electron-microscopic analysis. Knockout mice were provided by B.G., S.E.M., and B.A. The authors thank the subjects for their participation in this research and Matthew Young and Margit Burmeister for helpful insight. This work was supported by the “Institut National de la Santé et de la Recherche Médicale,” the University of Montpellier1, the Deafness Research Foundation (M.M.L.), the University of Michigan Biomedical Research Council (M.M.L.), the National Institute on Deafness and Communication Disorders K23 DC00161 (M.M.L.) and RO1-DC003544 (RJHS), the German Research Foundation (DFG NO 800/1-1 to R.N. and Center for Molecular Physiology of the Brain to T.M.), and the German Federal Ministry of Education and Research (Bernstein Center grant 01GQ0433 to T.M.). All the electron-microscopic analysis were performed at the “Centre de Ressources en Imagerie Cellulaire” (CRIC, Montpellier, France) with the assistance of Chantal Cazevieille.
References
- 1.Pleis J.R., Lethbridge-Cejku M. Summary health statistics for U.S. adults: National Health Interview Survey, 2005. Vital Health Stat. 2006;10:1–153. [PubMed] [Google Scholar]
- 2.Gates G.A., Couropmitree N.N., Myers R.H. Genetic associations in age-related hearing thresholds. Arch. Otolaryngol. Head Neck Surg. 1999;125:654–659. doi: 10.1001/archotol.125.6.654. [DOI] [PubMed] [Google Scholar]
- 3.Cruickshanks K.J., Wiley T.L., Tweed T.S., Klein B.E., Klein R., Mares-Perlman J.A., Nondahl D.M. Prevalence of hearing loss in older adults in Beaver Dam, Wisconsin. The Epidemiology of Hearing Loss Study. Am. J. Epidemiol. 1998;148:879–886. doi: 10.1093/oxfordjournals.aje.a009713. [DOI] [PubMed] [Google Scholar]
- 4.Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: Dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. doi: 10.1038/nrg2309. [DOI] [PubMed] [Google Scholar]
- 5.Greene C.C., McMillan P.M., Barker S.E., Kurnool P., Lomax M.I., Burmeister M., Lesperance M.M. DFNA25, a novel locus for dominant nonsyndromic hereditary hearing impairment, maps to 12q21–24. Am. J. Hum. Genet. 2001;68:254–260. doi: 10.1086/316925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs P.A., Glowatzki E., Moser T. The afferent synapse of cochlear hair cells. Curr. Opin. Neurobiol. 2003;13:452–458. doi: 10.1016/s0959-4388(03)00098-9. [DOI] [PubMed] [Google Scholar]
- 7.Nouvian R., Beutner D., Parsons T.D., Moser T. Structure and function of the hair cell ribbon synapse. J. Membr. Biol. 2006;209:153–165. doi: 10.1007/s00232-005-0854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khimich D., Nouvian R., Pujol R., Tom Dieck S., Egner A., Gundelfinger E.D., Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- 9.Roux I., Safieddine S., Nouvian R., Grati M., Simmler M.C., Bahloul A., Perfettini I., Le Gall M., Rostaing P., Hamard G. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 10.Varga R., Kelley P.M., Keats B.J., Starr A., Leal S.M., Cohn E., Kimberling W.J. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J. Med. Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasunaga S., Grati M., Cohen-Salmon M., El-Amraoui A., Mustapha M., Salem N., El-Zir E., Loiselet J., Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- 12.Seal R.P., Akil O., Yi E., Weber C.M., Grant L., Yoo J., Clause A., Kandler K., Noebels J.L., Glowatzki E. Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron. 2008;57:263–275. doi: 10.1016/j.neuron.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gras C., Amilhon B., Lepicard E.M., Poirel O., Vinatier J., Herbin M., Dumas S., Tzavara E.T., Wade M.R., Nomikos G.G. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat. Neurosci. 2008;11:292–300. doi: 10.1038/nn2052. [DOI] [PubMed] [Google Scholar]
- 14.Thirlwall A.S., Brown D.J., McMillan P.M., Barker S.E., Lesperance M.M. Phenotypic characterization of hereditary hearing impairment linked to DFNA25. Arch. Otolaryngol. Head Neck Surg. 2003;129:830–835. doi: 10.1001/archotol.129.8.830. [DOI] [PubMed] [Google Scholar]
- 15.Brown J., Fingert J.H., Taylor C.M., Lake M., Sheffield V.C., Stone E.M. Clinical and genetic analysis of a family affected with dominant optic atrophy (OPA1) Arch. Ophthalmol. 1997;115:95–99. doi: 10.1001/archopht.1997.01100150097016. [DOI] [PubMed] [Google Scholar]
- 16.Moser T., Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc. Natl. Acad. Sci. USA. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nouvian R. Temperature enhances exocytosis efficiency at the mouse inner hair cell ribbon synapse. J. Physiol. 2007;584:535–542. doi: 10.1113/jphysiol.2007.139675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindau M., Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- 19.Schmitz F., Konigstorfer A., Sudhof T.C. RIBEYE, a component of synaptic ribbons: A protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 20.Knirsch M., Brandt N., Braig C., Kuhn S., Hirt B., Munkner S., Knipper M., Engel J. Persistence of Ca(v)1.3 Ca2+ channels in mature outer hair cells supports outer hair cell afferent signaling. J. Neurosci. 2007;27:6442–6451. doi: 10.1523/JNEUROSCI.5364-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schug N., Braig C., Zimmermann U., Engel J., Winter H., Ruth P., Blin N., Pfister M., Kalbacher H., Knipper M. Differential expression of otoferlin in brain, vestibular system, immature and mature cochlea of the rat. Eur. J. Neurosci. 2006;24:3372–3380. doi: 10.1111/j.1460-9568.2006.05225.x. [DOI] [PubMed] [Google Scholar]
- 22.Wenthold R.J., Yokotani N., Doi K., Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies. Evidence for a hetero-oligomeric structure in rat brain. J. Biol. Chem. 1992;267:501–507. [PubMed] [Google Scholar]
- 23.Niedzielski A.S., Wenthold R.J. Expression of AMPA, kainate, and NMDA receptor subunits in cochlear and vestibular ganglia. J. Neurosci. 1995;15:2338–2353. doi: 10.1523/JNEUROSCI.15-03-02338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsubara A., Laake J.H., Davanger S., Usami S., Ottersen O.P. Organization of AMPA receptor subunits at a glutamate synapse: A quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J. Neurosci. 1996;16:4457–4467. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vissavajjhala P., Janssen W.G., Hu Y., Gazzaley A.H., Moran T., Hof P.R., Morrison J.H. Synaptic distribution of the AMPA-GluR2 subunit and its colocalization with calcium-binding proteins in rat cerebral cortex: An immunohistochemical study using a GluR2-specific monoclonal antibody. Exp. Neurol. 1996;142:296–312. doi: 10.1006/exnr.1996.0199. [DOI] [PubMed] [Google Scholar]
- 26.Eybalin M., Caicedo A., Renard N., Ruel J., Puel J.L. Transient Ca2+-permeable AMPA receptors in postnatal rat primary auditory neurons. Eur. J. Neurosci. 2004;20:2981–2989. doi: 10.1111/j.1460-9568.2004.03772.x. [DOI] [PubMed] [Google Scholar]
- 27.Trigueiros-Cunha N., Renard N., Humbert G., Tavares M.A., Eybalin M. Catecholamine-independent transient expression of tyrosine hydroxylase in primary auditory neurons is coincident with the onset of hearing in the rat cochlea. Eur. J. Neurosci. 2003;18:2653–2662. doi: 10.1046/j.1460-9568.2003.02989.x. [DOI] [PubMed] [Google Scholar]
- 28.Borgdorff A.J., Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- 29.Eybalin M., Renard N., Aure F., Safieddine S. Cysteine-string protein in inner hair cells of the organ of Corti: Synaptic expression and upregulation at the onset of hearing. Eur. J. Neurosci. 2002;15:1409–1420. doi: 10.1046/j.1460-9568.2002.01978.x. [DOI] [PubMed] [Google Scholar]
- 30.Nemzou N.R.M., Bulankina A.V., Khimich D., Giese A., Moser T. Synaptic organization in cochlear inner hair cells deficient for the CaV1.3 (alpha1D) subunit of L-type Ca2+ channels. Neuroscience. 2006;141:1849–1860. doi: 10.1016/j.neuroscience.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 31.Almqvist J., Huang Y., Laaksonen A., Wang D.N., Hovmoller S. Docking and homology modeling explain inhibition of the human vesicular glutamate transporters. Protein Sci. 2007;16:1819–1829. doi: 10.1110/ps.072944707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reimer R.J., Edwards R.H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch. 2004;447:629–635. doi: 10.1007/s00424-003-1087-y. [DOI] [PubMed] [Google Scholar]
- 33.Cruickshanks K.J., Klein R., Klein B.E., Wiley T.L., Nondahl D.M., Tweed T.S. Cigarette smoking and hearing loss: The epidemiology of hearing loss study. JAMA. 1998;279:1715–1719. doi: 10.1001/jama.279.21.1715. [DOI] [PubMed] [Google Scholar]
- 34.Gabriel S.B., Schaffner S.F., Nguyen H., Moore J.M., Roy J., Blumenstiel B., Higgins J., DeFelice M., Lochner A., Faggart M. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 35.Kim T.B., Isaacson B., Sivakumaran T.A., Starr A., Keats B.J., Lesperance M.M. A gene responsible for autosomal dominant auditory neuropathy (AUNA1) maps to 13q14–21. J. Med. Genet. 2004;41:872–876. doi: 10.1136/jmg.2004.020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starr A., Isaacson B., Michalewski H.J., Zeng F.G., Kong Y.Y., Beale P., Paulson G.W., Keats B.J., Lesperance M.M. A dominantly inherited progressive deafness affecting distal auditory nerve and hair cells. J. Assoc. Res. Otolaryngol. 2004;5:411–426. doi: 10.1007/s10162-004-5014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Ballesteros M., del Castillo F.J., Martin Y., Moreno-Pelayo M.A., Morera C., Prieto F., Marco J., Morant A., Gallo-Teran J., Morales-Angulo C. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum. Mutat. 2003;22:451–456. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- 38.Puel J.L. Chemical synaptic transmission in the cochlea. Prog. Neurobiol. 1995;47:449–476. doi: 10.1016/0301-0082(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 39.Fremeau R.T., Burman J., Qureshi T., Tran C.H., Proctor J., Johnson J., Zhang H., Sulzer D., Copenhagen D.R., Storm-Mathisen J. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc. Natl. Acad. Sci. USA. 2002;99:14488–14493. doi: 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herzog E., Gilchrist J., Gras C., Muzerelle A., Ravassard P., Giros B., Gaspar P., El Mestikawy S. Localization of VGLUT3, the vesicular glutamate transporter type 3, in the rat brain. Neuroscience. 2004;123:983–1002. doi: 10.1016/j.neuroscience.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 41.Gras C., Herzog E., Bellenchi G.C., Bernard V., Ravassard P., Pohl M., Gasnier B., Giros B., El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J. Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schafer M.K., Varoqui H., Defamie N., Weihe E., Erickson J.D. Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J. Biol. Chem. 2002;277:50734–50748. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- 43.Wojcik S.M., Rhee J.S., Herzog E., Sigler A., Jahn R., Takamori S., Brose N., Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc. Natl. Acad. Sci. USA. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brandt A., Striessnig J., Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henry K.R., Chole R.A. Genotypic differences in behavioral, physiological and anatomical expressions of age-related hearing loss in the laboratory mouse. Audiology. 1980;19:369–383. doi: 10.3109/00206098009070071. [DOI] [PubMed] [Google Scholar]
- 46.Obholzer N., Wolfson S., Trapani J.G., Mo W., Nechiporuk A., Busch-Nentwich E., Seiler C., Sidi S., Sollner C., Duncan R.N. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J. Neurosci. 2008;28:2110–2118. doi: 10.1523/JNEUROSCI.5230-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajan I., Cline H.T. Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J. Neurosci. 1998;18:7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oestreicher E., Knipper M., Arnold A., Zenner H.P., Felix D. Neurotrophin 3 potentiates glutamatergic responses of IHC afferents in the cochlea in vivo. Eur. J. Neurosci. 2000;12:1584–1590. doi: 10.1046/j.1460-9568.2000.00049.x. [DOI] [PubMed] [Google Scholar]
- 49.Blaesse P., Ehrhardt S., Friauf E., Nothwang H.G. Developmental pattern of three vesicular glutamate transporters in the rat superior olivary complex. Cell Tissue Res. 2005;320:33–50. doi: 10.1007/s00441-004-1054-8. [DOI] [PubMed] [Google Scholar]
- 50.Boulland J.L., Qureshi T., Seal R.P., Rafiki A., Gundersen V., Bergersen L.H., Fremeau R.T., Edwards R.H., Storm-Mathisen J., Chaudhry F.A. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J. Comp. Neurol. 2004;480:264–280. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- 51.Gillespie D.C., Kim G., Kandler K. Inhibitory synapses in the developing auditory system are glutamatergic. Nat. Neurosci. 2005;8:332–338. doi: 10.1038/nn1397. [DOI] [PubMed] [Google Scholar]
- 52.Roehm P.C., Hansen M.R. Strategies to preserve or regenerate spiral ganglion neurons. Curr. Opin. Otolaryngol. Head Neck Surg. 2005;13:294–300. doi: 10.1097/01.moo.0000180919.68812.b9. [DOI] [PubMed] [Google Scholar]
- 53.Botstein D., Risch N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.