

Abstract
EGFRvIII is a mutant epidermal growth factor receptor found in glioblastoma, and in carcinoma of the breast, ovary, and lung. The mutant receptor has a deletion in its extracellular domain that results in the formation of a new, tumor-specific extracellular sequence. Mice were immunized with a synthetic peptide corresponding to this sequence and purified EGFRvIII. A single chain antibody variable domain (scFv) phage display library of 8 × 106 members was made from the spleen of one immunized mouse. A scFv specific for EGFRvIII was isolated from this library by panning with successively decreasing amounts of synthetic peptide. This was used to make an immunotoxin by fusing the scFv DNA sequence to sequences coding for domains II and III of Pseudomonas exotoxin A. Purified immunotoxin had a Kd of 22 nM for peptide and a Kd of 11 nM for cell-surface EGFRvIII. The immunotoxin was very cytotoxic to cells expressing EGFRvIII, with an IC50 of 1 ng/ml (16 pM) on mouse fibroblasts transfected with EGFRvIII and an IC50 of 7–10 ng/ml (110–160 pM) on transfected glioblastoma cells. There was no cytotoxic activity at 1000 ng/ml on the untransfected parent glioblastoma cell line. The immunotoxin was completely stable upon incubation at 37°C for 24 h in human serum. The combination of good affinity, cytotoxicity and stability make this immunotoxin a candidate for further preclinical evaluation.
Keywords: EGFRvIII, glioblastoma, cancer therapy, Pseudomonas exotoxin A
Immunotoxins are therapeutic agents for cancer that consist of a targeting molecule linked to a cytotoxic agent (1). Antibodies, or genetically engineered antibody variable domains (Fvs), are usually used for targeting. Our laboratory has focused on using the protein Pseudomonas exotoxin A as a cytotoxic agent. Pseudomonas exotoxin A is a three-domain protein (2): domain I binds to the α2 macroglobulin receptor, which internalizes the toxin (3); domain II mediates translocation of the toxin to the cell cytosol; domain III ADP ribosylates elongation factor 2 leading to arrest of protein synthesis and cell death. The toxin can be converted to a cancer therapeutic by replacing domain I with binding domains that are selective for cancer cells. Recently our laboratory completed a phase I trial of an immunotoxin made with an antibody attached to domains II and III of Pseudomonas exotoxin A (4). The antibody was specific for a Lewis Y-related carbohydrate antigen that is overexpressed in many cancers. Tumor regressions were seen in several patients with advanced breast and colorectal cancer, demonstrating that immunotoxins made with Pseudomonas exotoxin A do have activity against solid tumors in humans.
The immunotoxin used in this phase I trial was made using a whole mouse monoclonal antibody chemically coupled to toxin. Disadvantages of this type of immunotoxin are the large size (≈200 kDa) which can result in poor tumor penetration (5), the heterogeneity of the linkage between antibody and toxin, and the difficulty of producing the conjugates in large amounts. To overcome these problems we have made wholly recombinant immunotoxins in which cloned Fvs are used for targeting (6, 7). Previously this has been done by cloning variable domains from specific hybridomas and converting them to single chain Fvs (scFvs) (8, 9) or disulfide-linked Fvs (10) by genetic engineering methods. Problems with this approach were that many antibody variable domains functioned poorly as scFvs, either because of instability due to a weak VH–VL association, or poor binding due to problems such as interference by the linker sequence. Here we have used phage display technology to bypass the hybridoma step and isolate scFvs directly. In phage display, peptides or proteins are expressed on the surface of phage as fusion proteins (11). This allows the selection and amplification of phage clones with specific binding activities.
Immunotoxins have been made that recognize a wide variety of cell-surface targets on cancer cells. Typically these are tumor-associated antigens—i.e., antigens that are overexpressed on cancer cells relative to normal cells. This study concerns the development of immunotoxins that target a mutant epidermal growth factor receptor (EGFR) which is only expressed on tumor cells. This mutant receptor was first detected in human glioblastoma cells. Early work on the genetic changes that take place in these cells showed that the EGFR gene is often amplified in these cells (12). Further studies showed that the gene is also frequently rearranged (13–15). The most common rearrangement was a deletion of exons 2–7. This deletion is in-frame, and a new glycine codon is formed at the deletion junction. The mutant receptor, designated EGFRvIII, is expressed on the cell surface and contains a new tumor-specific protein sequence in its extracellular domain. It has also been shown to have constitutive protein kinase activity (16). EGFRvIII cDNA has the properties of an oncogene; it transforms 3T3 mouse fibroblasts (15) and enhances the in vivo malignancy of human glioblastoma cells (16). EGFRvIII has been found in 50–60% of glioblastomas (17, 18). More recently it has also been shown to be present in 70–80% of breast and ovarian cancers (17, 18), and 16% of non-small cell lung cancers (19). The frequent expression of EGFRvIII in human tumors make it an attractive target for therapeutics. A peptide corresponding to the tumor-specific extracellular sequence is immunogenic in animals, and both polyclonal (13) and monoclonal antibodies (17, 20) have been raised that are specific for this sequence and for the mutant receptor.
Previously we have shown that EGFRvIII-specific antibodies chemically coupled to domains II and III of Pseudomonas exotoxin A were cytotoxic to cells expressing EGFRvIII, but had little or no activity against cells expressing wild-type EGFR (21). We next attempted to make wholly recombinant toxins that used only antibody Fv domains for targeting (I.A.J.L., C.N.P., D.D.B., and I.P., unpublished data). The variable regions of two EGFRvIII-specific antibodies were cloned and used to make immunotoxins. These were active, but in both cases had reduced binding compared with the original antibody, and reduced cytotoxicity compared with the whole antibody immunotoxins. In this study, we have used phage display to isolate a high-affinity EGFRvIII-specific Fv domain directly from an immunized mouse. This Fv was used to make an immunotoxin that had greatly improved cytotoxicity compared with the recombinant immunotoxins made previously.
MATERIALS AND METHODS
Mouse Immunization.
Ajax mice were immunized s.c. with 30 μg of peptide-3/keyhole limpet hemocyanin conjugate in complete Freund’s adjuvant. Peptide 3 has as its sequence the first 13 amino acids of the EGFRvIII N terminus plus a carboxyl-terminal cysteine (17). After 80 days, mice were boosted with 30 μg of peptide-3/keyhole limpet hemocyanin conjugate in incomplete Freund’s adjuvant containing 1 mg of Salmonella Minnesota (Ribi Immunochem) per ml. Mice were given three further s.c. boosts at ≈80-day intervals with 20 μg of peptide-3/keyhole limpet hemocyanin conjugate and 20 μg of affinity-purified EGFRvIII in incomplete Freund’s adjuvant. After 5.5 months, mice were given a boost i.p. with 20 μg of peptide-3/keyhole limpet hemocyanin conjugate and 30 μg of affinity-purified EGFRvIII without adjuvant. This was repeated 2 months later. Spleens were harvested 5 days after the final boost.
Library Construction.
Splenocytes were purified on a Ficoll cushion and poly(A)+ RNA was purified using a FastTrack mRNA isolation kit from Invitrogen. A scFv library was constructed in the vector pCANTAB5E using the recombinant phage antibody system from Pharmacia following the manufacturer’s protocol. One-third of the final ligation reaction was used to transform TG1 cells by electroporation.
Synthetic Peptide.
The synthetic peptide LEEKKGNYVVTDHSGGK-biotin was synthesized by Peptide Technologies (Gaithersburg, MD). The first 13 amino acids correspond to the N terminus of EGFRvIII and contain the tumor-specific deletion junction sequence. The SGG sequence is intended to function as a flexible spacer. The carboxyl-terminal residue is a lysine with biotin attached to the ɛ-amino group.
Panning Procedure.
Escherichia coli TG1 (Stratagene) were electroporated and grown in 10 ml 2× YT medium (16 g bacto-tryptone/10 g bacto-yeast extract/5 g NaCl per liter in H2O) plus 2% glucose with shaking for 1 h at 37°C. Ampicillin (final concentration, 100 μg/ml) and M13KO7 helper phage (4 × 1010 plaque-forming units) were then added, and the culture was grown for one more hour. Cells were then pelleted and resuspended in 10 ml 2× YT medium containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. The culture was then incubated overnight with shaking at 37°C. Phage were purified from the culture supernatant by precipitation three times with polyethylene glycol, resuspended in PBS, titered, and stored at −70°C until panning. The panning procedure used is a modification of the method described by Schier et al. (22). Phage (5 × 1011) in 0.5 ml PBS were mixed with 0.5 ml of 4% skim milk powder/0.05% Tween 20/PBS containing biotinylated peptide. This was mixed end-over-end at room temperature for 1 h and then added to 300 μl of Dynabeads M-280 streptavidin-coated magnetic beads (Dynal, Great Neck, NY) that had been blocked for 1 h at 37°C with 2% skim milk powder/0.025% Tween 20/PBS. After a 15-min incubation at room temperature, magnetic beads were captured with a magnet and washed six times with 0.05% Tween 20 in PBS and four times with PBS. Beads were resuspended in 600 μl PBS and half of this was used directly to reinfect log phase TG1 cells.
Phage and scFv ELISA.
Immulon 4 plates were coated overnight at 4°C with 200 μl per well 10 μg/ml streptavidin (Pierce). Plates were then washed three times with 0.05% Tween 20 in PBS. A total of 200 μl per well of 1 μM EGFRvIII peptide was added for 15 min at room temperature, and the plate was washed again. A total of 200 μl per well of blocking buffer (2% milk powder in PBS) was added for 1 h at room temperature. Phage or periplasm (see below) in blocking buffer was added for 1 h at room temperature. The plate was then washed three times as above. For phage ELISA, 200 μl per well of antiM13 antibody-horse radish peroxidase conjugate (Pharmacia) in blocking buffer, diluted according to the manufacturer’s instructions, was added; for periplasm, 200 μl per well of anti-Etag antibody-horse radish peroxidase conjugate (Pharmacia) in blocking buffer was added (1:8000 dilution). After 1 h at room temperature, plates were washed, and 200 μl per well of 2,2′-azinobis-[3-ethylbenzothizoline-6-sulfonic acid] (ABTS) substrate solution was added. After 15 min, absorbance in wells was determined by using an ELISA plate reader.
Periplasm Preparation.
E. coli HB2151 was infected with ELISA-positive phage. 5 ml of an overnight culture (from a single colony) was used to start a 1 l culture in 2× YT medium containing 2% glucose and 100 μg/ml ampicillin. This was grown to an A600 of 0.9, at which point cells were pelleted and resuspended in 1 liter of 2× YT medium containing 1 mM isopropyl β-d-thiogalactoside and 100 μg/ml ampicillin. After 3 h of growth at 37°C, cells were pelleted and resuspended in 20 ml of ice-cold TES buffer (0.2 M Tris·HCl/0.5 mM EDTA/0.5 M sucrose, pH 8.0). Thirty-five milliliters of cold water was added and cells were incubated on ice for 30 min. Cells were then removed by centrifugation, and the supernatant was recentrifuged at 14,000 rpm in a Sorvall SS34 rotor for 30 min. The supernatant was dialyzed overnight against 2 liters of PBS and stored at −70°C.
Sequencing.
Sequencing was done as described (23) by using an Applied Biosystems model 373A DNA sequencing system.
scFv Immunotoxin Plasmid Construction, Expression, and Purification.
A scFv immunotoxin was made as described by fusing the scFv to domains II and III of Pseudomonas exotoxin A (7). The scFv immunotoxin was expressed under control of the T7 promoter in E. coli BL21(λ DE3). Purification of inclusion bodies, refolding, and immunotoxin purification by ion-exchange and size-exclusion chromatography were all as described (7).
Surface Plasmon Resonance.
Binding to EGFRvIII peptide was measured by surface plasmon resonance (24) using the Pharmacia BIAcore 1000 with upgrade system. A total of 1800–2400 resonance units of streptavidin (Pierce) were coupled to the surface of carboxylated dextran-coated CMS research grade chips at pH 4.8 using the amide coupling reagents provided with the BIAcore system. Biotinylated EGFRvIII peptide was then bound to the streptavidin by flowing 10 or 100 nM solutions of peptide over the chip. Streptavidin binding sites were not saturated with the lower concentration of peptide. Solutions of MR1 immunotoxin (12.5, 25, or 50 μg/ml) were then passed over the chip to measure binding. Regeneration was done with 30 mM phosphoric acid. Nonspecific binding was determined using a streptavidin chip without peptide. Binding kinetics were analyzed by using BIAcore 1000 with upgrade software.
Cell Binding Assays.
MR1 immunotoxin (6 μg) was labeled with Bolton–Hunter reagent and purified on a PD10 column (Pharmacia) equilibrated with PBS containing 0.5% gelatin. The specific activity was 9 × 105 cpm/μg. Binding assays were performed by incubating NR6M cells at 4°C with varying amounts of labeled immunotoxin in DMEM containing 20 mM Hepes (pH 7.4) and 0.2% gelatin. After 2 h, cells were washed four times with 0.2% BSA in PBS to remove unbound immunotoxin. Bound immunotoxin was solubilized by incubation for 30 min at room temperature with 2% Triton X-100/0.2% BSA in PBS and counted in a γ counter. Nonspecific binding was determined by measuring binding in the presence of 10 μM EGFRvIII peptide. The binding dissociation constant Kd was determined by Scatchard analysis, using a least squares fit to a straight line.
Cell Culture and Cytotoxicity Assays.
The culture of NR6M cells was as described (21). U87MG and U87MG.ΔEGFR human glioblastoma cells were from W. Cavenee (University of California at San Diego) (16). They were grown in RPMI 1640 medium containing 10% fetal bovine serum (supplemented with 375 μg/ml G418 for the transfectant). Cytotoxicity assays were done as described (21), except that glioblastoma cells were labeled for 6 h with [3H]leucine.
RESULTS
Immunization, Library Construction, and Panning.
Mice were immunized with EGFRvIII peptide coupled to keyhole limpet hemocyanin and boosted with peptide and purified EGFRvIII receptor as described. Ajax mice were used because they showed a better response to peptide immunization than other mouse strains. Immunized mice showed high titers of antibody against EGFRvIII when tested by ELISA against EGFRvIII peptide. A scFv phage library was made from the spleen of one mouse, which had an endpoint serum titer in ELISA against EGFRvIII peptide of more than 1/100,000 14 days after the next to last antigen boost. A test transformation of the final ligation reaction showed that a library of 8 × 106 members could be made. One third of this ligation reaction (2.4 × 106 members) was used to transform E. coli for panning. The panning system used was chosen to select for a scFv with a high affinity for EGFRvIII. Panning was done in solution to avoid problems with selection on the basis of avidity that have been reported to occur with scFv libraries (22). Two rounds of panning were done with a high concentration of peptide (1 μM) to select for EGFRvIII binding phage. The number of phage captured on the magnetic beads increased 400-fold with the second round of panning (see Table 1). We then panned with successively decreasing concentrations of peptide (1 nM, 0.1 nM, and 0.01 nM) to select for phage with the highest affinity for EGFRvIII. The number of phage captured with 0.01 nM peptide was very low (Table 1), so we chose to analyze the phage captured from the previous round done with 0.1 nM peptide.
Table 1.
Panning of scFv library with EGFRvIII peptide
| Round | Concentration peptide, nM | No. of phage panned | No. of phage eluted |
|---|---|---|---|
| 1 | 1000 | 5 × 1011 | 9 × 104 |
| 2 | 1000 | 5 × 1011 | 3.7 × 107 |
| 3 | 1 | 5 × 1011 | 2.1 × 106 |
| 4 | 0.1 | 5 × 1011 | 9 × 105 |
| 5 | 0.01 | 2.5 × 1011 | 2.4 × 104 |
Characterization of Phage Clones.
Single colonies from the titration of captured phage were used to rescue 20 individual phage clones. These were analyzed by phage ELISA. None showed binding to streptavidin in the absence of EGFRvIII peptide. Nineteen of 20 showed binding in the presence of peptide. Ten ELISA-positive phage were used to prepare phagemids. The PCR was used to make scFv DNA from each of these. All of these had the fragments of the same size after digestion with BstNI restriction enzyme (not shown), suggesting that the clones were the same or very similar. Three of these were sequenced and found to be identical. This sequence has been deposited in the GenBank data base. The scFv coded for by this sequence was designated MR1 (for mutant receptor). These results show that, after four rounds of panning, we had selected for a phage population that consisted predominately of one clone. A single clone was used to make a crude periplasmic preparation of scFv protein with a C-terminal epitope tag (see Materials and Methods). This also showed specific binding to EGFRvIII peptide in an ELISA assay.
ScFv Immunotoxin Expression and Purification.
The scFv sequence was used to construct a plasmid for expression of a scFv immunotoxin (7). In this, the scFv sequence is fused to DNA for domains II and III of Pseudomonas exotoxin A. The version of Pseudomonas exotoxin A used here, PE38KDEL, has a modified C terminus which enhances its cytotoxicity (25). With this plasmid, expression is driven by the T7 promoter. MR1 immunotoxin was expressed as inclusion bodies in E. coli and could be detected as the major band on SDS/PAGE of solubilized whole cell. Inclusion bodies were purified, solubilized in 6 M guanidine HCl, and refolded (26). Refolded protein was purified by Q-Sepharose and MonoQ ion-exchange chromatography, followed by size exclusion chromatography. The yield of refolded immunotoxin was ≈2% (expressed as mg protein after MonoQ chomatography/mg inclusion body protein). The immunotoxin migrated as a monomer on size exclusion chromatography and migrated as a single band of the expected size (Mr 63,599) on SDS/PAGE (Fig. 1).
Figure 1.
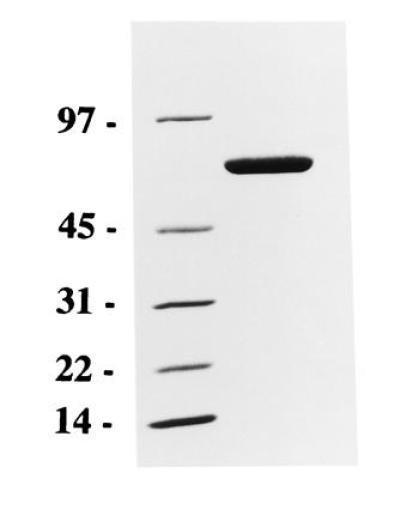
SDS/PAGE of purified MR1 immunotoxin. MR1 immunotoxin (5 μg) was electrophoresed on a 4–20% gradient gel. Migration of molecular weight markers (in kDa) is shown on the left.
Binding.
Binding of MR1 immunotoxin to synthetic peptide was measured by surface plasmon resonance (24). Streptavidin was first coupled to the chip, and the biotinylated peptide was then bound to this. This chip could be regenerated repeatedly because of the very high affinity of biotin-streptavidin binding. Values for on rates and off rates were determined for three different concentrations of immunotoxin and two different concentrations of peptide on the chip. Calculated Kd values were consistent under the different conditions used. kon was 1.28 × 105 (±0.15 × 105) M−1·sec−1. koff was 2.77 × 10−3 (±0.18 × 10−3) sec−1. The Kd calculated from these is 22 (±4) nM. (Each value is the mean of six determinations ± SD.)
We also measured binding of iodinated MR1 immunotoxin to the cell line NR6M (Fig. 2A). This is a Swiss 3T3 cell line selected for lack of expression of mouse EGFR and transfected with EGFRvIII cDNA (21). The labeled immunotoxin bound to NR6M cells, and binding could be competed by EGFRvIII peptide, showing that it was specific. Scatchard analysis of this data (Fig. 2B) gave a Kd of 11 nM. Number of sites per cell was 3 × 105 in agreement with previously published values (21).
Figure 2.
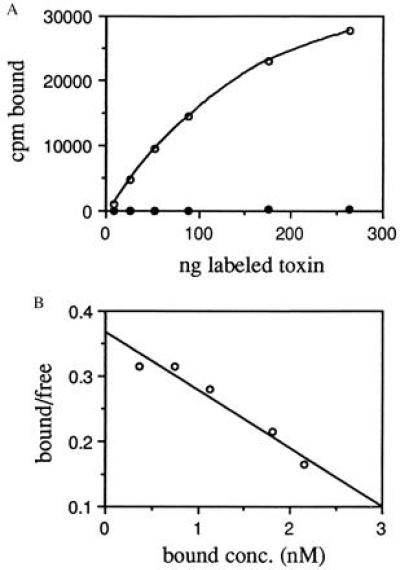
Binding of MR1 immunotoxin to NR6M cells. MR1 immunotoxin was labeled with 125I and binding to live cells at 4°C was assayed. (A) Binding in the absence (○) or presence (•) of 10 μM EGFRvIII peptide. (B) The same data replotted for Scatchard analysis. The slope was 0.088 nM−1, giving a Kd of 11 nM; the regression coefficient was 0.947.
Cytotoxicity.
The cytotoxicity of MR1 immunotoxin was first assayed using NR6M cells. To assess cytotoxicity, we have used inhibition of protein synthesis by Pseudomonas exotoxin as a surrogate measure. We have found previously that this corresponds well with cytotoxicity. MR1 immunotoxin inhibited 50% of protein synthesis at a concentration of 1 ng/ml (16 pM) when cells were exposed to immunotoxin for 24 h (Fig. 3A and Table 2). The IC50 was 30 ng/ml after cells were exposed for 2 h to immunotoxin, and 0.5–0.6 ng/ml after a 48-h exposure. This shows that the immunotoxin remains active in tissue culture for greater than 24 h. The stability of the immunotoxin in human serum was also evaluated; MR1 immunotoxin was incubated at 20 μg/ml in human serum at 37°C for various periods of time and then assayed for activity (Fig. 3B). There was no loss of activity after 24 h, showing that the scFv is very stable. The cytotoxicity of MR1 immunotoxin could be inhibited by 5 μM EGFRvIII peptide, showing that it was specific (Fig. 3C).
Figure 3.
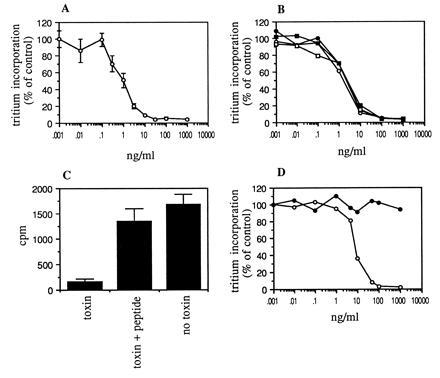
(A) Cytotoxicity of MR1 immunotoxin to NR6M cells. Cells were exposed for 24 h to immunotoxin, and incorporation of [3H]leucine was measured. (B) Stability of MR1 immunotoxin. Immunotoxin was incubated at 20 μg/ml in human serum for 0 h (○), 4 h (•), 8 h (□), or 24 h (▪) at 37°C. Cytotoxic activity was then measured as in A. (C) Inhibition by peptide. Cytotoxicity assays were performed with 10 ng/ml MR1 immunotoxin in the presence or absence of 5 μM EGFRvIII peptide. (D) Cytotoxicity of MR1 immunotoxin to human glioblastoma cells transfected with EGFRvIII. ○, U87MG.ΔEGFR transfectant; •, U87MG untransfected parent cell line. Each data point shown is the mean of triplicate determinations; error bars show SEM.
Table 2.
Cytotoxicity of immunotoxins on different cell lines
| Immunotoxin | Cell lines
|
||
|---|---|---|---|
| NR6M | U87MG.ΔEGFR | U87MG | |
| MR1scFvPE38KDEL | 1(16) | 7–10 (110-160) | >1000 |
| 2 h | 30(480) | ||
| 48 h | 0.5–0.6 (8-10) | ||
| MR1scFvPE38REDLK | 3–4 (48-64) | 40(640) | >1000 |
The concentration of immunotoxin at which 50% inhibition of protein synthesis occurs (IC50) is given in ng/ml with the value in pM in parentheses. All values are for 24-h exposure to immunotoxin unless stated otherwise.
The cytotoxicity of MR1 immunotoxin was also tested on U87MG cells transfected with EGFRvIII (16). U87MG is a human glioblastoma line and represents a more relevant cell line to assess the therapeutic potential of MR1 immunotoxin. The transfectants express 2 × 105 normal EGFRs and 4 × 105 EGFRvIII per cell by FACS analysis (C. Wikstrand and D.D.B., unpublished observations). The IC50 of MR1 immunotoxin on these cells was 7–10 ng/ml (110–160 pM) (Fig. 3D and Table 2). No cytotoxicity was seen on the untransfected parent cell line at concentrations up to 1000 ng/ml. This clearly demonstrates the specificity of MR1 immunotoxin for the mutant EGFR.
We also made a version of MR1 immunotoxin that had the naturally occurring carboxyl terminus of Pseudomonas exotoxin A, REDLK, instead of KDEL. This was used to assess the influence of the KDEL modification on the activity of MR1 immunotoxin. MR1 immunotoxin with REDLK carboxyl terminus was 3- to 4-fold less active on both NR6M cells and U87MG transfectants (Table 2).
DISCUSSION
The mutant EGFR EGFRvIII is a promising target for cancer therapy, because it has a tumor-specific extracellular sequence and it occurs frequently in a number of different tumor types. Here we have used phage display to directly isolate an EGFRvIII-specific scFv from an immunized mouse spleen. The immunotoxin made with this scFv had a higher binding affinity than the recombinant immunotoxins made previously. The Kd was 22 nM for the peptide and 11 nM for the cell surface receptor. The numbers are very similar, suggesting that scFv interacts with the peptide and receptor N-terminal sequence in the same way. The two values were determined by very different techniques; therefore we cannot tell if the small difference in affinity is due to differences in methodologies, or represents a true difference in affinity of the scFv for the receptor. (We have considered the latter possibility because boosts during immunization were done with both peptide and purified EGFRvIII.) ScFvs with Kd values of as low as 0.3 nM have been reported in the literature (27); it may therefore be possible to make even better EGFRvIII-specific immunotoxins, either by isolating new scFvs with higher affinities, or by improving the affinity of MR1.
MR1 immunotoxin also had better cytotoxic activity than the recombinant immunotoxins made previously, with an IC50 of 16 pM on transfected mouse fibroblasts. MR1 immunotoxin was 7- to 10-fold less active on the human glioblastoma cell transfectants. Since the two cell lines express similar levels of EGFRvIII, a difference in receptor numbers cannot explain this result. One difference between the two cell lines is that the glioblastoma cells express both wild-type and mutant receptor. We considered the possibility that the wild-type receptor interacts with the mutant receptor and decreases its rate of internalization. We found that addition of EGF during the assay, which would promote internalization of wild-type EGFRs, did not enhance the cytotoxicty of immunotoxins targeting EGFRvIII. Also U87MG cells tended to be more resistant to killing by a number of other immunotoxins targeting ubiquitously expressed receptors. On this basis we feel that the lower sensitivity of the glioblastoma cells is due to events downstream of receptor binding and internalization. This would include steps such as proteolytic processing, retrograde transport to the endoplasmic reticulum, and transport to the cytosol. However MR1 immunotoxin is still quite cytotoxic to the glioblastoma transfectants, and shows no cytotoxicity to glioblastoma cells that do not express EGFRvIII.
MR1 immunotoxin was also very stable. Cytotoxicity in tissue culture was proportional to the length of time the cells were exposed to immunotoxin over at least a 48-h period. There was no loss of activity after incubation at 37°C for 24 h in human serum. This contrasts with our findings with another scFv immunotoxin, which lost almost all of its activity after 24 h when assayed under the same conditions (10). We have found that many scFvs made from monoclonal antibodies are unstable, and that this can limit their antitumor activity in animal models (10). Differences in the stability of scFvs are mainly due to variations in the strength of the VH–VL interface in different antibodies. It seems likely that phage display selects directly for stable scFv. Standard phage rescue procedures involve overnight incubations at 37°C, which probably inactivate many unstable scFv. The process of secretion of scFv gene III fusion proteins across the bacterial inner membrane during phage assembly may also favor more stable scFvs. The stability of MR1 may be related to its 11-amino acid heavy chain CDR3, which is relatively long compared with the average length of 9 amino acids for mice (28). It has been shown that there is a correlation (albeit a weak one) between HCDR3 length and the surface area of the VH–VL interface (29).
In summary, MR1 immunotoxin is stable and is cytotoxic to cells expressing EGFRvIII, including human glioblastoma cells. We have previously shown that another Pseudomonas exotoxin A-based immunotoxin, LMB7, is active in a nude rat model of carcinomatous meningitis when administered intrathecally (30). MR1 immunotoxin appears to be a good candidate for testing as a therapeutic agent for primary brain cancer using a similar animal model. MR1 scFv may also be useful for targeting of radioisotopes or other therapeutic agents to EGFRvIII-expressing tumors.
Acknowledgments
A.K.-H. was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG), Germany.
Footnotes
References
- 1.Pastan I, Chaudhary V, FitzGerald D J. Annu Rev Biochem. 1992;61:331–354. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- 2.Allured V S, Collier R J, Carroll S F, McKay D B. Proc Natl Acad Sci USA. 1986;83:1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kounnas M Z, Morris R E, Thompson M R, Fitz-Gerald D J, Strickland D K, Saelinger C B. J Biol Chem. 1992;267:12420–12423. [PubMed] [Google Scholar]
- 4.Pai L H, Wittes R, Setser A, Willingham M C, Pastan I. Nat Med. 1996;2:350–353. doi: 10.1038/nm0396-350. [DOI] [PubMed] [Google Scholar]
- 5.Yokota T, Milenic D E, Withlow M, Schlom J. Cancer Res. 1992;52:3402–3408. [PubMed] [Google Scholar]
- 6.Chaudhary V K, Queen C, Junghans R P, Waldmann T A, FitzGerald D J, Pastan I. Nature (London) 1989;339:394–397. doi: 10.1038/339394a0. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann U, Pai L H, FitzGerald D J, Willingham M, Pastan I. Proc Natl Acad Sci USA. 1991;88:8616–8620. doi: 10.1073/pnas.88.19.8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bird R E, Hardman K D, Jacobson J W, Johnson S, Kaufman B M, Lee S-M, Lee T, Pope S H, Riordan G S, Withlow M. Science. 1988;242:423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 9.Huston J S, Levinson D, Mudgett-Hunter M, Tai M S, Novotny J, Margolies M N, Ridge R J, Bruccoleri E H, Crea R, Oppermann H. Proc Natl Acad Sci USA. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiter Y, Pastan I. Clin Cancer Res. 1996;2:245–252. [PubMed] [Google Scholar]
- 11.O’Neil K T, Hoess R H. Curr Opin Struct Biol. 1995;5:443–449. doi: 10.1016/0959-440x(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 12.Libermann T A, Nusbaum H R, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield M D, Ullrich A, Schlessinger J. Nature (London) 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 13.Humphrey P A, Wong A J, Vogelstein B, Zalutsky M R, Fuller G N, Archer G E, Friedman H S, Kwatra M M, Bigner S H, Bigner D D. Proc Natl Acad Sci USA. 1990;87:4207–4211. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugawa N, Ekstrand A J, James C D, Collins V P. Proc Natl Acad Sci USA. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamazaki H, Ohba Y, Tamaoki N, Shibuya M. Jpn J Cancer Res. 1990;81:773–779. doi: 10.1111/j.1349-7006.1990.tb02644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa R, Ji X-D, Harmon R C, Lazar C S, Gill G N, Cavenee W K, Huang H-J S. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wikstrand C J, Hale L P, Batra S K, Hill M L, Humphrey P A, Kurpad S N, McClendon R E, Moscatello D, Pegram C N, Reist C J, Traweek S T, Wong A J, Zalutsky M R, Bigner D D. Cancer Res. 1995;55:3140–3148. [PubMed] [Google Scholar]
- 18.Moscatello D K, Holgado-Madruga M, Godwin A K, Ramirez G, Gunn G, Zoltick P W, Biegel J A, Hayes R L, Wong A J. Cancer Res. 1995;55:5536–5539. [PubMed] [Google Scholar]
- 19.Garcia de Palazzo I E, Adams G P, Sundareshan P, Wong A J, Testa J R, Bigner D D, Weiner L M. Cancer Res. 1993;53:3217–3220. [PubMed] [Google Scholar]
- 20.Hill D, Rowlinson-Busza G, Gullick W J. Int J Cancer. 1995;63:537–543. doi: 10.1002/ijc.2910630414. [DOI] [PubMed] [Google Scholar]
- 21.Lorimer I A J, Wikstrand C J, Batra S K, Bigner D D, Pastan I. Clin Cancer Res. 1995;1:859–864. [PubMed] [Google Scholar]
- 22.Schier R, Bye J, Apell G, McCall A, Adams G P, Malmqvist M, Weiner L M, Marks J D. J Mol Biol. 1996;255:28–43. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 23.Lorimer I A J, Pastan I. Nucleic Acids Res. 1995;23:3067–3068. doi: 10.1093/nar/23.15.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlsson R, Michaelsson A, Mattsson L. J Immunol Methods. 1991;145:229–240. doi: 10.1016/0022-1759(91)90331-9. [DOI] [PubMed] [Google Scholar]
- 25.Seetharam S, Chaudhary V K, FitzGerald D, Pastan I. J Biol Chem. 1991;266:17376–17381. [PubMed] [Google Scholar]
- 26.Buchner J, Pastan I, Brinkmann U. Anal Biochem. 1992;205:263–270. doi: 10.1016/0003-2697(92)90433-8. [DOI] [PubMed] [Google Scholar]
- 27.Vaughan T J, Williams A J, Pritchard K, Osbourn J K, Pope A R, Earnshaw J C, McCafferty J, Hodits R A, Wilton J, Johnson K S. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 28.Wu T T, Johnson G, Kabat E A. Proteins Struct Funct Genet. 1993;16:1–7. doi: 10.1002/prot.340160102. [DOI] [PubMed] [Google Scholar]
- 29.Stanfield R L, Takimoto-Kamimura M, Rini J M, Profy A T, Wilson I A. Structure (London) 1993;1:83–93. doi: 10.1016/0969-2126(93)90024-b. [DOI] [PubMed] [Google Scholar]
- 30.Pastan I H, Archer G E, McLendon R E, Friedman H S, Fuchs H E, Wang Q-C, Pai L H, Herndon J, Bigner D D. Proc Natl Acad Sci USA. 1995;92:2765–2769. doi: 10.1073/pnas.92.7.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]