


Abstract
The ribosome is a complex molecular machine that offers many potential sites for functional interference, therefore representing a major target for antibacterial drugs. The growing number of high-resolution structures of ribosomes from different organisms, in free form and in complex with various ligands, provides unique data for structural and comparative analyses of RNA structures. We model the ribosome structure as a network, where nucleotides are represented as nodes and intermolecular interactions as edges. As shown previously for proteins, we found that the major functional sites of the ribosome exhibit significantly high centrality measures. Specifically, we demonstrate that mutations that strongly affect ribosome function and assembly can be distinguished from mild mutations based on their network properties. Furthermore, we observed that closeness centrality of the rRNA nucleotides is highly conserved in the bacteria, suggesting the network representation as a comparative tool for the ribosome analysis. Finally, we suggest a global topology perspective to characterize functional sites and to reveal the unique properties of the ribosome.
INTRODUCTION
The ribosome is a large complex of proteins and ribosomal RNA (rRNA) that is responsible for protein biosynthesis in all organisms. The ribosome is made up of two subunits: a small subunit (30S) and a large subunit (50S). In Escherichia coli, the small subunit consists of the 16S rRNA (1542 nt) and 21 proteins, whereas the large subunit contains the 23S rRNA (2904 nt), the 5S rRNA (120 nt) and 33 proteins. The small and the large ribosomal subunits associate into an active 70S complex, which catalyzes protein synthesis. The small subunit contains the decoding center (A-site), known to mediate the correct interaction between the tRNA anticodon and the mRNA, which is being translated. The large subunit contains the peptidyl transferase center (PTC), which catalyzes the peptide bond formation in the growing polypeptide (1). It is well established that the major functional sites in the ribosome that are involved in peptide bond formation are composed mainly of rRNAs, which is in the heart of the catalytic process (2). Though the ribosome is commonly considered a ribozyme (1), the involvement of a protein in the catalytic site has been demonstrated in E. coli (3).
High resolution structures of 30S and 50S ribosomal subunits have been solved by X-ray crystallography. These include the 30S subunit from the eubacteria Thermus thermophilus (4,5), the 50S subunit from the archaeon Haloarcula marismortui (6) and the eubacteria Deinococcus radiodurans (7). Several high-resolution structures of the 70S ribosome are also available, including the E. coli ribosome at 3.5 Å (8), and T. thermophilus at 2.8 Å (9) and 3.7 Å (10). The 70S ribosome structures provide unique information on the interface between the subunits, as well as the conformation of the active site in the context of the entire ribosome complex. Overall, the growing number of high-resolution structures of ribosomes from different organisms, in the free form as well as in complex with various ligands and antibiotics, provides a unique data set for structural and comparative analysis of the ribosome, specifically of the rRNA (4,6,8,9).
Different computational methods, which are based on force field, have been used to study macromolecular structures, specifically proteins (11). However, the computational cost of these methods for studying long and short range interactions in a large-scale system such as the ribosome is extremely high. Therefore, coarse grained methods have been developed (12,13). Furthermore, graph theory has been found to be a useful tool to investigate different properties of macromolecules such as folding, stability, function and dynamics (14). These studies have predominately concentrated on protein structures. In order to model structure as a graph (network), several representations have been developed, ranging from coarse representation, in which each node represents a secondary structure element (15), to finer modeling methods, in which each node represents an atom (16).
A common methodology to represent a macromolecular structure as a network considers amino acids/bases as nodes and inter-residue interactions as edges (17–20). In protein structures, it has been shown that the network of amino acid interactions is highly clustered and has properties of a small-world network. Such a network is characterized by the presence of a small number of central nodes (21). Interestingly, the central nodes defined by the small-world network description were found to be associated with key residues in protein folding and dynamics (18,22–25), functional sites such as enzymes catalytic sites, ligand-binding sites (17,26–29) and hot spots in protein–protein interactions (20,30).
Previously, a small-world network approach has been applied to study the conformational space of tRNA secondary structure (31). In addition, a network approach has been utilized for RNA structure characterization, in which different types of interactions (i.e. Watson–Crick, Hoogsteen and Sugar-edge) were applied to present the complexity of the structure (32). Here, we represented the rRNA 3D structure as a network with nucleotides as nodes and inter-nucleotides interactions as edges. We revealed that the rRNA structure-derived network fits the model of geometric random graphs with characteristics of a small-world network. Though the network parameters were directly derived from the structure of the ribosome complex, they were not found to simply correlate with classical structural parameters (e.g. solvent surface accessibility). The lack of strong correlation between structural properties and network parameters suggests that the latter can provide extra insights on the ribosome structure–function relationship. Specifically, we found that nucleotides with significantly high centrality values in the network correspond to the major functional regions in the small and large subunits of the ribosome. Additionally, we observed that rRNA mutations that cause a strong deleterious effect to the ribosome function, exhibit high centrality. In summary, applying a network-based analysis, we show that critical sites in the bacterial ribosome exhibit high values of local and global structural parameters. Moreover, these functional sites can be identified based solely on the network parameters without considering evolutionary information.
MATERIALS AND METHODS
Network analysis
The ribosome structure was presented as an undirected graph (network), in which nodes represent nucleotides or amino acids, and edges represent contacts. In order to generate the network, all atomic contacts were calculated using the CSU program (33). Nucleotides or amino acids were considered to be in contact if at least one of the corresponding atoms was in surface complementarity, as defined in ref. (33).
Two parameters were calculated to characterize the network: the average shortest path length, and the average clustering coefficient. The average shortest path length of a network with N nodes is defined by Equation (1):
| 1 |
where, Lij is the shortest path length between nodes i and j.
The average clustering coefficient of a network with N nodes is defined by Equation (2):
| 2 |
where, Ci is the clustering coefficient of node i, defined as the fraction of contacts that exist among its nearest neighbors relative to the maximum contacts among all neighbors.
In order to test the network properties, we calculated the average shortest path length and the average clustering coefficient for random and regular networks with the same number of nodes, and the same average number of edges (degree) (24). In a random network, Lrand ∼ N/ln K and Crand ∼ K/N, while for a regular network, Lreg = N(N + K − 2)/[2K(N − 1)] and Creg = 3(K − 2) / [4(K − 1)], N denotes number of nodes and K number of edges.
For each node in the network, we calculated three centrality measures, i.e. degree, closeness and betweennness. The degree of a node i is defined as the number of edges connected to i. The closeness of a node i is defined as the inverse average length of the shortest paths to all other nodes in the graph (34). The closeness of node i is given in Equation (3):
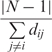 |
3 |
where, N is the total number of nodes in the network, and dij is the shortest path length to node i.
The betweenness of a node i is defined by the number of the shortest paths that cross node i. The betweenness of node i is given in Equation (4):
| 4 |
where, gjik is the number of the shortest paths from j to k that pass through i.
The shortest path length calculations are based on a modified form of Dijkstra's algorithm (35). Network parameters were calculated with the igraph package version 0.1.2 using GNU R statistical software (http://cneurocvs.rmki.kfki.hu/igraph), and the network python package (https://networkx.lanl.gov).
Statistical analysis
Enrichment of mutations within high centrality nucleotides was evaluated based on the Hyper Geometric Distribution using the Fisher's exact test. In addition, we tested the enrichment in 100 random sets of nucleotides of the same size of each mutations dataset and expended the tested set to include the contacting nucleotides, as applied for the original datasets. The proportion of nucleotides that were randomly sampled from the large and small ribosomal subunit was as in the mutation datasets.
Ribosome structure analysis
In order to define the ribosomal subunits interface, the solvent accessible surface area of each nucleotide in the 16S and the 23S rRNA was calculated using the POPS server (36,37). A nucleotide was considered to be in the interface if it had at least one atom that lost solvent accessibility upon complex formation (PDB codes 2AVY and 2AW4).
The nucleotides of the tunnel were defined using the 3V Channel Extractor (http://geometry.molmovdb.org/3v) with a large probe radius of 9.0 Å and a small probe radius of 3.4 Å. Using the 3V Channel Extractor, an output file with the tunnel surface represented as water atoms was generated. We considered a nucleotide to be located in the tunnel if at least one of its atoms was located at a distance <5 Å to the computed water oxygen atom.
Accessible surface area of each nucleotide was calculated using the POPS server (36,37). POPS is based on a fast algorithm for calculating solvent accessible surface areas of large macromolecular assemblies, such as the ribosome. Nucleotides were defined as interface nucleotides if their solvent accessible surface area decreased upon subunit association. The accessible surface area of each nucleotide in the 70S E. coli rRNA (PDB codes 2AVY and 2AW4), and the 16S and 23S rRNA were calculated separately.
Theoretical B-factors were downloaded from the iGNM V1.2 database (http://ignm.ccbb.pitt.edu). The GNM program calculated the theoretical B-factors (1JJ2) for a 1 nt model using a cutoff of 7.3 Å for aa, and 19 Å for nucleotide.
The following nucleotides in the 16S rRNA were defined as the A site: 1405, 1406, 1407, 1408, 1409, 1410, 1490, 1491, 1492, 1493, 1494, 1495 and 1496. The following nucleotides in the 23S rRNA were defined as the PTC nucleotides: 2061, 2062, 2063, 2064, 2251, 2252, 2439, 2450, 2451, 2452, 2453, 2492, 2493, 2494, 2503, 2506, 2507, 2553, 2554, 2555, 2573, 2583, 2584, 2585, 2602 and 2603.
Evolutionary rate
Aligned sequences and a guide tree for both the 16S/18S and the 23S/28S were downloaded from the ARB-SILVA database (release 92) (http://www.arb-silva.de) (38). The 16S/18S alignment comprises high-quality sequences with a minimum length of 1200 bases for Bacteria and Eukarya and 900 bases for Archaea. The 23S/28S alignment comprises high quality rRNA sequences with a minimum length of 1900 bases. Positional variability in bacteria was calculated with the ARB package using parsimony function (38), including 168131 and 4115 sequences for the 16S and 23S rRNA, respectively. The evolutionary rate was calculated for each column as the mutations sum divided by the mutations frequencies sum.
Mutation data
Datasets 1, 2
The two datasets are based on mutation data, collected from various studies. The ribosomal RNA mutation database in E. coli (16SMDB and 23SMDB) was downloaded (http://server1.fandm.edu/departments/Biology/Databases/RNA.html) (39). We further extended the database by including data from recent studies (40–43). Dataset 1 includes mutations with strong effect on the ribosome function were selected by using a keyword search (strong, lethal, severe and deleterious) excluding mutations with mild or moderate effect. Further, the mutations were manually filtered to include only single point mutations. In addition, those mutations which had their phenotypic effect tested with the presence of antibiotics were excluded. To avoid redundancy, we included mutations with a minimal space of 3 nt in the primary sequence. Additionally, we excluded mutations that appeared in Datasets 3 and 4. In total, Dataset 1 included 44 nt: 25 nt in the 16S rRNA and 19 in the 23S rRNA. The 16S rRNA mutated nucleotides included positions: 13, 18, 517, 529, 571, 627, 643, 702, 770, 787, 792, 865, 914, 922, 967, 981, 1200, 1207, 1401, 1409, 1414, 1418, 1483, 1491 and 1498. The 23S rRNA mutated nucleotides included positions: 1832, 1836, 1849, 1896, 1916, 1926, 1932, 1940, 1946, 1955, 1960, 1972, 1979, 1984, 2252, 2504, 2507, 2580 and 2584 (Supplementary Table S5). Dataset 2 includes mutations with mild effect of the bacteria function. In total, Dataset 2 included 30 nt: 18 nt in the 16S rRNA and 12 in the 23S rRNA. The 16S rRNA mutated nucleotides included positions: 531, 534, 618, 624, 631, 634, 641, 645, 651, 912, 966, 1203, 1341, 1351, 1388, 1397, 1404 and 1518. The 23S rRNA mutated nucleotides included positions: 1067, 1098, 1914, 1921, 1940, 1951, 1979, 2249, 2254, 2477, 2561 and 2661.
Dataset 3, 4
These two sets were derived from two recent studies, which applied random mutagenesis procedure on E. coli 16S and 23S rRNA genes (44,45). In these studies, 53 and 77 mutations in the 16S and 23S rRNA, respectively, were classified according to their phenotypic severity. The 16S and 23S rRNA included 50 and 69 base substitutions, and 3 and 8 deletions, respectively. Among the 16S base substitutions, 13 mutations were classified as strong, 17 as mild and 20 as moderate. In the 23S base substitutions, 12 mutations were classified as strong, 34 as mild and 23 as moderate. Overall, the datasets included 25 positions (13 + 12) that were classified as strong and 51 positions (17 + 34) that were classified as mild.
The 16S rRNA mutated nucleotides with strong deleterious phenotype (Dataset 3) included the following mutations: Y516G, C518U, C519U, A520G, G521A, G973A, G1058A, G1068A, A1111U, C1208G, C1395U, U1406C and U1495C. The 16S rRNA mutated nucleotides with mild effect included: U49C, A51G, G57A, A161G, G299A, A373G, A389G, C536U, G568C, C614A, A622G, U684C, A1014G, C1054U, A1055G, U1073C and U1085C. The 23S rRNA mutated nucleotides with strong deleterious phenotype included: U860C, G864A, U2249A, G2250A, U2438C, G2446A, A2450G, A2451U/G, A2453G, C2499U, G2603A, and G2664A. The 23S rRNA mutated nucleotides with mild effect (Dataset 4) included: G46A, A49G, G55A, G58A, G123A, G215A, A324G, U328C, A330G, A371G, G380A, C420U, G425A, G481A, U558A, G578A, G656U, G778A, G1259A, C1297U, A1342G, C1345U, U1397G, G1421A, A1572C, U1602C, A1614G, U1683C, U1765G, C1800U, U1995C, G2027A, G2641A and U2898C.
Throughout this work, nucleotides were presented according to the E. coli sequence numbering.
RESULTS AND DISCUSSION
Ribosomal RNA exhibits small-world properties
Previously, it has been shown that amino acid network of a model protein structure have the unique properties of a small-world network (25). To study the properties of the ribosome network, we investigated the topological properties of four rRNA networks, including the small ribosomal subunit (PDB code 1J5E), two large ribosomal subunits (PDB codes 1NKW, 1JJ2) and the rRNA of the entire ribosome structure (PDB codes 2AVY, 2AW4). We presented the nucleotides and amino acids of the ribosome as nodes in the graph. Two nodes were connected by an edge when at least one atom from 1 nt/aa was in contact with at least one atom from a neighboring node. The inter-atomic contacts were defined based on surface complementarity, calculated with the CSU software (33).
The topological characteristics of a network are generally defined by two major parameters: the clustering coefficient (C) and the average shortest path length (L) (see Materials and Methods section). The clustering coefficient is the probability that any two neighbors of a given node in a graph are directly connected to each other, whereas the average path length is defined as the average of the minimal distance between all pairs of nodes in the graph. Different than random networks, small-world networks are characterized by a small number of highly central nodes (21). In comparison to random networks of the same size, small-world networks have a significantly higher clustering coefficient (Csmall-world ≫ Crand) and an average shortest path length higher or in the same order of magnitude (Lsmall-world ≥ Lrand or Lsmall-world ≈ Lrand) (24). To test whether the ribosome structures have characteristics of small-world networks, we calculated the values of L and C of the ribosome networks, and compared to their equivalent values in regular and random networks with the same number of nodes and an average number of edges. The values of L and C of four rRNAs networks are presented in Table 1. Notably, the C values calculated for the ribosome structures were orders of magnitude larger than in randomly generated networks, while the L values were in the same order of magnitude as the theoretical random networks. In other words, rRNA structure can be described as a network of interacting nucleotides that have an intermediate value of L and C, a property which characterizes the class of small-world networks (21). In all four rRNA networks we analyzed, the clustering coefficient values as well as the average path lengths scaled with the values measured for protein structures (19,46).
Table 1.
Topological properties of four rRNA structures
| rRNA structure | Nodes | Edges | Average shortest path length |
Clustering coefficient |
||||
|---|---|---|---|---|---|---|---|---|
| Lrand | Lobserved | Lreg | Crand | Cobserved | Creg | |||
| T. thermophilus (30S) | 1494 | 5389 | 3.7 | 12.5 | 104 | 0.0048 | 0.50 | 0.63 |
| D. radiodurans (50S) | 2766 | 10 673 | 3.9 | 11.6 | 179.6 | 0.0028 | 0.47 | 0.64 |
| H. marismortui (50S) | 2754 | 10 447 | 3.9 | 11.9 | 181.2 | 0.0028 | 0.48 | 0.64 |
| E. coli (70S) | 4371 | 16 049 | 4.2 | 15.5 | 198.2 | 0.0017 | 0.48 | 0.63 |
Average shortest path length and clustering coefficient were calculated for the small ribosomal subunit of T. thermophilus (PDB code IJ5E), the large ribosomal subunit of D. radiodurans (PDB code 1NKW), the large ribosomal subunit of H. marismortui (PDB code 1JJ2) and the structure of the 70S ribosome from E. coli (PDB codes 2AVY and 2AW4). The C and L of the random and the regular networks were calculated as described in text.
Small-world and scale-free properties have been found in a large number of real world networks. However, the small-world property is less studied in networks that are not scale-free (47). To study the network properties of the ribosome, we selected the 2.4 Å high-resolution structure of the large ribosome subunit from H. marismortui (PDB code 1JJ2). We compared the 23S rRNA network extracted from 1JJ2 to five random graph models (48): Erdös-Rényi random graphs (‘er’) (49), random graphs with the same degree distribution as the data model network (‘er-dd’), 3D geometric random graphs (‘geo’) (50), Barabási-Albert-type scale-free networks (‘sf’) (51) and stickiness-index-based networks (‘sticky’) (52). Based on this comparison (Supplementary Table S1), we found that the rRNA structure-derived network fits best with the geometric random graph model both by global and local properties. Overall, the rRNA structure-derived network fits the geometric random graph model but is highly clustered as typical for small-world networks.
Centrality measures and unique properties of the ribosome
Network analyses of protein structures have concluded that high centrality measures are associated with key (functional) positions, such as: enzyme catalytic sites, ligand-binding sites (17,27,28) as well as hot spots in protein–protein interactions (20,30). The major parameters examined in these studies included both local and global properties of the protein structures. In an attempt to identify prominent nucleotides that have a central role in the rRNA structure, we calculated three centrality measures for all nucleotides (nodes) in the network, i.e. degree, closeness and betweenness. Degree is a local measure that is defined as the number of edges directly connected to a node, whereas closeness and betweenness are based on the global centrality of the node. Closeness reflects the average length of the shortest path connecting a node to all other nodes in the network. Betweenness reflects the frequency in which a node appears on the shortest path between any two nodes in the network.
Closeness, betweenness and degree distributions have been utilized to compare network topologies. Among them, networks with small-world properties, such as protein–protein interaction network (53) and protein structure-based network (26). The distribution of the three centrality measures of all nucleotides in the 23S rRNA network (1JJ2) is presented in Figure 1. As previously observed in protein structures (17,26), the closeness parameter in the rRNA ribosome network showed a bell-shaped distribution (Figure 1A). Additionally, in accordance with previous observations in protein structures (19,25,54), the degree parameter of the rRNA structure follows a Poisson distribution (Figure 1C). The latter observation is different from other small-world networks such as regulatory networks that are scale-free (51), namely, a small number of nodes having very high degree values (51,55). Important to note that based on a visual investigation of wiring diagrams of the 16S rRNA, it has been suggested that the ribosome has characteristics of a scale-free network (32). Nevertheless, the observed distribution of the degree values in our nucleotide network (Figure 1C) indicates that the probability of highly connected nodes is relatively low. This result is expected based on the limited binding capacity of a given nucleotide, due to the excluded-volume effect in the RNA (56). Unlike the distribution of the degree parameter, the betweenness centrality parameter displays a power law distribution (Figure 1B), meaning that the majority of nucleotides demonstrate low values of betweenness, while only a few possess high values.
Figure 1.

Closeness, betweenness and degree distributions calculated for the structure-based network of the large ribosomal subunit (PDB code 1JJ2). The presented distributions correspond to the 23S rRNA nucleotides. (A) Closeness, (B) betweenness and (C) degree.
We further calculated the Pearson correlation coefficient for the different parameters in the 23S rRNA network (PDB code 1JJ2, 2754 nt). Our results indicate a weak positive correlation between the parameters. The correlation between closeness and betweenness is 0.42 (P < e-15), while the correlation between degree and closeness, and between degree and betweenness is 0.54 and 0.50, respectively (P < e-15). The relative weak correlation between the network parameters suggest that each parameter can be applied independently to describe specific nodes (nucleotides) in the network, i.e. a node in the network can have a small number of neighbors (low degree) but can still be located in a central position in the structure (high closeness and betweenness). A notable example is nt 2602 in the 23S rRNA (E. coli numbering throughout), which has a high closeness value in E. coli 70S (z-score = 1.7, P < 0.05) but has a very low degree, i.e. in contact with only two neighbors. Moreover, the betweenness of this nucleotide was zero, meaning that there is no short path that passes through this position. Interestingly, nt 2602 is the only nucleotide in the PTC that has zero betweenness. The unique global and local properties of nt 2602 are consistent with the different conformations observed in various functional complexes of the large subunit with different antibiotics and substrate analogs (57–60).
Though this study focuses on the unique properties of the rRNA, since the ribosome consists of both the rRNA and proteins, it was intriguing to examine the influence of the ribosomal proteins on the network properties of the rRNA. Thus, we analyzed the centrality measures separately for the 23S rRNA structure and for the 50S subunit, which comprises the 23S rRNA, the 5S rRNA and the ribosomal proteins. Both networks are based on the atomic coordinates of the 2.4 Å resolution structure from H. marismortui (PDB code 1JJ2). The first network included 2754 nodes and 10 447 edges (Table 1), while the second network, including the ribosomal proteins, was comprised of 6577 nodes and 33 013 edges. Interestingly, high correlations between the rRNA and the 50S subunit were obtained for closeness (r = 0.96, P < e-15) and betweenness (r = 0.72, P < e-15). While a lower correlation was measured for the degree parameter (r = 0.63, P < e-15). Similarly, we compared between the T. thermophilus (PDB code 1J5E) 16S rRNA network (Table 1) and the 30S structure that comprises the 16S rRNA and the ribosomal proteins (3850 edges and 18 929 edges). The Pearson correlation coefficient of the 16S rRNA nucleotides was 0.91, 0.86 and 0.67 (P < e-15) for closeness, betweenness and degree, respectively. The high correlations in the network parameters, specifically the closeness, between the rRNA and the entire ribosome networks of both the 30S and the 50S suggest that the proteins do not change the network properties. These results reinforce that the rRNA network can be analyzed independently.
Comparative analysis of the rRNA structure using centrality measures
We further compared the centrality values of ribosomal networks from different organisms. When comparing between the 70S rRNA of E. coli (8) (PDB codes 2AVY and 2AW4) and the 70S of T. thermophilus (9) (PDB codes 2J00 and 2J01) we observed extremely high correlations for both closeness (r = 0.98, P < e-15,) and degree (r = 0.87, P < e-15), though a lower correlation was observed for betweenness (r = 0.7, P < e-15). Similar results were obtained when comparing the 23S rRNA network from bacteria to the 23S rRNA from archaea, as well as for the 16S rRNA from different bacteria (E. coli and T. thermophilus) (Supplementary Table S2). Here again, among the three centrality parameters closeness was the most conserved, while betweenness tended to be more variable.
A comparison between the network parameters from different organisms is shown for a stretch of consecutive nucleotides in the central domain V of the 23S rRNA (Figure 2). The fine-tuned correlation in the closeness values (Figure 2A) is notable when comparing the 70S structures from E. coli (8) and T. thermophilus (9) as well as to the H. marismortui 50S structure (6). Conversely, the betweenness (Figure 2B) and the degree (Figure 2C) parameters show notable variations among the different structures. For example, variations in the betweenness and degree parameters between the two E. coli structures were observed in the region between nucleotides U2473 and C2475 in helix H89. These variations may correspond to the movement of the L1 arm, as observed in the two ribosome structures (8).
Figure 2.

The correlation between the centrality measures in different organisms is presented for the central domain V, nt 2433–2610. (A) Closeness, (B) betweenness and (C) degree. Network parameters are based on the rRNA network calculated for the E. coli 70S (2AVY, 2AW4: blue line), (2AW7, 2AWB: cyan line), T. thermophilus 70S (2J00, 2J01: red line) and H. marismortui 50S (1JJ2: black line).
Centrality and functionality
The most intriguing question when studying the structural properties of the ribosome is to what extent these properties correspond to functionality. Given that evolutionary conservation is indicative of functionality, we computed the correlation between the network parameters and the evolutionary rate of the 23S rRNA sequence. Evolutionary rate in bacteria was calculated using the ARB software, based on aligned sequences and a guide tree downloaded from the SILVA database (38) (see Materials and Methods section). Our results show a weak negative correlation between the evolutionary rate and closeness (PDB codes 2AVY and 2AW4) in the 16S and 23S rRNA (r = 0.44, P < e-15). These results are consistent with earlier studies in proteins in which the closeness was found to be in correlation with evolutionary conservation, with values ranging between r = 0.24 and r = 0.62 (17).
To test the conjecture that the network parameters can give new insight into the ribosome function, we focused on two major functional sites, the PTC and the nascent peptide exit tunnel. The PTC is the catalytic center, located on the interface side of the large subunit at a cavity that opens into a roughly cylindrical tunnel, which is ∼100 Å long and spans the entire body of the large ribosomal subunit. Quite expected, we found that the PTC nucleotides (defined in the Materials and Methods section) demonstrated higher than average closeness values (the average z-score for all PTC nucleotides was 1.47). Moreover, when compared to all nucleotides in the entire ribosome complex, the closeness values of the PTC nucleotides were significantly higher (Figure 3). This result is consistent with the observation that the catalytic sites in enzymes are located in regions with high closeness (17,27,61).
Figure 3.

Closeness versus solvent accessible surface area (SASA) calculated for the E. coli 70S rRNA. The Pearson coefficient shows a weak negative correlation (r = 0.51, P < e-15). The A-site nucleotides of the 16S rRNA are colored green. The PTC nucleotides are colored red. Notably, the PTC nucleotides demonstrate high closeness values independent of the solvent accessibility.
In order to examine whether high closeness of the ribosomal nucleotides simply reflect their low accessibility to the solvent, we calculated the Pearson correlation between the solvent accessible surface area of each nucleotide in the 23S rRNA (36,37) and the network parameters. The correlations for closeness, betweenness and degree were −0.57, −0.37 and −0.65, respectively (in all cases the P-value was highly significant P < e-15). As shown in Figure 3, overall the closeness and the surface accessibility demonstrated a weak negative correlation. Nevertheless, it can be noticed that the high closeness of the PTC is independent of the surface accessibility.
The outstanding high closeness centrality values observed here for the PTC is consistent with the high evolutionary conservation previously shown for this region (57,58,62–64). To test the ability of each one of the parameters: evolutionary rate, solvent accessibility, closeness, betweenness and degree to exclusively characterize the PTC nucleotides, we applied a ROC curve analysis (65) for each property independently (Figure 4). As described in the Materials and Methods section, the PTC nucleotides were labeled as ‘positives’ and the remaining nucleotides in the 70S ribosome (16S and 23S) were labeled as ‘negatives’. The ROC curves show the true positive rate versus the false positive rate. To quantify the classification performance, we compared the area under the curve (AUC) of the different ROC plots. As can be noticed (Figure 4) the closeness centrality better classifies the PTC nucleotides (AUC = 0.95) compared to all other structural parameters, as well as to the evolutionary conservation rate (AUC = 0.87). Remarkably, for 100% sensitivity, i.e. all 26 nt comprises the PTC nucleotides being classified correctly, the closeness achieved 78% accuracy while the evolutionary rate achieved only 48% accuracy. Additionally, our results show that the solvent accessibility parameter did not improve the PTC nucleotides’ identification. This latter result differs from observations in proteins showing that solvent accessibility improves the identification of enzyme catalytic residue significantly (17,26). Taken together, our results indicate that the ribosome major catalytic site can be accurately identified by a global topological measure (closeness) with no requirement for evolutionary information.
Figure 4.

ROC curves classifying the PTC nucleotides. The structural parameters are based on the E. coli 70S network (2AVY, 2AW4). As shown, the PTC nucleotides were better classified using the closeness parameter (red line, AUC = 0.95) rather than the evolutionary rate (black line, AUC = 0.87), solvent accessibility (blue line, AUC = 0.74), degree (green line, AUC = 0.71) and betweenness (magenta line, AUC = 0.67).
In addition to the PTC, other functional sites in the ribosome such as the A-site in the 30S (Figure 3) and the exit tunnel in the 50S (Supplementary Table S4) demonstrated high closeness values. Generally, the 23S rRNA nts were found to have higher closeness values compared to the 16S rRNA nucleotides, with a maximum z-score of 2.37 and 1.85, respectively. Nevertheless, it can be noticed that both the PTC and the A-site nucleotides encompass relatively high closeness values compared to the rest of the 70S nucleotides (Figure 3). Additionally, the exit tunnel nucleotides (50S) were found to possess high closeness values with the highest values for the tunnel nucleotides facing the PTC (Supplementary Table S4). Further, we examined the number of contacts (degree) of the tunnel nucleotides (PDB code 1JJ2). Interestingly, the high degree values, like the closeness values, were independent of the solvent accessibility. Overall, the average degree of the tunnel nucleotides (123 nt) was found to be significantly higher (8.3) than the average degree of all 23S rRNA nucleotides (7.6) (P = 0.001, t-test).
When examining the distribution of degree values along the tunnel by dissecting it into 10 Å wide slices (5 Å overlap) starting from the PTC (nt 2451), we found that the highest degree values in the tunnel were within a distance of 20–30 Å from the PTC (Supplementary Table S3). The distribution of the degree values along the ribosome tunnel could be indicative of the tunnel dynamics. To better understand the correspondence between the tunnel dynamics and the degree we compared the profile of the degree in the tunnel with a theoretical B-factor calculated with the Gaussian Network Model (GNM) method (66,67). GNM has been introduced as an efficient tool to explore the dynamics of proteins and large biomolecular structures such as the ribosome (12,54,68). In the GNM, a structure is represented as a network of N nodes corresponding to the α-carbon atoms of proteins and selected atoms of the nucleotides (see Materials and methods section). In accordance with the degree distribution, the theoretical B-factor confirmed that the center of the tunnel (15–25 Å from nt 2451) is more rigid, but its flexibility increases as it reaches the tunnel exit. Based on the crystallographic B-factor values for the RNA atoms in the H. marismortui large ribosomal subunit, an upper bound estimate of nucleotide thermal fluctuations of 0.63 Å was suggested (6). It has been argued that random structural fluctuations of this magnitude will not allow drastic changes in the tunnel dimensions, which would be required in order for the nascent polypeptide to form tertiary structures (69). Our network analysis, demonstrating high closeness of the tunnel nucleotides and a relatively high degree of nucleotides that construct the tunnel wall (Supplementary Tables S3 and S4, Figure S1) supports this hypothesis (69). The relatively high degree in a central region within a macromolecule is quite expected and is consistent with other observations in globular proteins showing that the residues in the protein core are characterized by a higher mean degree than the surface residues (18).
As shown, the PTC nucleotides have significantly high closeness values among all the ribosome nucleotides. Notably, the top ranked nucleotides in the 70S rRNA only partially overlap with the PTC nucleotides. This can also be observed in the exit tunnel profile (Supplementary Table S4), which shows that the highest average closeness is within a distance of 5–15 Å from nt 2451. To identity the nucleotides with the highest closeness in the rRNA, we ranked the closeness values of nucleotides in the E. coli 70S rRNA (8) (PDB codes 2AVY and 2AW4) and the T. thermophilus 70S rRNA (9) (PDB codes 2J00 and 2J01). Interestingly, among the top ten nucleotides with the highest closeness values in each of the ribosome networks (from different bacteria), 7 nt were common (1938, 2439, 2584, 2591, 2604, 2605 and 2608). Interestingly, these nucleotides only partially overlap the PTC nucleotides. This result is consistent with previous observations in proteins (26) showing that the most central residues fall near the center of the structure, which often does not overlap with the catalytic residues. Notably, in the ribosome networks, among the top ranked nucleotides there were no nucleotides that directly interact with the ribosome substrate, such as nt 2062, 2573, 2585, 2602 and 2609 (62). This could be partially explained by the conformation variations observed for these nucleotides in different ribosome structures (57,62,70). Interestingly, among the ten top ranked nucleotides in the two bacteria (70S) and in the archaeon (50S) was nt A2439 with extremely high closeness. This nt (A2439, H74) is located in the symmetrical region of the ribosome (62), and forms an A- to P-region contact through its ribose (A2439 is bulged with U2586, H93). The high closeness values in the vicinity of the PTC both in the bacteria (70S), and in archaea (50S), may suggests that the spatial organization of the rRNA nucleotides is required to enable signal transmission between remote locations in the ribosome. This hypothesis is supported by the observation of a 2-fold symmetry axis within domain V, which suggests that the PTC resulted from an early gene duplication event and may indicate that domain V is the most ancient part of the primitive ribosome (62,70).
Network properties of critical positions in the ribosome
As demonstrated in Table 1, the network of interacting nucleotides in the 70S rRNA is highly clustered (Table 1). Consequently, there is a relatively small number of nucleotides that are highly central and serve as interconnection hubs between all nucleotides in the structure. A fascinating conjecture is that the latter nucleotides have a central role in the function or stability of the ribosome complex. Here, we focused on functional information derived from mutagenesis studies, including directed mutations and random mutations, which have been reported to specifically affect ribosome function or subunits association (39–45,71).
The first dataset we tested included 44 mutations, which have been reported to have strong, lethal, severe or deleterious effect on ribosome function, activity, growth, association or translation fidelity (see Materials and Methods section). We defined the mutated nucleotides and their structural neighbors as experimental set 1. Overall, the nucleotides of the experimental set 1 represented 7% of the ribosomal nt (308/4371). As a negative control, we extracted from various studies a dataset of 30 mutations that have mild effect of the bacteria function (see Materials and Methods section, Supplementary Table S5). The mild mutations and their structural neighbors were defined as experimental set 2. Overall, the nucleotides of the experimental set 2 represented 5% of the ribosomal nt (212/4371).
The two additional datasets were based on a random mutants library of rRNA gene (44,45). The first dataset included deleterious mutation with strong (25 nt) effect on E. coli growth. The second dataset included mutations with mild (51 nt) effect on E. coli growth (44,45). Further, we defined the mutated nucleotides and their structural neighbors as experimental sets. Overall, the nucleotides of experimental set 3, derived from the strong deleterious mutations represented 4% of the ribosomal nt (185/4371), and the nucleotides of experimental set 4 derived from the mutations with mild phenotype represented 10% of the ribosomal nt (422/4371).
Next, we defied three subsets of nucleotides that encompass high centrality, i.e. nucleotides with the top 5% values of closeness, betweenness and degree, in the E. coli 70S rRNA (PDB codes 2AVY and 2AW4). Their exact position is presented on the ribosome secondary structure diagram (Supplementary Figures S4 and S5). In order to calculate the correspondence of the experimental sets to the network parameters, we calculated the fraction of the experimental sets nucleotides among the nucleotides with high values (top 5%) of closeness, betweenness and degree (Figure 5). To evaluate the enrichment of the mutations among nucleotides with the high centrality values, we applied the Hyper Geometric Distribution analysis using the Fisher's exact test. Additionally, to test the significance of the results, we selected 100 random sets with equal number of nucleotides as in each one of the experimental sets (see Materials and Methods section) and tested their overlap with the subsets of nucleotides with high centrality values. As shown in Figure 5A, the set of mutations with strong deleterious effects on bacterial growth (experimental set 1), was significantly enriched with nucleotides with high values of closeness and betweenness (Figure 5A, black bars). An enrichment (relatively to randomly selected nucleotides) was also observed in experimental set 3 (Figure 5C) but was not observed in the control sets (experimental set 2, 4), which comprises mutations with mild effect on bacterial growth (Figure 5B and D). Interestingly, the significant enrichment of deleterious mutations among highly central nucleotides was found in two independent sets of data, which were obtained from different sources. By examining different thresholds for high centrality definition, we obtained that in all thresholds (Supplementary Figure S2) there was an enrichment of nucleotides with strong deleterious mutation (experimental set 1, 3) among nucleotides with high closeness centrality. Here again, we did not observe a significant enrichment for the nucleotides with the mild phenotype (experimental set 2, 4), relatively to randomly selected nucleotides (Figure 5, Figure S2).
Figure 5.

A histogram demonstrating the enrichment of mutated nucleotides among three sets of nucleotides: (A) experimental set 1, (B) experimental set 2, (C) experimental set 3 and (D) experimental set 4. High centrality refers to nucleotides with significantly high values (top 5%) for closeness, betwenness and degree. Black bars represent nucleotides with high centrality values (top 5%). The gray bars represent subset of ribosomal nucleotides that do not possess high centrality. White bars represent randomly selected equal sized subsets of nucleotides. The enrichment of mutations that cause strong deleterious effect on ribosome function is statistically significant in the strong deleterious mutations (A and C) but not in the mild mutations (B and D).
It has been shown (44,45) that evolutionary conservation (72) is high for both strong deleterious mutations and for a large fraction of the mild mutations. In order to quantify the ability of evolutionary rate (38), and the closeness centrality to classify strong deleterious mutations we applied a ROC curve analysis (65). Strong deleterious mutations comprising nucleotides in experimental set 1 and 3 were labeled as ‘positives’ (433) and the remaining nucleotides in the 16S and 23S rRNA were labeled as ‘negatives’. Our results showed (Supplementary Figure S3) that in the 16S rRNA, the performance of the closeness-based classifier was better (AUC = 0.77) than the evolutionary rate classifier (AUC = 0.67). An overall higher performance was observed in the 23S rRNA for both the closeness (AUC = 0.85) and the evolutionary rate classifiers (AUC = 0.84).
In summary, we have shown that deleterious mutations, in contrast to mild mutations, tend to have high closeness values in the rRNA. Furthermore, with the closeness parameter derived from a single ribosome structure we were able to distinguish the strong deleterious mutations from mutations which do not affect the ribosome function independent of the evolutionary conservation of the mutated nucleotides. Quite strikingly, in the small subunit the closeness parameter achieved even better performance compared to the evolution-based parameter, which is traditionally used for defining functionality.
CONCLUSION
Here, we have applied a network approach to study the unique properties of the bacterial and the archaeal rRNA. We show that the different ribosome structures have properties of a small-world network and that the network properties correlate weakly with structural properties of the ribosome as well as with evolutionary conservation. Interestingly, the active sites of the ribosome demonstrated high centrality measures independent of the surface accessibility and evolutionary conservation, reinforcing their unique structural properties. Most prominently, we show that the network parameters can point out nucleotides with critical functional roles. Specifically, we found that the major functional nucleotides in the ribosome demonstrate significantly high centrality values in both global and local properties. In addition, we were able to distinguish mutations in nucleotides, which have deleterious effect on the ribosome function or assembly from those which cause mild effect, based on their network centrality. We suggest that the closeness centrality measures can be used to classify functional and critical positions in the ribosome, independent of their evolutionary conservation. In conclusion, we present a new approach to characterize essential positions in the bacterial ribosome, which can also be exploited as a tool for structural comparative analysis of large RNA structures.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Ilana Agmon and Anat Bashan for their help with ribosome structure analysis. Thanks to Alexander Mankin for the support and helpful suggestions during the course of the project and for valuable comments on the article. Many thanks to Ron Pinter for the helpful advice on the network analysis and for critical remarks on the article. This work was funded by the by the Israeli Science Foundation ISF grant number 923/05 granted to Y.M.G. Funding to pay the Open Access publication charges for this article was provided by the Israel Science Foundation Grant number 923/05.
Conflict of interest statement. None declared.
REFERENCES
- 1.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008;9:242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 2.Ramakrishnan V. Ribosome structure and the mechanism of translation. Cell. 2002;108:557–572. doi: 10.1016/s0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- 3.Maguire BA, Beniaminov AD, Ramu H, Mankin AS, Zimmermann RA. A protein component at the heart of an RNA machine: the importance of protein l27 for the function of the bacterial ribosome. Mol. Cell. 2005;20:427–435. doi: 10.1016/j.molcel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 5.Wimberly BT, Brodersen DE, Clemons W.M., Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 6.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 7.Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, et al. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001;20:1829–1839. doi: 10.1093/emboj/20.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 9.Selmer M, Dunham CM, Murphy F.V.4th, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 10.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 11.Cheatham TE., III Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr. Opin. Struct. Biol. 2004;14:360–367. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Rader AJ, Bahar I, Jernigan RL. Global ribosome motions revealed with elastic network model. J. Struct. Biol. 2004;147:302–314. doi: 10.1016/j.jsb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Trylska J, Tozzini V, McCammon JA. Exploring global motions and correlations in the ribosome. Biophys. J. 2005;89:1455–1463. doi: 10.1529/biophysj.104.058495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vishveshwara S, Brinda KV, Kannan N. Protein structure: insights from graph theory. J. Theor. Comp. Chem. 2002;1:187–211. [Google Scholar]
- 15.Grindley HM, Artymiuk PJ, Rice DW, Willett P. Identification of tertiary structure resemblance in proteins using a maximal common subgraph isomorphism algorithm. J. Mol. Biol. 1993;229:707–721. doi: 10.1006/jmbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Protein flexibility predictions using graph theory. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- 17.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 18.Atilgan AR, Akan P, Baysal C. Small-world communication of residues and significance for protein dynamics. Biophys. J. 2004;86:85–91. doi: 10.1016/S0006-3495(04)74086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bagler G, Sinha S. Network properties of protein structures. Physica A Stat. Mech. Appl. 2005;346:27–33. [Google Scholar]
- 20.del Sol A, O’Meara P. Small-world network approach to identify key residues in protein-protein interaction. Proteins. 2005;58:672–682. doi: 10.1002/prot.20348. [DOI] [PubMed] [Google Scholar]
- 21.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 22.Taylor TJ, Vaisman II. Graph theoretic properties of networks formed by Delaunay tessellation of protein structures. Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys. 2006;73:041925. doi: 10.1103/PhysRevE.73.041925. [DOI] [PubMed] [Google Scholar]
- 23.Dokholyan NV, Li L, Ding F, Shakhnovich EI. Topological determinants of protein folding. Proc. Natl Acad. Sci. USA. 2002;99:8637–8641. doi: 10.1073/pnas.122076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vendruscolo M, Dokholyan NV, Paci E, Karplus M. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;65:061910. doi: 10.1103/PhysRevE.65.061910. [DOI] [PubMed] [Google Scholar]
- 25.Bode C, Kovacs IA, Szalay MS, Palotai R, Korcsmaros T, Csermely P. Network analysis of protein dynamics. FEBS Lett. 2007;581:2776–2782. doi: 10.1016/j.febslet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Chea E, Livesay DR. How accurate and statistically robust are catalytic site predictions based on closeness centrality? BMC Bioinform. 2007;8:153. doi: 10.1186/1471-2105-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.del Sol A, Fujihashi H, Amoros D, Nussinov R. Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci. 2006;15:2120–2128. doi: 10.1110/ps.062249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibert B, Bredesen DE, del Rio G. Improved prediction of critical residues for protein function based on network and phylogenetic analyses. BMC Bioinform. 2005;6:213. doi: 10.1186/1471-2105-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Z, Bowen D, Southerland WM, Del Sol A, Pan Y, Nussinov R, Ma B. Ligand binding and circular permutation modify residue interaction network in DHFR. PLoS Comput. Biol. 2007;3:e117. doi: 10.1371/journal.pcbi.0030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.del Sol A, Fujihashi H, O’Meara P. Topology of small-world networks of protein-protein complex structures. Bioinformatics. 2005;21:1311–1315. doi: 10.1093/bioinformatics/bti167. [DOI] [PubMed] [Google Scholar]
- 31.Wuchty S. Small worlds in RNA structures. Nucleic Acids Res. 2003;31:1108–1117. doi: 10.1093/nar/gkg162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lescoute A, Westhof E. The interaction networks of structured RNAs. Nucleic Acids Res. 2006;34:6587–6604. doi: 10.1093/nar/gkl963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 34.Freeman LC. Centrality is social networks I: conceptual clarification. Soc.Networks. 1979;1:215–239. [Google Scholar]
- 35.Brandes U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001;25:163–177. [Google Scholar]
- 36.Cavallo L, Kleinjung J, Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraternali F, Cavallo L. Parameter optimized surfaces (POPS): analysis of key interactions and conformational changes in the ribosome. Nucleic Acids Res. 2002;30:2950–2960. doi: 10.1093/nar/gkf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Triman KL. Mutational analysis of the ribosome. Adv. Genet. 2007;58:89–119. doi: 10.1016/S0065-2660(06)58004-6. [DOI] [PubMed] [Google Scholar]
- 40.Kim HM, Yeom JH, Ha HJ, Kim JM, Lee K. Functional analysis of the residues C770 and G771 of E. coli 16S rRNA implicated in forming the intersubunit bridge B2c of the ribosome. J. Microbiol. Biotechnol. 2007;17:1204–1207. [PubMed] [Google Scholar]
- 41.Liiv A, Karitkina D, Maivali U, Remme J. Analysis of the function of E. coli 23S rRNA helix-loop 69 by mutagenesis. BMC Mol. Biol. 2005;6:18. doi: 10.1186/1471-2199-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liiv A, O’Connor M. Mutations in the intersubunit bridge regions of 23 S rRNA. J. Biol. Chem. 2006;281:29850–29862. doi: 10.1074/jbc.M603013200. [DOI] [PubMed] [Google Scholar]
- 43.Pulk A, Maivali U, Remme J. Identification of nucleotides in E. coli 16S rRNA essential for ribosome subunit association. RNA. 2006;12:790–796. doi: 10.1261/rna.2275906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yassin A, Fredrick K, Mankin AS. Deleterious mutations in small subunit ribosomal RNA identify functional sites and potential targets for antibiotics. Proc. Natl Acad. Sci. USA. 2005;102:16620–16625. doi: 10.1073/pnas.0508444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yassin A, Mankin AS. Potential new antibiotic sites in the ribosome revealed by deleterious mutations in RNA of the large ribosomal subunit. J. Biol. Chem. 2007;282:24329–24342. doi: 10.1074/jbc.M703106200. [DOI] [PubMed] [Google Scholar]
- 46.Greene LH, Higman VA. Uncovering network systems within protein structures. J. Mol. Biol. 2003;334:781–791. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 47.Barnett L, Paolo ED. Spatially embedded random networks. Phys. Rev. 2007;76:056115. doi: 10.1103/PhysRevE.76.056115. [DOI] [PubMed] [Google Scholar]
- 48.Milenkovic T, Lai J, Przulj N. GraphCrunch: a tool for large network analyses. BMC Bioinform. 2008;9:70. doi: 10.1186/1471-2105-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erdos P, Renyi A. On random graphs. Publ. Math. 1959;6:290–279. [Google Scholar]
- 50.Penrose M. Random Geometric Graphs. Oxford: Oxford University Press; 2003. [Google Scholar]
- 51.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 52.Przulj N, Higham D. Modelling proteinprotein interaction networks via a stickiness index. J. Roy. Soc. Inter. 2006;3:711–716. doi: 10.1098/rsif.2006.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hormozdiari F, Berenbrink P, Przulj N, Sahinalp SC. Not all scale-free networks are born equal: the role of the seed graph in PPI network evolution. PLoS Comput. Biol. 2007;3:e118. doi: 10.1371/journal.pcbi.0030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newman ME, Girvan M, Farmer JD. Optimal design, robustness, and risk aversion. Phys. Rev. Lett. 2002;89:028301. doi: 10.1103/PhysRevLett.89.028301. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez A. Excluded-volume effects on the stacking of RNA base pairs. Phys. Rev. A. 1991;44:R7910–R7912. doi: 10.1103/physreva.44.r7910. [DOI] [PubMed] [Google Scholar]
- 57.Bashan A, Agmon I, Zarivach R, Schluenzen F, Harms J, Berisio R, Bartels H, Franceschi F, Auerbach T, Hansen HA, et al. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Mol. Cell. 2003;11:91–102. doi: 10.1016/s1097-2765(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 58.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 59.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 60.Schmeing TM, Seila AC, Hansen JL, Freeborn B, Soukup JK, Scaringe SA, Strobel SA, Moore PB, Steitz TA. A pre-translocational intermediate in protein synthesis observed in crystals of enzymatically active 50S subunits. Nat. Struct. Biol. 2002;9:225–230. doi: 10.1038/nsb758. [DOI] [PubMed] [Google Scholar]
- 61.Tang YR, Sheng ZY, Chen YZ, Zhang Z. An improved prediction of catalytic residues in enzyme structures. Protein Eng. Des. Sel. 2008;21:295–302. doi: 10.1093/protein/gzn003. [DOI] [PubMed] [Google Scholar]
- 62.Agmon I, Bashan A, Zarivach R, Yonath A. Symmetry at the active site of the ribosome: structural and functional implications. Biol. Chem. 2005;386:833–844. doi: 10.1515/BC.2005.098. [DOI] [PubMed] [Google Scholar]
- 63.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 64.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 65.Metz CE. Basic principles of ROC analysis. Semin. Nucl. Med. 1978;8:283–298. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- 66.Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 67.Yang LW, Liu X, Jursa CJ, Holliman M, Rader AJ, Karimi HA, Bahar I. iGNM: a database of protein functional motions based on Gaussian Network Model. Bioinformatics. 2005;21:2978–2987. doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chennubhotla C, Rader AJ, Yang LW, Bahar I. Elastic network models for understanding biomolecular machinery: from enzymes to supramolecular assemblies. Phys. Biol. 2005;2:S173–S180. doi: 10.1088/1478-3975/2/4/S12. [DOI] [PubMed] [Google Scholar]
- 69.Voss NR, Gerstein M, Steitz TA, Moore PB. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 2006;360:893–906. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 70.Agmon I, Auerbach T, Baram D, Bartels H, Bashan A, Berisio R, Fucini P, Hansen HA, Harms J, Kessler M, et al. On peptide bond formation, translocation, nascent protein progression and the regulatory properties of ribosomes. Derived on 20 October 2002 at the 28th FEBS Meeting in Istanbul. Eur. J. Biochem. 2003;270:2543–2556. doi: 10.1046/j.1432-1033.2003.03634.x. [DOI] [PubMed] [Google Scholar]
- 71.Triman KL, Peister A, Goel RA. Expanded versions of the 16S and 23S ribosomal RNA mutation databases (16SMDBexp and 23SMDBexp) Nucleic Acids Res. 1998;26:280–284. doi: 10.1093/nar/26.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}