

The crystal structure of an N-terminally (25 residues) truncated fragment of selenophosphate synthetase (SPS-ΔN) from Aquifex aeolicus has been determined at 2.0 Å resolution.
Keywords: SelD, selenocysteine, selenophosphate, selenouridine
Abstract
Selenophosphate synthetase (SPS) catalyzes the activation of selenide with ATP to synthesize selenophosphate, the reactive selenium donor for biosyntheses of both the 21st amino acid selenocysteine and 2-selenouridine nucleotides in tRNA anticodons. The crystal structure of an N-terminally (25 residues) truncated fragment of SPS (SPS-ΔN) from Aquifex aeolicus has been determined at 2.0 Å resolution. The structure revealed SPS to be a two-domain α/β protein, with domain folds that are homologous to those of PurM-superfamily proteins. In the crystal, six monomers of SPS-ΔN form a hexamer of 204 kDa, whereas the molecular weight estimated by ultracentrifugation was ∼63 kDa, which is comparable to the calculated weight of the dimer (68 kDa).
1. Introduction
Selenophosphate synthetase (SPS) is an enzyme that is responsible for the activation of selenide with ATP to synthesize selenophosphate. Selenophosphate is the essential reactive selenium donor for the biosynthesis of the 21st amino acid selenocysteine (Sec; Leinfelder et al., 1990 ▶; Glass et al., 1993 ▶). The ∼37 kDa SPS protein is the product of the selD gene in bacteria and the corresponding protein is conserved in archaea/eukarya that possess Sec-synthesis/incorporation systems. Animals, including humans, possess two SPS homologues (SPS1 and SPS2). It is intriguing that some of the bacterial SPSs, all known archaeal SPSs and eukaryal SPS2 species are themselves Sec-containing proteins (selenoproteins), in which a catalytically important Cys residue in the N-terminal segment is replaced by Sec (Fleischmann et al., 1995 ▶; Low et al., 1995 ▶; Bult et al., 1996 ▶). According to sequence similarity, the SPS proteins are classified as members of the PurM superfamily (Anand et al., 2004 ▶; Morar et al., 2006 ▶). The selenophosphate synthesized by SPS is also utilized in the sulfur/selenium-exchange reaction to obtain the 2-selenouridine modification, such as 5-methylaminomethyl-2-selenouridine (mnm5Se2U) in tRNA anticodons, from the 2-thiouridine modification (Leinfelder et al., 1990 ▶; Veres et al., 1990 ▶, 1992 ▶).
SPS exhibits selenide water dikinase activity (EC 2.7.9.3), by which selenide and ATP are stoichiometrically converted to selenophosphate, orthophosphate and AMP (Veres et al., 1994 ▶),
The phosphate moiety of selenophosphate is derived from the ATP γ-phosphate, while the orthophosphate is from the β-phosphate (Ehrenreich et al., 1992 ▶). Mutational studies on Escherichia coli SPS have revealed that Cys17 and Lys20 in the N-terminal glycine-rich segment are crucial for activity (Kim et al., 1992 ▶, 1993 ▶). Based on isotope-exchange experiments under conditions excluding selenide, the reaction was hypothesized to involve a phosphorylated enzyme intermediate (Mullins et al., 1997 ▶; Walker et al., 1998 ▶). The Cys17 residue was presumed to be the phosphorylation site or the site that accepts selenide (Kim et al., 1992 ▶; Veres et al., 1994 ▶; Liu & Stadtman, 1997 ▶). In contrast, some reports argued against the essential role of the Cys residue (or the corresponding Sec residue in Sec-containing SPSs; Kim et al., 1997 ▶; Lacourciere & Stadtman, 1999 ▶). Others had speculated that the Sec (UGA) codon is embedded in the SPS gene as a translation control (Guimaraes et al., 1996 ▶; Kim et al., 1997 ▶; Tamura et al., 2004 ▶). Thus, the role of the Cys/Sec residue in catalysis and the reaction mechanisms still remain elusive and their solution awaits a crystallographic analysis of SPS.
As the initial step of our structural study, we determined the crystal structure of the N-terminally truncated inactive fragment of SPS (SPS-ΔN) from Aquifex aeolicus. The SPS protein is composed of the typical PurM-like α/β-fold. Interestingly, six monomers of SPS-ΔN form a hexamer of ∼204 kDa in the crystal, despite the fact that SPS-ΔN exists as a dimer in solution.
2. Materials and methods
2.1. Expression and purification of SPS-ΔN
ORF aq_1030, corresponding to an N-terminally truncated form of SPS (SPS-ΔN, amino-acid residues 26–336), was amplified from A. aeolicus genomic DNA (Deckert et al., 1998 ▶) by PCR and was cloned into vector pET25b (Novagen). E. coli strain BL21-CodonPlus(DE3)-RIL (Stratagene) was transformed with this vector and the protein was overexpressed. The harvested cells were suspended in buffer A (20 mM Tris–HCl pH 8.0, 2 mM dithiothreitol) and were disrupted using an ultrasonic homogenizer. After removing the cell debris by centrifugation, the lysate was heated at 343 K for 30 min to denature the host E. coli proteins.
The supernatant was applied onto a HiTrap Q column (GE Healthcare Biosciences) equilibrated with buffer A and the protein was eluted with a linear gradient of 0.0–1.0 M NaCl. The peak fraction was supplemented with 1.2 M ammonium sulfate and applied onto a Resource Phe column (GE Healthcare Biosciences). The protein was eluted with a reverse gradient of 1.2–0.0 M ammonium sulfate. The protein fraction was dialyzed against buffer A and was purified using a 0.0–1.0 M NaCl gradient on a HiTrap Q column (GE Healthcare Biosciences). The peak fraction was further purified on a HiLoad Superdex 75 column (GE Healthcare Biosciences) equilibrated with buffer A containing 150 mM NaCl. The purified protein was concentrated to 9.9 mg ml−1 using an Amicon Ultra centrifugal filter device (Millipore).
2.2. Crystallization and data collection
Crystal Screen and Crystal Screen II (Hampton Research) were used to determine the initial crystallization conditions for SPS-ΔN. Initial crystals were obtained with Crystal Screen II reagent No. 43 and the conditions were further refined. The crystals used for data collection were obtained by mixing 1 µl protein solution with 1 µl of a reservoir solution containing 100 mM Na HEPES pH 7.1, 50% 2-methyl-2,4-pentanediol, 200 mM ammonium phosphate and 50 mM ammonium sulfate and equilibrating this mixture against 500 µl reservoir solution at 293 K. X-ray data were collected from flash-cooled crystals at 90 K using synchrotron radiation at SPring-8 BL41XU (Harima, Japan). The data were processed with the HKL-2000 program (Otwinowski & Minor, 1997 ▶; Table 1 ▶). General handling of the scaled data was performed using the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). The SPS-ΔN crystals belong to space group C2221, with unit-cell parameters a = 93.2, b = 165.2, c = 167.7 Å. The Matthews coefficient (Matthews, 1968 ▶) and the solvent content were calculated to be 3.1 Å3 Da−1 and 60.8%, respectively, assuming the presence of three SPS-ΔN molecules in the asymmetric unit.
Table 1. Crystallographic data and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Native | Ethylmercury(II) chloride derivative |
|---|---|---|
| Data processing | ||
| Space group | C2221 | C2221 |
| Unit-cell parameters (Å) | a = 93.2, b = 165.2, c = 167.7 | a = 92.3, b = 163.9, c = 165.5 |
| Resolution (Å) | 50–2.0 | 50–3.3 |
| Unique reflections | 82314 | 18004 |
| Completeness (%) | 93.9 (94.7) | 93.4 (93.7) |
| Mean I/σ(I) | 18.3 (3.7) | 12.0 (2.8) |
| Rmerge† (%) | 9.7 (42.9) | 15.6 (65.2) |
| Phasing | ||
| No. of Hg sites | 9 | |
| FOM | 0.23 | |
| FOM after density modification | 0.62 | |
| Refinement | ||
| Resolution (Å) | 50–2.0 | |
| Reflections | 82004 | |
| Rcryst‡ (%) | 21.2 | |
| Rfree‡ (%) | 23.9 | |
| No. of protein atoms | 7077 | |
| No. of ligand/ion atoms | 15 | |
| No. of solvent atoms | 441 | |
| R.m.s.d. bonds (Å) | 0.15 | |
| R.m.s.d. angles (°) | 1.7 | |
| R.m.s.d. improper angles (°) | 1.3 |
R
merge = 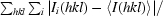
 , where I(hkl) is the observed intensity of reflection hkl.
, where I(hkl) is the observed intensity of reflection hkl.
R
cryst and R
free = 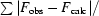
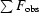 , where the crystallographic R factor is calculated including and excluding refinement reflections, respectively. In each refinement, the free reflections consisted of 5% of the total number of reflections.
, where the crystallographic R factor is calculated including and excluding refinement reflections, respectively. In each refinement, the free reflections consisted of 5% of the total number of reflections.
2.3. Structure determination and refinement
The crystal structure of SPS-ΔN was solved by the single isomorphous replacement (SIR) method. Several heavy-atom compounds were tested for the production of isomorphous heavy-atom derivatives, but only mercury derivatives were successful. The best mercury derivative was obtained by soaking the crystals for 15 h in reservoir solution containing 5 mM ethylmercury(II) chloride. The derivative data set was collected in the same manner as the native data set. Using the SOLVE program (Terwilliger & Berendzen, 1999 ▶), nine Hg positions (three sites per monomer) were determined, which facilitated the initial phase calculation. Phase improvement was carried out with the RESOLVE program (Terwilliger, 2000 ▶), which yielded an interpretable electron-density map. For model building, 74% and 42% of the amino-acid main chains and side chains, respectively, were automatically placed with the RESOLVE program (Terwilliger, 2003a ▶,b ▶) and the remaining parts were manually built using the O program (Jones et al., 1991 ▶). The model contains three SPS-ΔN molecules in the asymmetric unit. Structure refinement was carried out against the native data to 2.0 Å resolution using the CNS program (Brünger et al., 1998 ▶). After a number of cycles of manual revisions of the model using O, followed by simulated-annealing, positional and temperature-factor refinement using CNS, the R and R free values converged to 21.2% and 23.9%, respectively (Table 1 ▶). Amino-acid residues 26, 27, 105 and 106 in chain A, 26, 27, 42, 43 and 104–107 in chain B, and 26, 27, 43, 44 and 104–106 in chain C were excluded from the final model as the electron density was weak for these regions.
2.4. Ultracentrifugation analysis
To estimate the oligomeric state of SPS-ΔN in solution, the molecular weight was analyzed by ultracentrifugation. A sedimentation-equilibrium experiment was performed using an analytical ultracentrifuge (Optima XL-I, Beckman Coulter). Six-channel centrepieces were used, with each channel filled with 100 µl of sample or reference solution. The protein was dissolved in 20 mM Tris–HCl buffer pH 7.0 and concentrations of 0.7, 0.4 and 0.2 mg ml−1 were examined. An eight-position rotor (An-50 Ti) was rotated at 9000, 11 000 and 13 000 rev min−1 (5300g, 8000g and 11 200g, respectively) at 293 K. Data were collected after 12, 14 and 16 h at each speed by measuring the absorbance at 280 nm. The protein samples were then completely sedimented at 40 000 rev min−1 (106 000g) for 6 h to generate the baseline. Data were analyzed using a partial specific volume of 0.7498 ml g−1 and a solvent density of 1.00494 g ml−1 to determine the molecular weight using the XL-A/XL-I data-analysis software v.6.03.
3. Results and discussion
3.1. Structure determination
We started our crystallographic analysis with an N-terminally truncated fragment of SPS from A. aeolicus (ORF aq_1030, SPS-ΔN) as it was difficult to develop an overexpression system for the full-length SPS in the early stages of our study. The 25 N-terminal residues are missing in SPS-ΔN and therefore it lacks the catalytically important Cys/Sec and Lys residues (Sec13 and Lys16, respectively, in A. aeolicus SPS, corresponding to Cys17 and Lys20, respectively, in E. coli SPS). The SPS-ΔN crystal structure was solved by the single heavy-atom isomorphous replacement method with a mercury derivative and was refined against the native data to 2.0 Å, with crystallographic R work and R free factors of 21.2% and 23.9%, respectively (Table 1 ▶). The final model contains three nearly identical monomers in the asymmetric unit [the r.m.s.d.s from chain A for chains B and C (287 Cα atoms) are 0.26 and 0.37 Å, respectively]. The final model lacks the disordered regions: residues 26, 27, 105 and 106 in chain A, 26, 27, 42, 43 and 104–107 in chain B and 26, 27, 43, 44 and 104–106 in chain C.
3.2. Crystal structure of SPS-ΔN
SPS-ΔN is a mixed α/β protein consisting of two domains: the N- and C-terminal domains (residues 26–156 and 157–336, respectively; Fig. 1 ▶). The N-terminal domain consists of a six-stranded mixed β-sheet flanked by two α-helices. The C-terminal domain is composed of a seven-stranded mixed β-sheet, eight α-helices and a 310-helix. A structural homology analysis with the DALI program (Holm & Sander, 1998 ▶) indicated that the protein fold is the typical PurM-like fold (Table 2 ▶). SPS-ΔN shares high structural similarities with aminoimidazole ribonucleotide synthetase (PurM; Li et al., 1999 ▶), formylglycinamide ribonucleotide amidotransferase (PurL; Anand et al., 2004 ▶; Morar et al., 2006 ▶), thiamine-monophosphate kinase (ThiL) and [NiFe] hydrogenase maturation protein (HypE; Watanabe et al., 2007 ▶), which are members of the PurM superfamily, with Z scores of 15.3–26.2 and r.m.s.d.s of 2.5–3.9 Å, despite the low levels of sequence identity (10–20%; Table 2 ▶).
Figure 1.

The crystal structure of A. aeolicus SPS-ΔN. (a) Overall structure of the SPS-ΔN monomer shown in a ribbon representation. The α-helices and the β-strands are coloured red and yellow, respectively. The molecular graphics were prepared with PyMOL (http://www.pymol.org). (b) The same structure as in (a), but with the N- and C-terminal domains coloured blue and cyan, respectively. The N-terminal segment (residues 26–43), which mediates most of the intermolecular interactions in the hexamer, is highlighted in magenta. (c) Distribution of the surface electrostatic potential.
Table 2. Structural alignment statistics of SPS-ΔN (chain A) with other proteins belonging to the PurM superfamily as evaluated using the DALI algorithm.
| Protein | PDB code | Z† | R.m.s.d.‡ | LALI§ | LSEQ2¶ | %IDE†† |
|---|---|---|---|---|---|---|
| [NiFe] hydrogenase maturation protein (HypE; Thermococcus kodakaraensis) | 2z1e | 26.2 | 2.5 | 256 | 298 | 20 |
| Thiamine monophosphate kinase (ThiL; A. aeolicus) | 1vqv | 25.3 | 2.9 | 255 | 287 | 18 |
| Aminoimidazole ribonucleotide synthetase (PurM; E. coli) | 1cli | 21.1 | 3.2 | 256 | 341 | 16 |
| Formylglycinamide ribonucleotide amidotransferase (PurL; Thermotoga maritima) | 2hs3 | 19.6 | 3.7 | 247 | 602 | 18 |
| Formylglycinamide ribonucleotide amidotransferase (PurL; Salmonella typhimurium) | 1t3t | 15.3 | 3.9 | 250 | 1283 | 11 |
Z score; the strength of the structural similarity in standard deviations above the expected value.
Positional root-mean-square deviation of superimposed Cα atoms (in Å).
Total number of equivalent residues.
Length of the entire chain of the equivalent structure.
Percentage sequence identity over equivalent positions.
3.3. Unique oligomerization state in the crystal
The crystallographic asymmetric unit contains three SPS-ΔN monomers. A twofold symmetry operation around the b axis generates three more SPS-ΔN monomers and the six monomers form a hexamer of ∼204 kDa (Fig. 2 ▶). The hexamer adopts a trimer-of-dimers configuration. The N-terminal domains form a huge central β-barrel, consisting of 21 β-strands with threefold noncrystallographic symmetry (3 × 7 strands). The β-strands are mostly in an antiparallel configuration, except for a parallel interaction between two strands (residues 94–98 and 127–132). The diameter and the length of the β-barrel are ∼35 and ∼65 Å, respectively. The N-terminal segment (residues 26–43) forms a long antiparallel β-sheet with the corresponding segment of another subunit (Fig. 2 ▶ c) and also forms a short antiparallel β-sheet with a strand (residues 129–131) in the other subunit (Fig. 2 ▶ d). Thus, the N-terminal segment mediates most of the intermolecular interactions in the hexamer.
Figure 2.

Oligomerization state of SPS-ΔN in the crystal. (a) The SPS-ΔN hexamer in the crystal. One SPS-ΔN monomer is coloured as in Fig. 1 ▶(b) and the other five monomers are coloured pink, orange, yellow, green and slate, respectively. The crystallographic twofold axis (b axis) is shown as a black ellipse in the middle. (b) The same structure as in (a), but viewed from the direction rotated by 90° around the horizontal axis. (c, d) Two possible dimer units in the hexamer are shown.
However, this hexameric oligomerization differs from those observed in other members of the PurM superfamily. The crystal structures of PurM, ThiL and HypE revealed that they all form homodimers by creating a smaller barrel in the middle (Li et al., 1999 ▶; Watanabe et al., 2007 ▶). PurL is a monomeric protein with two intramolecular repeats which form a pseudo-dimer in the same configuration as PurM (Anand et al., 2004 ▶; Morar et al., 2006 ▶). To address this discrepancy, we carried out an ultracentrifugation analysis in order to determine the molecular weight of SPS in solution (Fig. 3 ▶). A sedimentation-equilibrium experiment under physiological conditions provided an estimated molecular weight of ∼63.0 kDa, which is close to the calculated weight of the SPS-ΔN dimer (68.2 kDa). Based on the results of the analytical ultracentrifugation and the structural organization of the other PurM-superfamily proteins, it is most likely that the hexamer results from crystal packing and does not reflect a physiological oligomerization state. The lack of the N-terminal short segment (residues 1–25) in SPS-ΔN might be one of the causes of the crystallographic oligomerization, as the N-terminal region is principally involved in the hexamerization. However, although it is less likely, the possibility remains that the hexameric state is related to some physiological function other than selenophosphate synthesis.
Figure 3.

Determination of the molecular weight by analytical ultracentrifugation. Data obtained at a protein concentration of 0.4 mg ml−1 and a rotation speed of 11 000 rev min−1 (8000g) are shown. The absorbance gradient at 280 nm in the centrifugal cell after attaining sedimentation equilibrium is shown in the bottom panel. The open circles are experimental values and the solid line represents a fit to a single ideal species. The top panel shows the residuals, which represent the difference between the fit and experimental values as a function of the radial position.
Supplementary Material
PDB reference: selenophosphate synthetase, 2zau, r2zausf
Acknowledgments
We thank Drs N. Shimizu and M. Kawamoto for supporting our data collection at SPring-8 beamline BL41XU. We thank E. Fusatomi for supporting our protein preparation. We are grateful to Dr K. Stetter for the generous gift of A. aeolicus genomic DNA. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative, the National Project on Protein Structural and Functional Analyses.
References
- Anand, R., Hoskins, A. A., Stubbe, J. & Ealick, S. E. (2004). Biochemistry, 43, 10328–10342. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bult, C. J. et al. (1996). Science, 273, 1058–1073. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Deckert, G. et al. (1998). Nature (London), 392, 353–358.
- Ehrenreich, A., Forchhammer, K., Tormay, P., Veprek, B. & Böck, A. (1992). Eur. J. Biochem.206, 767–773. [DOI] [PubMed]
- Fleischmann, R. D. et al. (1995). Science, 269, 496–512. [DOI] [PubMed]
- Glass, R. S., Singh, W. P., Jung, W., Veres, Z., Scholz, T. D. & Stadtman, T. C. (1993). Biochemistry, 32, 12555–12559. [DOI] [PubMed]
- Guimaraes, M. J., Peterson, D., Vicari, A., Cocks, B. G., Copeland, N. G., Gilbert, D. J., Jenkins, N. A., Ferrick, D. A., Kastelein, R. A., Bazan, J. F. & Zlotnik, A. (1996). Proc. Natl Acad. Sci. USA, 93, 15086–15091. [DOI] [PMC free article] [PubMed]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res.26, 316–319. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kim, I. Y., Guimaraes, M. J., Zlotnik, A., Bazan, J. F. & Stadtman, T. C. (1997). Proc. Natl Acad. Sci. USA, 94, 418–421. [DOI] [PMC free article] [PubMed]
- Kim, I. Y., Veres, Z. & Stadtman, T. C. (1992). J. Biol. Chem.267, 19650–19654. [PubMed]
- Kim, I. Y., Veres, Z. & Stadtman, T. C. (1993). J. Biol. Chem.268, 27020–27025. [PubMed]
- Lacourciere, G. M. & Stadtman, T. C. (1999). Proc. Natl Acad. Sci. USA, 96, 44–48. [DOI] [PMC free article] [PubMed]
- Leinfelder, W., Forchhammer, K., Veprek, B., Zehelein, E. & Böck, A. (1990). Proc. Natl Acad. Sci. USA, 87, 543–547. [DOI] [PMC free article] [PubMed]
- Li, C., Kappock, T. J., Stubbe, J., Weaver, T. M. & Ealick, S. E. (1999). Structure, 7, 1155–1166. [DOI] [PubMed]
- Liu, S. Y. & Stadtman, T. C. (1997). Arch. Biochem. Biophys.341, 353–359. [DOI] [PubMed]
- Low, S. C., Harney, J. W. & Berry, M. J. (1995). J. Biol. Chem.270, 21659–21664. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Morar, M., Anand, R., Hoskins, A. A., Stubbe, J. & Ealick, S. E. (2006). Biochemistry, 45, 14880–14895. [DOI] [PMC free article] [PubMed]
- Mullins, L. S., Hong, S.-B., Gibson, G. E., Walker, H., Stadtman, T. C. & Raushel, F. M. (1997). J. Am. Chem. Soc.119, 6684–6685.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Tamura, T., Yamamoto, S., Takahata, M., Sakaguchi, H., Tanaka, H., Stadtman, T. C. & Inagaki, K. (2004). Proc. Natl Acad. Sci. USA, 101, 16162–16167. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2003a). Acta Cryst. D59, 38–44. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2003b). Acta Cryst. D59, 45–49. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Veres, Z., Kim, I. Y., Scholz, T. D. & Stadtman, T. C. (1994). J. Biol. Chem.269, 10597–10603. [PubMed]
- Veres, Z., Tsai, L., Politino, M. & Stadtman, T. C. (1990). Proc. Natl Acad. Sci. USA, 87, 6341–6344. [DOI] [PMC free article] [PubMed]
- Veres, Z., Tsai, L., Scholz, T. D., Politino, M., Balaban, R. S. & Stadtman, T. C. (1992). Proc. Natl Acad. Sci. USA, 89, 2975–2979. [DOI] [PMC free article] [PubMed]
- Walker, H., Ferretti, J. A. & Stadtman, T. C. (1998). Proc. Natl Acad. Sci. USA, 95, 2180–2185. [DOI] [PMC free article] [PubMed]
- Watanabe, S., Matsumi, R., Arai, T., Atomi, H., Imanaka, T. & Miki, K. (2007). Mol. Cell, 27, 29–40. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: selenophosphate synthetase, 2zau, r2zausf