

Summary
The generation of recombinant antibodies (Abs) using phage display is a proven method to obtain a large variety of Abs that bind with high affinity to a given antigen (Ag). Traditionally, the generation of single chain Abs depends on the use of recombinant proteins in several stages of the procedure. This can be a problem, especially in the case of cell surface receptors, because Abs generated and selected against recombinant proteins may not bind the same protein expressed on a cell surface in its native form and because the expression of some receptors as recombinant proteins is problematic. To overcome these difficulties, we developed a strategy to generate single chain Abs that does not require the use of recombinant protein at any stage of the procedure. In this strategy, stably transfected cells are used for the immunization of mice, measuring Ab responses to immunization, panning the phage library, high throughput screening of arrayed phage clones, and characterization of recombinant single chain variable regions (scFvs). This strategy was used to generate a panel of single chain Abs specific for the innate immunity receptor Toll-like receptor 2 (TLR2). Once generated, individual scFvs were subcloned into an expression vector allowing the production of recombinant Abs in insect cells, thus avoiding the contamination of recombinant Abs with microbial products. This cell-based system efficiently generates Abs that bind to native molecules on the cell surface, bypasses the requirement of recombinant protein production, and avoids risks of microbial component contamination.
Keywords: Toll-like receptor, antibody, phage-displayed antibody, cell surface antigen, flow cytometry
Introduction
Hybridoma1 and phage-display recombinant antibody systems2 are currently the predominant methods for obtaining monoclonal antibodies. Display of single chain variable fragment (scFv) antibodies on the surface of bacteriophage M13 has numerous advantages compared to traditional hybridoma technology. The number of phage antibodies that can be generated and screened is vast compared to the number of hybridoma cells that can be produced and examined in the same period of time, or from the same number of immunized animals. A process of re-assortment occurs during the heavy and light chain assembly that forms scFvs wherein the heavy chain from one antibody can join to the light chain from a different antibody, creating a unique antibody that was not present in the immune repertoire of the donor animal. This combinatorial re-assortment can create antibodies recognizing epitopes that might not have elicited an antibody response due to tolerance, as well as scFv antibodies binding to antigens that are too toxic to use as immunogens. Recombinant antibodies such as scFvs are produced and maintained using standard molecular biology techniques, which provide greater genetic stability than hybridomas and allow the re-engineering of scFvs into other formats.
Current methods for generating recombinant antibodies typically rely heavily on the use of recombinant proteins for several procedures, including immunizations, library enrichment, clone screening, and characterization of antibody specificity and affinity. However, for many cell surface molecules, the use of recombinant proteins for Ab generation may not be feasible. Some surface molecules, such as G protein coupled receptors, cannot be expressed in recombinant forms that retain their native conformation. Moreover, molecules with large extracellular domains may contain complex structures that are not properly formed during recombinant expression. Many standard screening practices, such as the immobilization of recombinant proteins on plastic, may significantly alter protein conformation. For these reasons, Abs selected on the basis of binding to a recombinant protein may not bind the same protein when it is in its native context.
Toll-like receptors (TLR) encompass a family of cell surface receptors that recognize conserved pathogen-associated molecular patterns3. TLRs appear to be structurally complex proteins that are not readily amenable to standard Ab production methods. To date, 13 mammalian TLRs have been identified that play important roles in modulating both innate and adaptive immune functions during infections, inflammatory events, and autoimmune diseases4; 5; 6; 7 TLRs appear to be vital for immune response, yet studies of TLR biochemistry have been severely limited due to a lack of reagents. Production of recombinant TLRs has proven to be difficult and most existing antibodies are either unreliable or not useful for biochemical analysis due to their failure to bind native TLR proteins (NIAID Biodefense Workshop Summary, http://www3.niaid.nih.gov/about/organization/dait/conferences.htm). To perform functional studies and explore the activity of TLRs in a physiologic setting, it is critical to have antibodies that recognize native TLRs expressed on the surface of cells.
Here, we describe a strategy to generate recombinant anti-TLR antibodies that specifically recognize the receptor protein in its native conformation. This strategy requires no recombinant protein, instead utilizing antigen expressed on the surface of stable transfectants for immunizations, measuring Ab responses in immunized mice, library panning, clone screening, and scFv clone characterization. This cell-based system was able to efficiently generate multiple scFvs that bind to cell-surface TLR2 while no such scFvs were generated using our standard protein-based methods. These results demonstrate that the use of an entirely cell-based strategy to generate recombinant Abs against cell surface proteins is feasible and suggest that such a strategy is applicable to wide variety of cell surface proteins.
Results
Immunization and its evaluation by flow cytometry or ELISA
To maximize Ab responses to native cell surface receptors, mice were immunized with cells from the 300.19 mouse tumor cell line stably transfected with hTLR2. This immunization strategy has been shown to result in robust Ab responses against transfected proteins and some tumor Ags8. To maximize the diversity of the immune response, the animals immunized included wild type BALB/c mice (mus musculus) and TLR2-deficient C57BL/6 mice9, which were expected to develop increased anti-hTLR2 immune responses due to a lack of tolerance to epitopes shared with murine TLR2.
After 6 rounds of immunization, anti-hTLR2 IgG titers were determined in two ways. First, consistent with standard methods, Ab titers were measured using ELISA assays against recombinant hTLR2. Second, to avoid the use of recombinant protein and evaluate the response of Abs that bind to native cell surface-expressed hTLR2, Ab titers were determined by measuring the intensity with which serum from immunized mice stained hTLR2-transfected cells in a flow cytometric assay. To eliminate the background caused by serum binding to molecules other than hTLR2, the staining intensity on non-transfected cells was subtracted from each Ab sample and dilution.
As shown in figure 1, both protein- and cell-based assays indicated robust serum antibody responses to hTLR2 in immunized mice with no signal detected in serum from non-immunized mice. The rate at which specific signal decreased in serial dilutions was similar between the assays. ELISA assays displayed a greater variability in signal among individual mice but it is not clear if this is biologically significant. Both assays identified WT mouse #4 as having the greatest response to immunization. These findings demonstrate that determination of serum Ab responses in a flow cytometric assay provides results comparable to protein-based ELISA assays and that immunization with transfected cells results in robust anti-hTLR2 Ab responses. The use of TLR2-deficient mice appears to offer no advantage in terms of overall Ab response.
Figure 1.
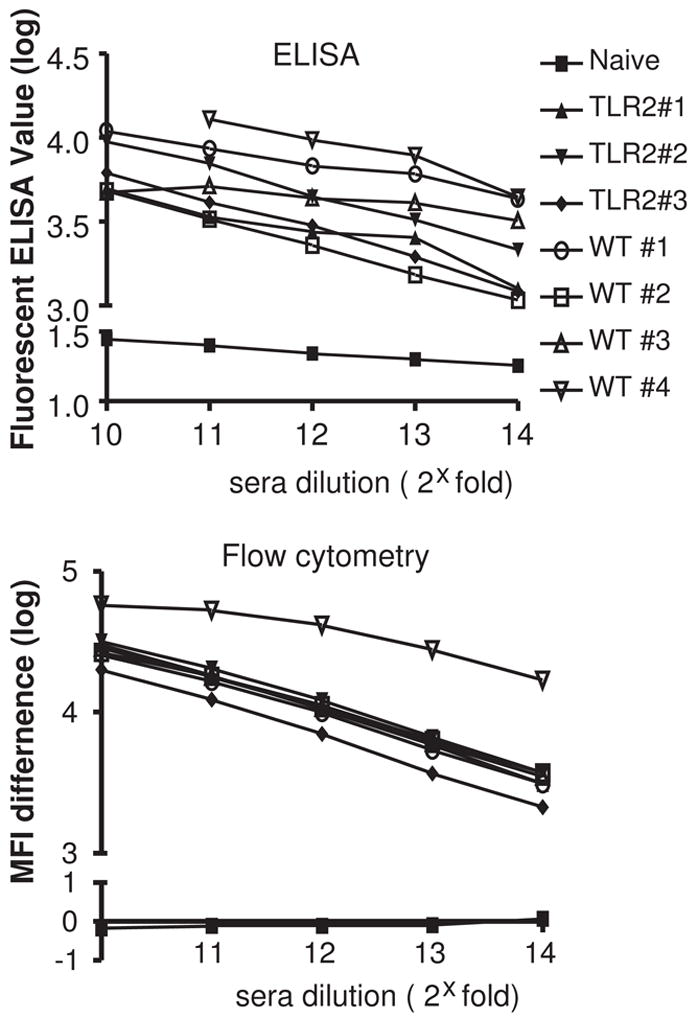
Protein-based or cell-based assays to evaluate mouse hTLR2-specific IgG responses after immunization. Sera from 3 TLR2−/− BL6 mice and 4 WT BL6 mice were harvested 7 days after the 5th boost. Serial 2-fold dilutions were assayed for binding against recombinant hTLR2 protein by ELISA (top) or against hTLR2-HEK cells by flow-cytometry (bottom). Both assays utilized labeled anti-mouse IgG as a secondary Ab to detected hTLR2-specific IgG binding. In the cell-based assay, hTLR2-specific binding was measured as the difference in MFI between hTLR2-HEK and control WT-HEK staining. Serum from non-immunized mice was used as a negative control (naïve).
Phage library construction
Spleens from six mice identified as having high titer immune responses were used for construction of an M13 phage library of scFv antibodies. Multiple mice were selected from different strains in an effort to increase diversity of the scFv phage clones obtained. After reverse transcription of spleen mRNA, separate PCR reactions were performed to specifically amplify the variable regions of the heavy (VH) and light chain (VL) genes from each mouse. The resulting VH and VL regions were pooled in equimolar amounts for assembly into scFv inserts, allowing the random association of heavy and light chains from different mice to maximize combinatorial reassortment. A theoretical complexity of 3x106 phage clones was obtained for this anti-hTLR2 scFv library.
Selection of anti-hTLR2 scFv clones using recombinant protein or stable hTLR2 transfectants
The use of recombinant protein to select phage displaying scFvs specific for that protein is simple and rapid using well-characterized methods10, often requiring only a single round to obtain 1000-fold enrichment for antigen-specific phage11. We performed parallel selections from our anti-hTLR2 scFv library to compare the efficacy of cell-based selections with recombinant protein selections for obtaining antibodies specific for cell surface hTLR2 as determined by flow cytometry measurements. A single round of selection for phage that bind to recombinant hTLR2 adsorbed to polystyrene was performed. Absorbance values from ELISA measurements using pools of phage from the selected phage versus the starting library demonstrated a high degree of enrichment for hTLR2 binding (Figure 2a). This enrichment is specific to hTLR2, since no enrichment was seen for binding to a control His-tagged recombinant protein.
Figure 2.
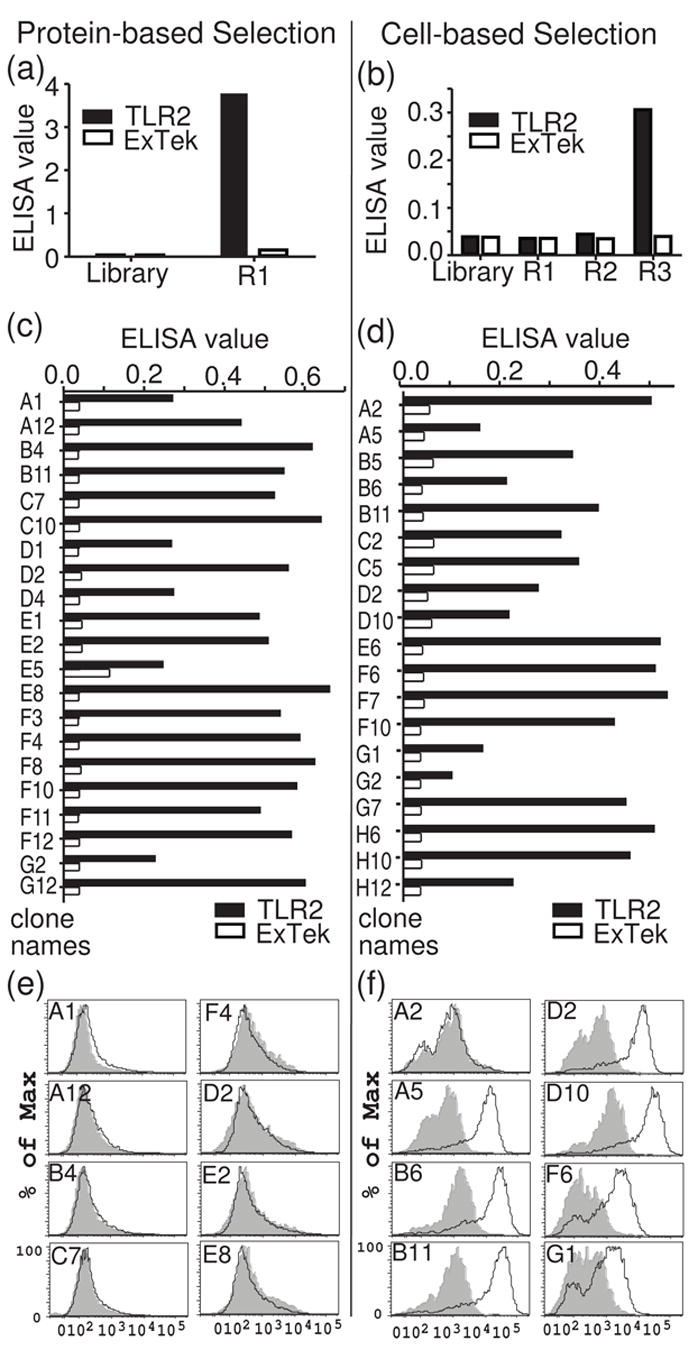
Comparison of protein-based and cell-based selections. (a, b) Equal numbers of phage (1010 phage) from the starting M13 library, after immunotube selection (a), or after three rounds of BRASIL selection (b) were assayed for binding to recombinant hTLR2 by ELISA. A labeled anti-M13 antibody was used for detection of bound phage. (c, d) Binding of individual phage clones to recombinant hTLR2 after round 1 of immunotube selection (c) or round 3 of BRASIL selection (d). Phage clones were prepared in 96-well plates and tested in ELISA assays against recombinant hTLR2-extracellular domain (TLR2) or a control protein (ExTek). (e, f) Binding of individual phage clones to cell-surface hTLR2 as assessed by flow cytometry. Unique ELISA-positive phage clones were incubated at 1012 phage/ml with hTLR2-HEK cells (black lines) or WT-HEK cells (gray shadow) and examined by flow cytometry (Y-axis: % of maximum cell number. X-axis: log scale of fluorescent intensity for phage staining in APC channel).
The cell-based selection procedure12 we employed relies on differential centrifugation to obtain phage clones that specifically recognize cell-surface antigen. The anti-hTLR2 scFv phage library was pre-cleared by incubating it with parental HEK293 cells in an aqueous solution, which was then layered over a non-polar organic phase and centrifuged. The HEK293 cells and any phage bound to the cells formed a pellet in the lower organic phase. The pre-cleared phage remaining in the aqueous phase were then incubated with hTLR2-HEK cells. Following washes to remove the bulk of unbound phage, the cells were again centrifuged through the non-polar organic phase. Pelleting the cells through this non-polar solution dissociated phage loosely bound due to non-specific interactions, reducing the background of irrelevant phage. Phage bound to the hTLR2-HEK cells were used to infect bacteria and generate enriched phage stocks for iterative rounds of selections. Three iterative rounds of enrichment were performed. To determine the degree of enrichment for hTLR2-specific phage, ELISA measurements were done to compare binding of pooled phage from each round of selection with the starting library. Three rounds of selection were required to obtain an appreciable degree of enrichment for hTLR2-binding phage clones (Figure 2b). The absorbance value for this third round of cell-based selection was not as high as that of the single round of enrichment with recombinant hTLR2.
Individual phage clones from round 1 of recombinant hTLR2 selection and round 3 of the cell-based selection were assayed for binding to recombinant hTLR2 by ELISA. Numerous phage clones from both selections showed a high degree of specific binding to hTLR2 (Figure 2 c and d). Clones from the cell-based selection showed absorbance values in ELISA assays that were comparable to those obtained by recombinant hTLR2 selection. This suggests that although the cell-based phage pool displays a lower degree of enrichment than the pool obtained by recombinant hTLR2 panning (Figure 2a vs. Figure 2b), the cell-based selection is nonetheless capable of identifying clones that bind with similar affinities in ELISA assays.
To determine the number of distinct phage clones obtained from each selection, the phage clones identified binding to hTLR2 in ELISA screens were examined by DNA fingerprinting of the scFv inserts. Screening of 96 protein-selected clones by ELISA identified 21 clones that display significant binding to recombinant hTLR2 (Figure 2C). Among these, 12 clones were unique (Table 1). Screening of 154 cell-selected clones by ELISA identified 19 that bound recombinant hTLR2. Of these, 8 clones proved to be unique (Table 1). One clone from each set of duplicates was used for further analysis.
Table 1.
Relative efficiencies of protein-based versus cell-based selection and screening methods.
| Initial screening-Raw phage | Fingerprint | Purified phage binding to cells | scFv binding to cells | |||
|---|---|---|---|---|---|---|
| Selected on | Screened by | Number of clones screened | Number of Unique clones | Number of clones positive | Number of clones positive | Number of clones positive |
| Protein | ELISA | 96 | 21 (21%) | 12/21 (57%) | 0/12 (0%) | No data |
|
|
||||||
| cells | ELISA | 154 | 19 (12%) | 8/19 (42%) | 7/8 (88%) | 5/5 (100%) |
| Flow cytometry | 168 | 78 (46%) | 9/23 (39%) | 8/9 (89%) | 5/5 (100%) | |
We next compared the ability of the hTLR2-binding clones obtained from these two selections to bind cell surface hTLR2. A concentrated, highly purified stock of M13 phage particles for each clone was prepared and used as the primary staining reagent in flow cytometry13; 14. As a negative control, we employed the anti-ExTek phage clone 1D6 (Lipes and Kenan, manuscript in preparation), which binds to the extracellular domain of Tie2. Since Tie2 is an endothelial cell-specific receptor tyrosine kinase15, this phage clone is well suited for use as a control with HEK cells. When tested by flow cytometry, none of the clones obtained by selection with recombinant protein bound to cell-surface hTLR2 (Fig 2e and 4b), despite binding to recombinant hTLR2 in ELISA assays. Performing a second round of selection with recombinant hTLR2 did not produce any additional clones, instead yielding multiple clones representing a small subset of the round one positive clones. In contrast, 7 of the 8 unique clones from the cell-based selection were positive for cell surface hTLR2 binding (Figure 2f, 4a and Table 1). These results demonstrate that cell-based selection is substantially more efficient than panning on protein-absorbed plates for enriching phage clones that bind native hTLR2 expressed on the cell surface.
Figure 4.
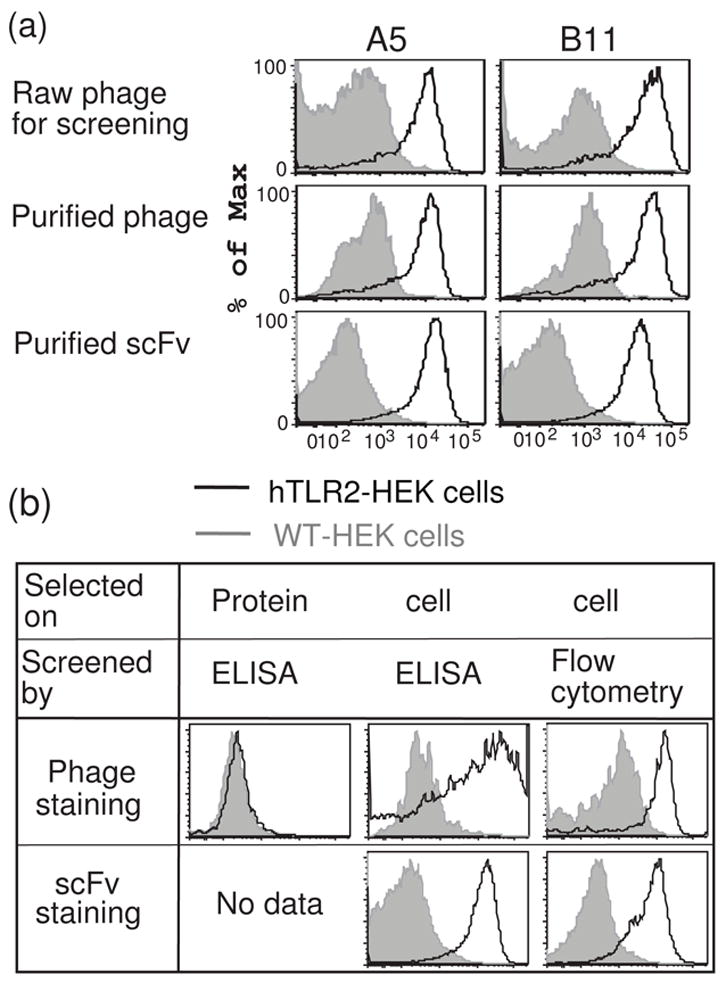
Representative histograms of flow-cytometric staining profiles obtained from individual clones at each stage of screening and validation. (a) A5 and B11, two clones that bind cell-surface hTLR2, demonstrate hTLR2-specific staining during flow cytometric screening of raw phage preparations (top), when prepared as purified phage (middle), and when expressed as purified scFvs (bottom). (b) Representative results of flow-cytometric validation assays of clones obtained by three distinct selection and screening methods. Validation includes assays of purified phage (top row) or purified scFv (bottom row) for binding to hTLR2-HEK cells (black lines) versus WT-HEK cells (gray shadow). Histograms are formatted as in figure 3a.
High throughput flow-based screening for positive clones
Having determined the efficiency of cell-based selection, we next sought to streamline the screening process and completely eliminate the requirement for recombinant TLR2 protein. To achieve this, we developed a 96-well plate flow cytometric screening procedure to directly identify phage clones that bind cell surface hTLR2. This procedure eliminates the need to perform screening by ELISA, removing one step from the previous screening method.
For this high-throughput assay, equal numbers of hTLR2-HEK cells and HEK cells stably transfected with yellow fluorescent protein (YFP-HEK cells) were mixed, stained with individual phage clones in 96-well plates, and subjected to flow cytometric analysis using an automatic plate-loader. The YFP-HEK cells served as an internal negative control for each sample. By combining target and negative control cells in every well, we ensured that both cell types were exposed to identical staining conditions and also lowered the sample number and the amount of staining reagent that was required. In pilot studies, we demonstrated clear separation of YFP+/hTLR2− and YFP−/hTLR2+ HEK cells in the FITC channel (Figure 3a). Detection of phage binding to cells was accomplished using an anti-M13 Ab and APC-conjugated-anti mouse IgG as secondary and tertiary reagents, respectively. The anti-Tie2 1D6 phage clone was used as a negative control. Previously identified phage clones demonstrated to bind cell surface hTLR2 were used as positive controls. In initial studies, we found that addition of serum to the raw phage preparations causes non-specific phage binding to cells (data not shown). Thus it was critical to use a serum-free staining buffer throughout the process. Employing this screening method to evaluate a total of 168 cell-selected phage clones, we identified 78 clones that bound specifically to hTLR2-HEK cells (Figure 3b and Table 1). The results shown in figure 3b are representative of all 96-well plates screened, revealing hTLR2-specific clones (e.g. A3 and A4 in this figure), non-binding clones (B4 and B5), and clones that bind to YFP-HEK cells (D5) in various ratios. By eliminating ELISA assays and directly screening with flow cytometry, we were able to rapidly identify large numbers of phage clones that specifically recognize cell surface hTLR2. 23 of these clones were subjected to fingerprinting analysis, resulting in the identification of 9 unique clones (Table 1). When prepared as purified phage, 8 of these 9 unique clones bind to native hTLR2 in a flow cytometric assay (Figure 4a). As discussed further below, these clones also bind to native hTLR2 when they are prepared as purified scFvs (Figure 4). These results demonstrate that flow-cytometric screening of raw phage preparations is a rapid and efficient method to identify Abs that bind to cell surface Ags.
Figure 3.
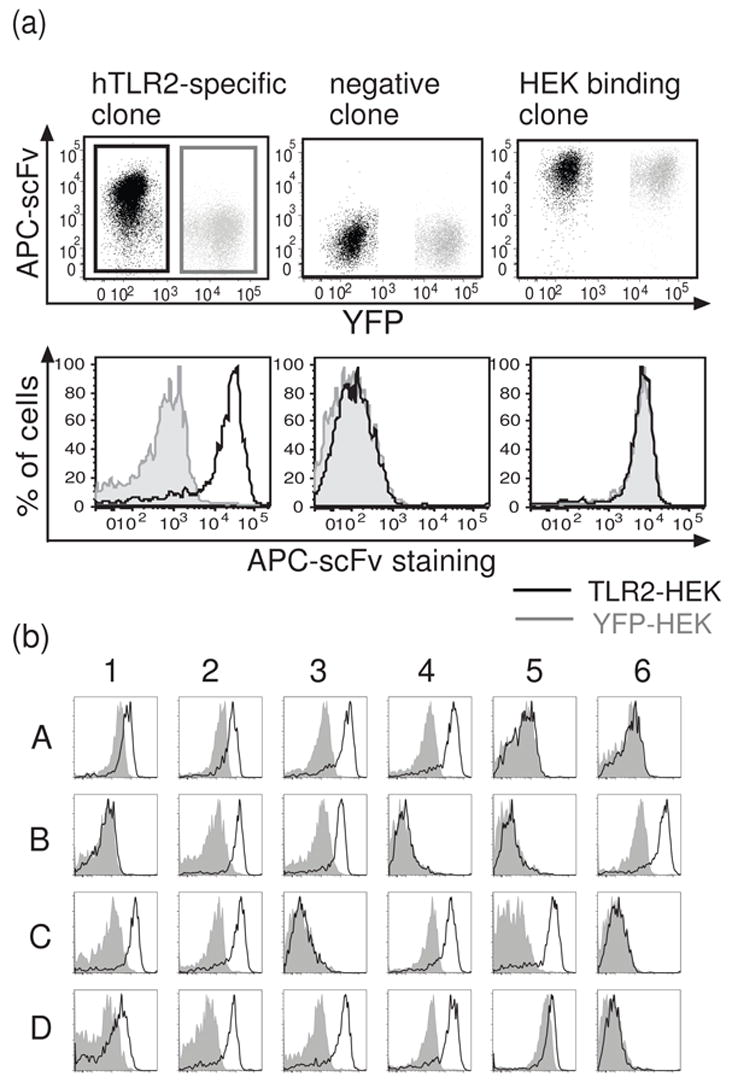
Flow cytometric screening of phage clones for binding to cell-surface hTLR2. (a) Representative scatter plots and histograms demonstrating typical results for hTLR2-specific clones (left), non-binding clones (center), and clones that bind to all HEK cells (right). Equal numbers of hTLR2-HEK and YFP-HEK cells were mixed and stained with raw phage preparations of individual clones. hTLR2-HEK cells (black gate) were distinguished from control YFP-HEK cells (gray gate) in the FITC channel as shown and the binding of phage to each population displayed as overlays of histograms (black line -hTLR2-HEK cells; gray shadow - YFP-HEK cells). (b) Typical results of flow cytometry-based screening. Results for 24 out of 96 wells in one screen of the cell-selected library are show. Histograms are formatted as in (a).
Expression of soluble scFv antibodies in Drosophila
The preceding flow cytometry measurements were performed using a normalized titer of purified phage, allowing us to make preliminary assessments as to which phage-conjugated scFv antibodies bind cell-surface hTLR2. The most promising candidates were chosen for expression as soluble scFvs. The simplest method for obtaining soluble scFvs is expression from the phagemid vector in a non-suppressor strain of E. coli (Escherichia coli). However, many scFvs either fail to express at acceptable levels in E. coli or form inclusion bodies of incorrectly folded non-functional protein. Furthermore, we found that scFvs purified from E. coli are prone to contamination with endotoxin (data not shown). This endotoxin contamination causes significant problems in functional assays involving TLRs or other components of the immune system since even slight levels of contamination with bacterial components may activate leukocytes and profoundly alter experimental results. Therefore, it is desirable to generate antibodies using a bacteria-free expression system.
We employed an insect cell expression system to overcome these problems. ScFv insert sequences were recovered by PCR amplification from positive phagemid clones, then ligated with topoisomerase into a donor plasmid between recombination sequences for a bacteriophage recombination enzyme. Corresponding recombination sequences were added to a Drosophila expression vector to allow rapid transfer of the scFv inserts into this vector for expression in S2 insect cells. To date, we have successfully expressed dozens of scFv antibodies against various antigens as secreted products in stably transfected S2 cells. There have been no instances in which an antibody failed to express in the Drosophila system, in contrast to the recognized scFv expression problems in the E. coli system. In the case of hTLR2, 5 out of 5 clones from both the protein-based selection and the cell-based selection specifically bind to hTLR2-HEK cells when expressed as scFvs by this method (Fig. 4, Table 1) and provoke no endotoxin-mediated stimulation of immune cells (data not shown). Typical yields for scFvs in the Drosophila expression system are 1– 2 mg/L of culture, and range from 0.5 to 5 mg/L. This is tenfold higher than the typical yield of 0.1– 0.2 mg/L from E. coli.
Staining of endogenous hTLR2 with scFvs
To determine whether scFvs selected against ectopically expressed hTLR2 would bind to the endogenous receptor on human cells, we examined hTLR2 expression on two human cell lines: THP-1 cells16 (a macrophage cell line) and CaCo-2 cells17 (an intestinal epithelial cell line). Both soluble scFvs tested, A5 and B11, demonstrate specific binding to both THP-1 and CaCo-2 cells, generating a robust signal at scFv concentrations of 0.1 ug/ml (Figure 5a). One scFv, B11, was also tested on freshly isolated bronchioalveolar cells obtained by lavage from a patient with known pulmonary inflammation. B11 demonstrates specific staining of an SSC-low cell population (Figure 5b). These results demonstrate that a cell-based system of phage selection and screening efficiently generates scFvs that bind specifically to endogenous cell surface proteins.
Figure 5.
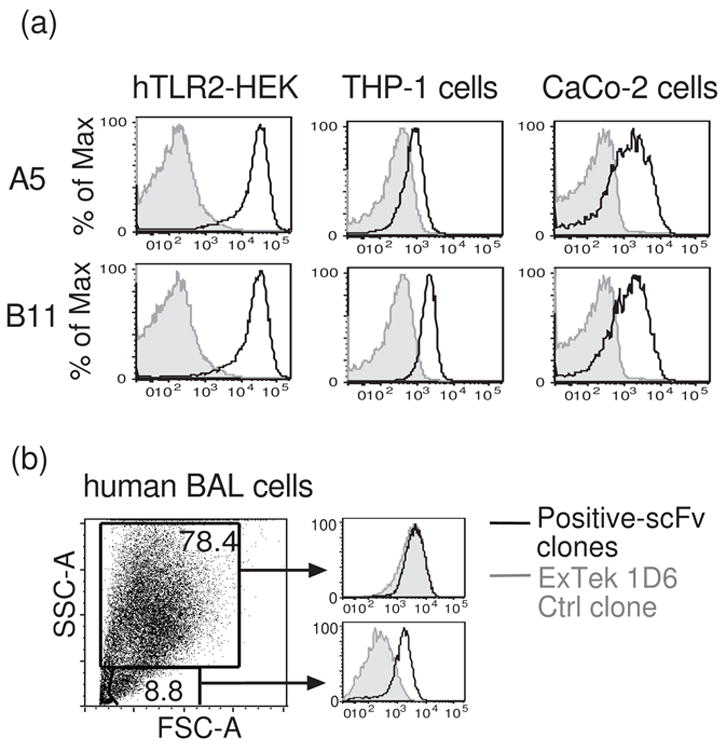
Anti-hTLR2 scFvs specifically bind to endogenous hTLR2. (a) Flow cytometric profiles of the binding of two scFv clones to human THP-1 and CaCo-2 cells. The binding of the clones to hTLR2-HEK cells is shown for comparison. (b) Binding of an anti-hTLR2 scFv to human bronchial alveolar lavage (BAL) cells obtained during pulmonary inflammation. Only small SSC-low cells demonstrate specific staining for hTLR2. In each panel, signals obtained with anti-hTLR2 scFvs (black lines) were compared with signals obtained with an anti-ExTek negative control (gray shadows). Histograms are formatted as in figure 3a.
Discussion
Here we describe a completely cell-based procedure for the generation of antibodies specific for cell-surface proteins (summarized in Fig. 6). Eliminating the need for purified antigen is an advantage for many membrane-associated proteins since these proteins are often difficult to express and purify. Furthermore, given the complex structures and post-translational modifications adopted by receptors with transmembrane domains, recombinant proteins may not recapitulate native conformations, especially when they are absorbed to plastic. Because of this, immunization and screening with recombinant antigen using standard techniques has a tendency to produce Abs recognizing only denatured epitopes. While such Abs may be useful as detection agents for ELISA and Western blot assays, they have no utility for cell based assays, receptor modulation experiments with cultured cells, or in vivo studies.
Figure 6.
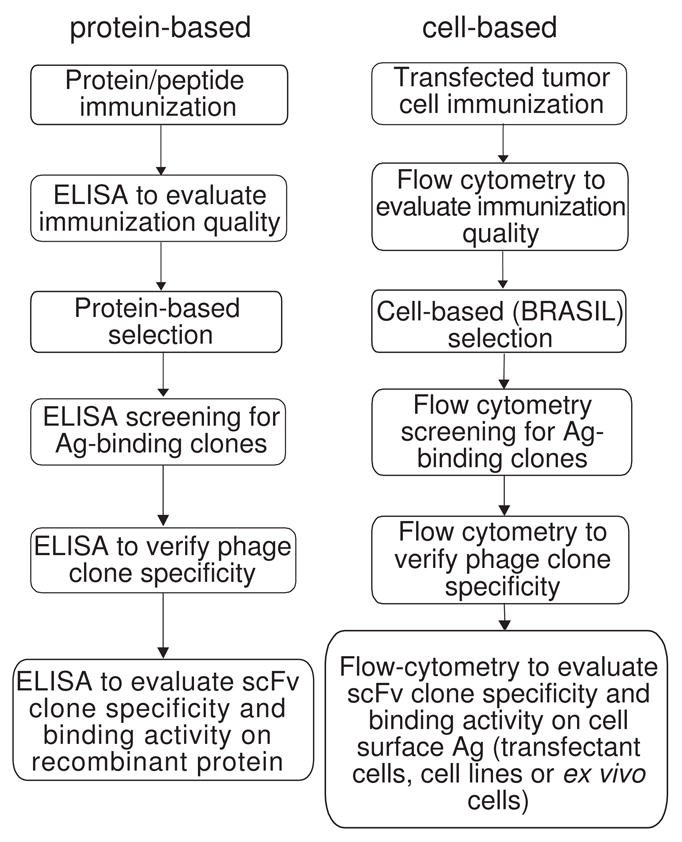
Comparison of the individual procedures involved in traditional protein-based scFv generation (left) with the cell-based protocol for scFv generation we now describe (right).
In our hands, immunization with stably transfected cells that express hTLR2 generated impressive responses. Such cell-based immunizations have previously been shown to work well in the generation of mAbs using hybridomas8. We were able to measure responses to immunization with flow cytometric assays using sera from the immunized animals, obtaining titers comparable to those determined by ELISA. Evaluating the immune response with both approaches allows determination of the extent of the polyclonal response for a full constellation of native and denatured epitopes. However, the ability to perform both immunizations and evaluations of Ab titers without purified protein is advantageous for many antigens.
A comparison of protein-based versus cell-based selections with the anti-hTLR2 scFv M13 phage library suggests that cell-based selections may provide a distinct advantage when it is critical to obtain Abs able to bind cell-surface antigens (Table. 1). Selection of phage antibodies with recombinant hTLR2 was rapid and yielded a population of Abs that bound with high specificity to the recombinant antigen. However, none of the antibody phage clones obtained from our selection with recombinant hTLR2 were able to bind the receptor in its native context on the cell surface. It may have been possible to identify such clones through an exhaustive screen of the clones from our recombinant protein selection. Alternatively, it may have been possible to obtain these clones by modifying the selection method in an effort to allow the protein to adopt a native structure. For example, immobilizing the recombinant protein via an N- or C-terminal tag to magnetic beads instead of adsorbing it to polystyrene may have been beneficial18. Nonetheless, the relative ease by which phage clones binding to cell-surface hTLR2 were found with cell-based screening illustrates this approach’s significant advantage of ensuring the selected antibodies will bind the target protein on cell surfaces, and therefore should be considered even when recombinant antigen is available. Combining other selection methods such as fluorescence activated cell sorting (FACS)13; 14 or magnetically activated cell sorting19 with our cell-based immunizations and screens may offer even greater selection efficiencies than the BRASIL selection method employed here.
Performing direct screens of selected antibody phage clones by flow cytometry in a 96-well high-throughput format further streamlined the identification of clones specific for cell-surface hTLR2. High throughput screening is important when the objective is to obtain Abs for a specific purpose such as receptor modulation or therapeutic applications since Abs with these capabilities would be typically expected to comprise a very small fraction of the total pool of antibodies. Here, we provide an optimized technique for such screens using 96-well preparations of individual phage clones. The use of an internal control cell line expressing YFP provides excellent discrimination of positive phage clones from background. Another critical finding from the optimization of this approach is that use of serum as a blocking agent causes non-specific binding of M13 phage to HEK cells. This background is eliminated by use of bovine serum albumin as the blocking agent.
Previous studies have utilized flow cytometry for phage selections, iteratively employing FACS to enrich for phage binding a subpopulation of cells 13; 14 and then individually screening resulting clones by flow cytometry. Screening of phage clones individually appears to be more labor intensive and expensive than the 96-well flow cytometric screening approach used here.
The cell-based procedure presented here for hTLR2 could be used to obtain Abs specific for other cell-surface antigens for which it has proven difficult to obtain antibodies by traditional methods. Such antigens include almost all TLRs and many cytokine and chemokine receptors. Surface receptors such as TLR5, TLR1, TLR6 have critical roles in modulating immune responses, but monoclonal antibody reagents against surface-expressed native epitopes for these receptors have yet to be developed. A cell-based selection for TLR5 performed using the methods developed here has yielded phage clones binding to TLR5-HEK cells in flow cytometric assays (manuscript in preparation), further indicating that cell-based methods may provide a useful approach to solve many long standing technical challenges in the field of immunology.
The methods developed here for rapidly shuttling scFv inserts into a vector for expression in S2 insect cells provides an advantage over expression in E. coli, given the sensitivity of immune system receptors such as TLRs to bacterial components such as lipopeptides and LPS. The yields of functional scFv protein we obtained from Drosophila were greater than those we typically obtain from E. coli, a finding others have noted 20. This insect cell expression system will also facilitate re-engineering anti-hTLR2 scFv antibodies into other antibody formats such as full-length IgG 21.
In conclusion, we developed a completely cell-based method that generates multiple monoclonal recombinant antibodies against native surface proteins, bypassing requirement for recombinant protein production. This method is an effective and efficient approach that can potentially be applied to obtain antibodies for many other structurally complex cell surface proteins.
Materials and Methods
hTLR2 expression constructs and cell lines
hTLR2-HA-pUNO plasmids were purchased from InvivoGen (San Diego, CA). The 300.19 mouse tumor cell line8 (kindly provided by Dr. T. Tedder, Duke University, Durham, NC) was transfected with hTLR2-HA-pUNO by electroporation and used for immunizations. HEK293 cells (ATCC, Manassas, VA) were transfected with hTLR2-HA-pUNO using Superfect (Quiagen, Valencia, CA) for use in library panning and flow cytometry assays. Expression of hTLR2 was verified by probing Western blots of cell extracts with anti-HA antibody. The YFP-HEK cells were kindly provided by Dr. R. Lefkowitz (Duke University). The human BAL cells were kindly provided by Dr. S. Palmer (DUMC).
Expression and purification of recombinant hTLR2 protein
The putative extracellular domain of human TLR2 (hTLR2-ECD) was expressed in Sf9 insect cells using a baculovirus vector construct, sTLR2-ECD-PVL1393 (a kind gift from Dr. Yoshio Kuroki, Sapporo Medical University School of Medicine, Japan) as described previously22; 23. The HIS-tagged soluble hTLR2-ECD was purified from culture medium by NTA agarose (Qiagen) following manufacturer’s instructions.
Flow cytometry
hTLR2-HEK and YFP-HEK cells were grown in MEM to 70% confluence, harvested into FACS buffer (PBS, 0.5% BSA, 10 mM EDTA, 10 mM HEPES) by gentle pipetting, washed, resuspended in FACS buffer, mixed in equal numbers, and plated in 96-well U-bottom plates at 5 x105 cells/well. Unless otherwise noted, all incubations were performed using 100ul volumes for 30 min on ice. To evaluate serum IgG titers, plates were centrifuged at 500g for 3 min, the supernatant poured off, and the cells resuspended in diluted mouse serum. After incubation, the cells were washed x2 in 200ul FACS buffer, resuspended in 0.5 ug/ml allophycocyanin-conjugated goat-anti-mouse IgG F(ab)′2 (Jackson ImmunoResearch Labs, West Grove, PA), incubated, washed x2 in 200ul FACS buffer, resuspended in 200ul FACS buffer and subjected to flow cytometric analysis using a BD LSRII flow cytometer. Data were analyzed using Flowjo software (TrecStar Inc, Ashland, OR). To determine hTLR2-specific binding, hTLR2-HEK and YFP-HEK cell populations were gated separately based on YFP fluorescence in the FITC channel, and the mean fluorescence intensity (MFI) of allophycocyanin was determined for each population. hTLR2-specific staining was calculated as the hTLR2-HEK MFI minus the YFP-HEK MFI.
The determination of hTLR2-specific binding by purified phage was performed as above except that 100 ul of 1012 phage/ml purified phage stocks (see below) were used as the primary staining reagent, 1 ug/ml mouse-anti-M13 mAb (Amersham) was used as the secondary reagent, and 0.5 ug/ml allophycocyanin-conjugated goat-anti-mouse IgG F(ab)′2 was used for tertiary staining. For high throughput screening, staining was performed as with purified phage except that mixtures of 100 ul FACS buffer and 100 ul unpurified phage supernatants, obtained from the corresponding wells of the rescued 96-well phage plate, were used as the primary staining reagent and an automatic plate loader was used to process the samples.
Staining of cells with purified biotinylated scFvs was performed on hTLR2-HEK, THP-116, and CaCo-2 17 cells (ATCC), and cells freshly obtained from lung transplant recipients by bronchoalveolar lavage (kindly provided by Dr. Scott Palmer of Duke University). The staining procedure was as above except that the staining buffer contained 5% normal human AB serum and 5% normal goat serum, 0.5 ug/ml biotinylated scFvs were used as the primary staining reagent, and 0.2 ug/ml allophycocyanin-conjugated streptavidin (BD Biosciences, San Jose, CA) was used as the secondary staining reagent.
Immunizations and sera harvest
Five WT BALB/c mice (Charles River Laboratories, Wilmington, MA) and five TLR2−/− C57BL/6 mice9 (The Jackson Laboratory, Bar Harbor, ME) were immunized subcutaneously at two sites on the back with 108 hTLR2-transfected 300.19 cells. The mice were boosted with 5x 107 cells IP at 14 day intervals. 7 days after the 5th boost, blood was collected and serum serially diluted in FACS buffer for analysis by ELISA or flow cytometry. ELISA measurements of serum IgG titers were performed as described previously24; 25.
scFv library construction
Spleens were harvested from immunized BALB/c and TLR2−/− C57BL/6 mice that displayed significant anti-hTLR2 IgG titers, homogenized in Trizol (Invitrogen, Carlsbad, CA), then enriched for mRNA using Dynal magnetic oligo(dT) beads (Invitrogen). The resulting mRNA was used for preparation of an anti-hTLR2 scFv library using the reagents supplied in the Recombinant Phage Antibody System (GE Healthcare, Buckinghamshire, UK). RNA from each spleen was reverse transcribed with random hexamers and used as template for separate PCR reactions for heavy and light chain variable fragments. The VH and VL PCR products were pooled in equimolar amounts for assembly and cloned into pCANTAB 5E. The library of resulting phagemids was transformed into TG-1 cells (Stratagene, La Jolla, CA) in thirty separate electroporations, performed with a BioRad Gene Pulser II at 1700 V, with a resistance of 200 ohms and capacitance of 25 uF. The electroporated bacteria were pooled and plated on 2xYT plates supplemented with 2% glucose and 0.1 mg/ml ampicillin (2xYT/AG). Colonies of phagemid clones were scraped into 2xYT and pooled to make library stocks.
Phage Rescue
Rescues with M13K07 (New England Biolabs, Ipswich, MA) were performed to obtain M13 phage particles for selections and screening. To obtain concentrated phage preparations, a 12 ml 2xYT/AG culture was inoculated from frozen stocks and grown at 37 °C to an OD600 of 0.7. 2x1011 M13K07 helper phage were added and the culture was shaken at 200 rpm for 30 minutes at 37°C. Bacteria were pelleted at 3500g for 10 minutes and the medium replaced with 50 ml of 2xYT supplemented with ampicillin and kanamycin. The culture was shaken at 37°C for 30 minutes, then at 30°C for 16 hours. The culture was centrifuged as above, and the supernatant subjected to multiple precipitations in 0.2 volumes of ice-cold 20% PEG-8000/2.5 M NaCl to purify and concentrate the phage particles. Phage particles were pelleted at 10,000g for 10 minutes. Following the last precipitation, the phage were resuspended in 1 ml PBS and stored at 4°C. Phage titers were determined by infections of TG-1 cells with serially diluted phage.
Individual phage clones were rescued for screening in sterile 2 ml/well 96-well plates (Continental Lab Products, San Diego, CA). 400 ul of 2xYT/AG in each well was inoculated with a phage clone and shaken at 200 rpm for 16 hours at 30°C. Aliquots from the wells of this master plate were used to inoculate a rescue plate containing 400 ul of 2xYT/AG supplemented with 2x1010 phage/ml of M13K07 helper phage. The rescue plate was shaken at 37°C for 4 hours. Bacteria were pelleted at 3000g for 10 minutes, resuspended in 400 ul of 2xYT/AK and shaken for 16 hours at 30°C. The bacteria were pelleted as above and the supernatant containing phage particles was transferred to a fresh plate for screening assays.
Immunotube selection
Recombinant hTLR2 (50 ug in 1 ml PBS) was adsorbed to NUNC immunotubes. After rinsing with PBS, this tube and a control tube were blocked with 4 ml of 2% milk in PBS (MPBS). An aliquot of 1012 freshly prepared M13 phage particles from the hTLR2 scFv library was pre-cleared for one hour at room temperature in the MPBS control tube then transferred to the target tube with hTLR2 for a one hour incubation. Unbound phage were removed with 20 washes of 4 ml PBS 0.1% Tween-20 followed by 20 washes with 4 ml PBS. A 1 ml aliquot of log-phase TG-1 bacteria was added to the target tube and incubated at 37 °C for 30 minutes to allow infection with the bound phage. The TG-1 cells were plated on 2xYT/AG plates and grown overnight at 30°C. Individual phagemid colonies were rescued in 96-well plates to produce phage particles for screens as detailed above.
Cell-based selection
Cell-based selection was performed using the biopanning and rapid analysis of selected interactive ligand (BRASIL) approach of Giodano et al.12 with slight modification. All solutions and incubations were at 4 °C unless otherwise noted. HEK293 and hTLR2-HEK cells were harvested with Versene (Invitrogen), washed in Dulbecco’s PBS and resuspended at 107 cells/ml in DMEM (Invitrogen)/1% BSA Fraction V (Sigma, St. Louis, MO). An aliquot of anti-hTLR2 scFv phage library containing 5x109 M13 phage particles was added to 150 ul of HEK293 cells suspension and rotated for two hours. This solution was layered on top of 200 ul of a mixture of dibutyl phthalate:cyclohexane (15:1) (Sigma) and spun at 10000g in a microfuge for 10 minutes. The aqueous phase containing pre-cleared phage was used to resuspend a cell pellet containing 1.5x106 hTLR2-HEK cells. This suspension was rotated for 1.5 hours. The cells were washed three times in 200 ul DMEM/1% BSA, then resuspended in 200 ul DMEM/ 1% BSA and layered over 200 ul of dibutyl phthalate:cyclohexane (15:1). After centrifugation (10000g x 10 min), the tube was flash frozen in liquid nitrogen. The cell pellet with bound phage in the tube tip was removed with a tube cutter (GeneMate). The cell pellet was resuspended in 10 ml of log-phase TG-1 cells. Following a 30 minute incubation at 37°C, the cells were concentrated by centrifugation and plated on two 15 cm 2xYT/AG plates. The plates were incubated for 16 hours at 30°C. The resulting colonies were scraped into 2xYT and used to make a glycerol frozen stock
DNA fingerprinting
ScFv inserts obtained by PCR from infected bacteria26 were subjected to restriction fragment length polymorphism or “DNA fingerprint” analysis. PCR was performed using 2ul of TG-1 culture, 200 uM dNTPs, 200 uM pCANTAB 5E primers (scFvGWfwd: GCGGCCCAGCCGGCC; scFvGWrev: CTGGAACCGCGTG), 3 units EasyA DNA polymerase (Stratagene), and manufacturer’s buffer in 50 ul. After amplification (95°C x 2 min, 30 cycles of 95°C x 30 sec, 58°C for 30 sec, 72°C for 1 min; and 72°C for 10 minutes), aliquots of the product were digested with Sau3A I or BstN I (New England Biolabs) then fractionated on 2% agarose TAE gels.
Expression of soluble scFvs in Drosophila system
ScFv inserts were amplified from pCANTAB 5E using above PCR conditions and ligated with topoisomerase into the Gateway entry vector pCR8Topo (Invitrogen). The Drosophila expression vector pMTBiP/V5His (Invitrogen) was converted to a Gateway destination vector by replacing the multiple cloning sequence with recombination sites for the bacteriophage enzyme LRII (Invitrogen) and a BirA Avitag site (Avidity, Denver, CO). After recombining scFv inserts into pMTBiP, plasmids were transfected into Schneider 2 (S2) cells using Cellfectin (Invitrogen). Stable tranfectants were obtained by co-transfection with plasmid pCoBlast (Invitrogen) and selection in 25 mg/ml blasticidin. Expression of scFvs was induced with 0.75 mM CuSO4 in Sf900-II serum-free medium (Invitrogen) for 36 to 48 hours at 28°C. ScFvs were purified from culture supernatant by ion metal affinity chromatography (Talon resin, Clontech, Mountain View, CA). Purity and quantity were assessed by SDS-PAGE and BCA assay (Pierce, Rockford, IL). To make biotinylated-scFvs, purified scFvs were subject to in vitro BirA biotinylation (Avidity) as described in manufacture’s instructions.
Acknowledgments
We thank John Whitesides, Patti McDermott, and Danielle King of Duke Human Vaccine Institute Flow Cytometry facility for their excellent technical assistance. This work was supported by NIH grant U19-AI056572.
Abbreviation used
- Ab
antibody
- Ag
antigen
- BSA
bovine serum albumin
- IgG
immunoglobulin G
- MFI
mean florescent intensity
- scFv
single-chain variable fragment
- TLR
toll-like receptor
- YFP
yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.McCafferty DP, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable fragments. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 3.Gay NJ, Gangloff M, Weber AN. Toll-like receptors as molecular switches. Nature Reviews Immunology. 2006;6:69–698. doi: 10.1038/nri1916. [DOI] [PubMed] [Google Scholar]
- 4.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann SHE. The contribution of immunology to the rational design of novel antibacterial vaccines. Nat Rev Micro. 2007;5:491–504. doi: 10.1038/nrmicro1688. [DOI] [PubMed] [Google Scholar]
- 6.Netea MG, van der Graaf C, Van der Meer JWM, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75:749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 7.Verstak B, Hertzog P, Mansell A. Toll-like receptor signalling and the clinical benefits that lie within. Inflammation Research. 2007;56:1–10. doi: 10.1007/s00011-007-6093-7. [DOI] [PubMed] [Google Scholar]
- 8.Kearney JF, Radbruch A, Liesegang B, Rajewsky K. A New Mouse Myeloma Cell Line that Has Lost Immunoglobulin Expression but Permits the Construction of Antibody-Secreting Hybrid Cell Lines. J Immunol. 1979;123:1548–1550. [PubMed] [Google Scholar]
- 9.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. Toll-Like Receptor 2 Is Required for Innate, But Not Acquired, Host Defense to Borrelia burgdorferi. J Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 10.Kontermann R, Dubel S. Antibody Engineering. Springer; New York: 2001. [Google Scholar]
- 11.Barbas CF, 3rd, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A. 1991;88:7978–82. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giordano RJ, Cardo-Vila M, Lahdenranta J, Pasqualini R, Arap W. Biopanning and rapid analysis of selective interactive ligands. Nat Med. 2001;7:1249–53. doi: 10.1038/nm1101-1249. [DOI] [PubMed] [Google Scholar]
- 13.de Kruif J, Terstappen L, Boel E, Logtenberg T. Rapid selection of cell subpopulation-specific human monoclonal antibodies from a synthetic phage antibody library. Proc Natl Acad Sci U S A. 1995;92:3938–42. doi: 10.1073/pnas.92.9.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lekkerkerker A, Logtenberg T. Phage antibodies against human dendritic cell subpopulations obtained by flow cytometry. Journal of Immunological Methods. 1999;231:53–63. doi: 10.1016/s0022-1759(99)00140-4. [DOI] [PubMed] [Google Scholar]
- 15.Sato TN, Qin Y, Kozak CA, Audus KL. Tie2, a putative protein tyrosine kinase from a new class of cell surface receptor. Growth Factors. 1993;9:99–105. [PubMed] [Google Scholar]
- 16.Tsuchiya SYM, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) International Journal of Cancer. 1980;26:171–6. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 17.Jumarie CMC. Caco-2 cells cultured in serum-free medium as a model for the study of enterocytic differentiation in vitro. J Cell Physiol. 1991;149:24–33. doi: 10.1002/jcp.1041490105. [DOI] [PubMed] [Google Scholar]
- 18.Lorimer I, Keppler-Hafkemeyer A, Beers R, Pegram C, Bigner D, Pastan I. Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: Targeting with a single chain antibody variable domain isolated by phage display. Proceedings of the National Academy of Sciences. 1996;93:14815–14820. doi: 10.1073/pnas.93.25.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegel DL, Chang TY, Russell SL, Bunya VY. Isolation of cell surface-specific human monoclonal antibodies using phage display and magnetically-activated cell sorting: applications in immunohematology. J Immunol Methods. 1997;206:73–85. doi: 10.1016/s0022-1759(97)00087-2. [DOI] [PubMed] [Google Scholar]
- 20.Reavy B, Ziegler A, Diplexcito J, Macintosh SM, Torrance L, Mayo M. Expression of functional recombinant antibody molecules in insect cell expession system. Protein Expression and Purification. 2000;18:221–228. doi: 10.1006/prep.1999.1191. [DOI] [PubMed] [Google Scholar]
- 21.Kirkpatrick RB, Ganguly S, Angelichio M, Griego S, Shatzman A, Silverman C, Rosenberg M. Heavy chain dimers as well as complete antibodies are efficiently formed and secreted from Drosophila via a BiP-mediated pathway. J Biol Chem. 1995;270:19800–19805. doi: 10.1074/jbc.270.34.19800. [DOI] [PubMed] [Google Scholar]
- 22.Iwaki D, Mitsuzawa H, Murakami S, Sano H, Konishi M, Akino T, Kuroki Y. The Extracellular Toll-like Receptor 2 Domain Directly Binds Peptidoglycan Derived from Staphylococcus aureus. J Biol Chem. 2002;277:24315–24320. doi: 10.1074/jbc.M107057200. [DOI] [PubMed] [Google Scholar]
- 23.O’Reilly DR, Miller LK, Luckow VA. Baculovirus Expression Vector: A Laboratory Manual. W. H. Freeman and Company; New York: 1992. pp. 139–179. [Google Scholar]
- 24.Nordone SK, Peacock JW, Kirwan SM, Staats HF. Capric acid and hydroxypropylmethylcellulose increase the immunogenicity of nasally administered peptide vaccines. AIDS Res Hum Retroviruses. 2006;22:558–68. doi: 10.1089/aid.2006.22.558. [DOI] [PubMed] [Google Scholar]
- 25.Bradney CP, Sempowski GD, Liao HX, Haynes BF, Staats HF. Cytokines as adjuvants for the induction of anti-human immunodeficiency virus peptide immunoglobulin G (IgG) and IgA antibodies in serum and mucosal secretions after nasal immunization. J Virol. 2002;76:517–24. doi: 10.1128/JVI.76.2.517-524.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gussow D, Clackson T. Direct clone characterization from plaques and colonies by the polymerase chain reaction. Nucleic Acids Research. 1989;17:4000–4003. doi: 10.1093/nar/17.10.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]