

Abstract
Organotrifluoroborates are robust reagents capable of withstanding ozonolysis of remote alkenes, thus providing a new route to oxo-substituted organotrifluoroborates. The primary ozonides initially generated upon ozonolysis can be reduced with Zn/AcOH to afford the carbonyl compounds. Alternatively, capture of the carbonyl oxides with either an appropriate N-oxide or H2O easily gives the desired oxo-substituted organotrifluoroborates. Both unsaturated alkyltrifluoroborates and -aryltrifluoroborates effectively participate in the reaction. The process provides oxo-functionalized organotrifluoroborates that cannot be prepared directly via either transmetalation or hydroboration protocols.
Introduction
Organoborons are normally regarded as relatively sensitive compounds that are used immediately upon preparation for synthetic transformations in which the carbon-boron bond is exploited as the key reactive functional group in the desired conversion. Synthetic sequences can be envisioned, however, in which it would be desirable to carry the boron moiety through several routine transformations on remotely embedded functional groups before taking advantage of its unique properties in new bond-forming events. Organotrifluoroborates provide an ideal platform to explore these possibilities.
The versatility and unique reactivity of organotrifluoroborates in a number of synthetically useful processes (e.g., Suzuki-Miyaura coupling reactions,1,2 rhodium-catalyzed 1,4-additions,3,4 and allylation of aldehydes5), makes them among the more useful organoboron reagents. Organotrifluoroborates are easily prepared in high yields from the corresponding boronic acids or boronic acid derivatives.6–8 The tetracoordinate nature of these species, fortified with exceptionally strong boron-fluorine bonds, provides extraordinary opportunities for manipulation of remote functional groups with retention of the trifluoroborate because the boron moiety is effectively immunized against a number of routine chemical operations. As one example, we have previously demonstrated that alkene epoxidation reactions can be performed in the presence of the organotrifluoroborate unit, creating a novel entry to potentially valuable and unique organic synthons.9 In this Note we describe the ozonolysis of unsaturated potassium- and tetrabutylammonium organotrifluoroborates, which not only demonstrates the resistance of the carbon-boron bond to oxidation by ozone, but also provides a unique access to oxo-substituted organoborons; reagents that are inaccessible by direct hydroboration or transmetalation protocols.
Ozonolysis of carbon-carbon double bonds provides an efficient method for the introduction of carbonyl functional groups into a host of organic substrates.10 In addition to having a well-understood mechanism,10,11 the variety of available reaction conditions makes ozonolysis a powerful and versatile method in organic synthesis.12 Even so, a single example exists in which this method has been used with an organometallic intermediate.13 Herein we describe our efforts toward ozonolysis of both potassium- and tetrabutylammonium organotrifluoroborates, providing carbonyl-containing organoborons.
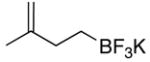
During early investigations of the ozonolysis of potassium 3-methylbut-3-enyltrifluoroborate 1 we discovered that treatment of the alkenyltrifluoroborate with ozone gave a secondary ozonide and a small fraction of carbonyl product without the cleavage of the carbon-boron bond. In fact, once formed, the ozonide demonstrated remarkable stability, resisting transformation to the carbonyl product 2 even under refluxing methyl sulfide.
To avoid this stable intermediate we employed a solvent participating protocol11 whereby a protic solvent was used in the reaction mixture to capture the carbonyl oxide intermediate before it could recyclize to form a secondary ozonide (Figure 1). Following a survey of several protic solvents, alkene 1 was ozonized successfully using water as a cosolvent (Table 1, entry 1). This presumably involved the conversion of the carbonyl oxide intermediate into the carbonyl product 2 (Procedure A), thereby avoiding formation of the stable secondary ozonide (Figure 1).11
Figure 1.
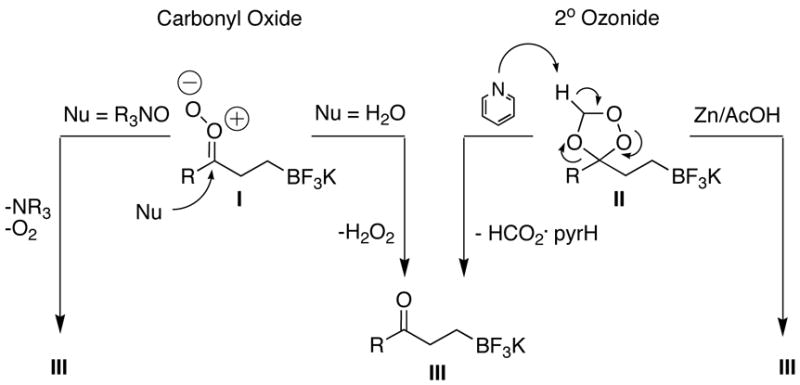
Transformation to Product from Carbonyl Oxides (I) vs. Secondary Ozonides (II).
Table 1.
Organotrifluoroborate Substrates Subjected to Ozonolysis
| entry | organotrifluoroborate | procedure | product | % isolated yield |
|---|---|---|---|---|
|
|
|||
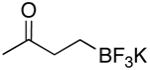
| 1 | 1 | A | 2 | 70 |
|
|
|||
| 2 | 3 | Ba | 4 | 76 |
|
|
|||
| 3 | 5 | Bb | 4 | 79 |
| 4 | 5 | Bc | 4 | 72 |
|
|
|||
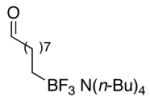
| 5 | 6 | Bd | 7 | 75 |
| 6 | 6 | D | 7 | 95 |
|
|
|
|||
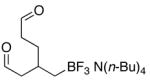
| 7 | 8 | Be | 9 | 79 |
|
|
|
|||
| 8 | 10 | Bb | 11 | 80 |
| 9 | 10 | Bd | 11 | 82 |
| 10 | 10 | C | 11 | 71 |
|
|
|||
| 11 | 12 | Ba | 13 | 89 |
| 12 | 12 | D | 13 | 95 |
A, Acetone/H2O, O3 −70 °C;
B, CH2Cl2, NMO (5 equiv), O3 −78 °C;
B, 50% DMF/Et3N, O3 0 °C, then (n-Bu4N)OH;
B, 40% DMF/CH2Cl2, O3 −78 °C, then (n-Bu4N)OH;
B, CH2Cl2, pyridine (3 equiv), pyridine N-oxide (2 equiv), O3 −78 °C;
B, CH2Cl2, pyridine (0.5 equiv), O3 −78 °C; C, CH2Cl2, O3 −78 °C, then pyridine (2 equiv); D, CH2Cl2, O3 −78 °C, Zn (5 equiv), AcOH (7 equiv).
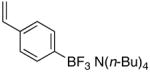
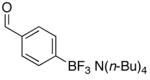
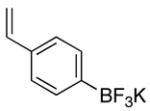
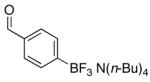
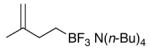
To determine the generality of this method, potassium 4-ethenylphenyltrifluoroborate 5 was subjected to the same conditions. Unfortunately, application of this method to the styrenyl substrate gave a 50:50 mixture of potassium 4-formylphenyltrifluoroborate and 4-carboxyphenyltrifluoroborate as products. The generation of the acid side product was suppressed by the addition of N-oxides, amines (which are transformed to N-oxides by exposure to ozone), or amides to the reaction mixture.14 Furthermore, the product was isolated by extraction with tetrabutylammonium hydroxide to give tetrabutylammonium 4-formylphenyltrifluoroborate 4 in good yields (Procedure B, Table 1, entries 3 and 4).
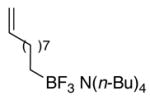
During the investigation of terminal alkenyltrifluoroborates, the method was extended to tetrabutylammonium decenyltrifluoroborate 6 and tetrabutylammonium 4-butenyltrifluoroborate 10. The installation of an aldehyde functional group was initially pursued utilizing 6. The tetrabutylammonium 8-trifluoroboratooctanal 7 was obtained in good yield by ozonolysis of the alkenyl substrate 6 with the addition of pyridine N-oxide and pyridine to the reaction mixture (Table 1, entry 5). The more densely functionalized γ-substituted aldehyde tetrabutylammonium 3-trifluoroboratopropanal 11 was accessed from 10 by the addition of either triethylamine or a combination of pyridine N-oxide and pyridine to the reaction mixture (Table 1, entries 8 and 9). Furthermore, aldehyde 11 was obtained by treatment of the secondary ozonide of 10 with pyridine (Procedure C), giving the product in good yield (71%). This latter protocol marks an interesting expansion of the overall ozonolysis transformation, as the use of pyridine in this acid/base reaction (see Figure 1) is one of only two methods found that effectively collapsed the very stable secondary ozonide formed from unsaturated organotrifluoroborates.
The second method found for the decomposition of the organotrifluoroborate secondary ozonides utilized a zinc/acetic acid slurry to reduce the tetrabutylammonium ozonide to the desired carbonyl product. Addition of this slurry following the introduction of sufficient ozone for complete conversion of the alkenyl substrate yielded the desired product for both organotrifluoroborates 6 (95%) and 12 (95%) (entries 6 and 12, respectively).
During the study of the tetrabutylammonium (cyclohex-3-enyl)methyltrifluoroborate substrate 8, good yields of the aliphatic dialdehyde tetrabutylammonium 4-formyl-2-(formylmethyl)butyltrifluoroborate 9 were achieved when pyridine was utilized in the reaction mixture (Procedure B), presumably bypassing the secondary ozonide intermediate with a mechanism closely paralleling that of the solvent participating protocol (Figure 1). Notably, the reaction of alkene 8 used substoichiometric pyridine (0.5 equiv) to give the highly sensitive dialdehyde product 9, suggesting that not only is the N-oxide effectively generated and used in situ, but also that, following the collapse of the intermediate, pyridine is regenerated. Interestingly, when the solvent participating conditions with water (Procedure A) were used on the aliphatic aldehyde precursors 8 and 6, the reaction became problematic and produced polymerized product.
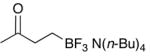
Finally, the tetrabutylammonium salt of substrate 1 was revisited (Table 1, entry 11). Using Procedure B it was demonstrated that in addition to their effectiveness in the installation of the aldehyde functional group, the use of N-oxides is effective in generating ketones as well, giving 13 in good yield (89%).
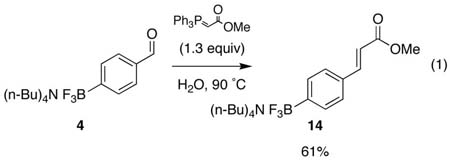
In summary, unsaturated organotrifluoroborates can be ozonized to the corresponding carbonyl moieties under standard conditions. This method presents a significant expansion of the chemistry of organoboron compounds, including the ability to introduce both aldehyde and ketone functional groups into organoboron reagents in a direct manner. These particular functional groups are not compatible with the traditional transmetalation or hydroboration routes to organoborons. Access to boronic acid or boronate ester substrates containing these carbonyl groups would thus require cumbersome protection/deprotection schemes or a complete alteration of synthetic strategy. Although to date we have been unable to cross-couple enolizable aldehyde-containing organotrifluoroborates, the aldehydes and ketones within the organotrifluoroborates can be further elaborated by carbonyl addition reactions9b as well as Wittig and Horner-Wadsworth-Emmons reactions (eq 1).9d,h
The organotrifluoroborates thus serve as robust surrogates for boronic acids in which remote functional groups can be elaborated to increase the molecular complexity of the organoboron reagent while maintaining the integrity of the carbon-boron bond for ultimate transformation by cross-coupling or other processes.15
Experimental Section
General Experimental Procedures A through D for the Ozonolysis of Potassium and Tetrabutylammonium Organotrifluoroborates. Preparation of Potassium 3-Oxobutyl Trifluoroborate (2) via Procedure A
To a solution of potassium trifluoroborate 1 (0.101 g, 0.643 mmol) in an acetone/H2O mixture (30% H2O in acetone, 7 mL) at −70 °C was applied a flow of ozone for 14 min. The solution was then degassed with N2 for 15 min followed by the addition of H2O (1 mL). This was allowed to warm to rt while stirring. Following solvent removal the resulting white solid was purified by dissolving in hot acetone and precipitating with Et2O, affording the trifluoroborate 2 as a white solid (0.072 g, 0.453 mmol, 70%).
Tetrabutylammonium 4-Formylphenyl Trifluoroborate (4) via Procedure Ba
To a solution of tetrabutylammonium 4-vinylphenyl trifluoroborate 3 (85 mg, 0.2 mmol) and 4-methylmorpholine N-oxide (100 mg, 1.0 mmol) in CH2Cl2 (3 mL) was applied a flow of ozone at −78 °C until the solution appeared light blue in color. The solution was degassed and then washed with H2O (3 ×5 mL) and brine to give the product 4 (63 mg, 0.15 mmol, 76%) following solvent removal.
Tetrabutylammonium 2-Formethyltrifluoroborate (11) via Procedure C
To a solution of tetrabutylammonium 4-butenyltrifluoroborate 10 (75 mg, 0.2 mmol) in CH2Cl2 (2 mL) was applied a flow of ozone at −78 °C until the solution turned blue in color. The solution was degassed followed by the addition of pyridine (158 mg, 0.4 mmol) with stirring which was continued 24 h. To this was added 1.0 M CuSO4 (3 mL) at 0 °C. The organic layer was extracted and washed with a pH 7 buffer, after which the solvent was removed to give aldehyde 11 as product (38 mg, 0.14 mmol, 71%).
Tetrabutylammonium 3-Oxobutyl Trifluoroborate (13) via Procedure D
To a solution of tetrabutylammonium trifluoroborate 12 (0.089 g, 0.235 mmol) in CH2Cl2 (10 mL) at −78 °C was applied a flow of ozone until the solution became blue in color. The reaction mixture was immediately degassed with N2 for 15 min after which Zn (0.060 g, 0.923 mmol) in AcOH (0.100 g, 1.67 mmol) was added dropwise with stirring. The solution was allowed to warm to rt, and stirring was continued for 2 h (the reaction progress was followed by 1H NMR). The reaction mixture was filtered to remove the Zn after which 1.0 M NaHCO3 was added over 15 min. The reaction mixture was extracted with CH2Cl2 (3 × 5 mL) and then the combined organic extracts were washed with H2O (3 × 5 mL), dried (MgSO4), and filtered. Solvent was removed to give trifluoroborate 13 as a colorless oil (0.085 g, 0.223 mmol, 95%).
(E)-Tetrabutylammonium 4-(3-Methoxy-3-oxoprop-1-enyl)phenyltrifluoroborate (14)
To a round bottomed flask containing tetrabutylammonium 4-formylphenyl trifluoroborate 4 (412 mg, 1.0 mmol) and methyl (triphenylphosphoranylidene) acetate (434 mg, 1.3 mmol) was added H2O (5 mL) with stirring. The temperature of the solution was raised to 90 °C for 2 h. Heating was ceased and the solution was allowed to reach room temperature. The reaction mixture was extracted with CH2Cl2 (3 × 10 mL) and the combined organic phases were then washed with NaOH (2M, 2 × 5 mL), H2O (2 × 10 mL) and dried (MgSO4). The resulting suspension was filtered and concentrated to give a white solid that was dissolved in CH2Cl2 (2 mL) and reprecipitated using Et2O. The white crystals were washed with Et2O to give product 14 (282 mg, 0.6 mmol, 61%), identical in every respect to material previously reported.9h
Supplementary Material
Experimental procedures, compound characterization data, and NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors wish to thank NIH (GM35249), Amgen, and the Merck Research Laboratories for their generous support of our program. We are grateful to Professor Mikael Bergdahl (San Diego State University) for making us aware of his aqueous protocol for Wittig reactions. Finally, Dr. Rakesh Kohli is acknowledged for obtaining HRMS data.
References
- 1.a) Darses S, Michaud G, Genet JP. Tetrahedron Lett. 1998;39:5045–5048. [Google Scholar]; b) Darses S, Genet JP, Brayer JL, Demoute JP. Tetrahedron Lett. 1997;38:4393–4396. [Google Scholar]; c) Xia M, Chen Z-C. Synth Commun. 1999;29:2457–2465. [Google Scholar]; d) Batey RA, Quach TD. Tetrahedron Lett. 2001;42:9099–9103. [Google Scholar]
- 2.a) Molander GA, Biolatto B. J Org Chem. 2003;68:4302–4314. doi: 10.1021/jo0342368. [DOI] [PubMed] [Google Scholar]; b) Molander GA, Biolatto B. Org Lett. 2002;4:1867–1870. doi: 10.1021/ol025845p. [DOI] [PubMed] [Google Scholar]; c) Molander GA, Rivero MR. Org Lett. 2002;4:107–109. doi: 10.1021/ol0169729. [DOI] [PubMed] [Google Scholar]; d) Molander GA, Ito T. Org Lett. 2001;3:393–396. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]
- 3.Batey RA, Thadani AN, Smil DV. Org Lett. 1999;1:1683–1686. [Google Scholar]
- 4.Pucheault M, Darses S, Genet JP. Eur J Org Chem. 2002:3552–3557. [Google Scholar]
- 5.Thadani AN, Batey RA. Org Lett. 2002;4:3827–3830. doi: 10.1021/ol026619i. [DOI] [PubMed] [Google Scholar]
- 6.Darses S, Michaud G, Genet J-P. Eur J Org Chem. 1999:1875–1883. [Google Scholar]
- 7.Vedejs E, Fields SC, Hayashi R, Hitchcock SR, Powell DR, Schrimpf MR. J Am Chem Soc. 1999;121:2460–2470. [Google Scholar]
- 8.Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020–3027. [Google Scholar]
- 9.a) Molander GA, Ribagorda M. J Am Chem Soc. 2003;125:11148–11149. doi: 10.1021/ja0351140. [DOI] [PubMed] [Google Scholar]; b) Molander GA, Ellis NM. J Org Chem. 2006;71:7491–7493. doi: 10.1021/jo061324u. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Molander GA, Figueroa R. Org Lett. 2006;8:75–78. doi: 10.1021/ol052549e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dambacher J, Zhao W, El-Batta A, Anness R, Jiang C, Bergdahl M. Tetrahedron Lett. 2005;46:4373–4477. [Google Scholar]; e) Molander GA, Ham J. Org Lett. 2006;8:2031–2034. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Molander GA, Ham J. Org Lett. 2006;8:2767–2770. doi: 10.1021/ol060826r. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Molander GA, Petrillo DE. J Am Chem Soc. 2006;128:9634–9635. doi: 10.1021/ja062974i. [DOI] [PubMed] [Google Scholar]; (h) Molander GA, Figueroa R. J Org Chem. 2006;71:6135–6140. doi: 10.1021/jo060863w. [DOI] [PubMed] [Google Scholar]
- 10.Criegee R. Angew Chem. 1975;87:765–771. [Google Scholar]
- 11.Bailey PS, editor. Ozonation in Organic Chemistry. Vol. 1. Academic Press; New York: 1978. pp. 381–394. [Google Scholar]
- 12.Belew JS, editor. Oxidation. Vol. 1. Dekker; New York: 1969. pp. 259–335. [Google Scholar]
- 13.Nicolaou KC, Li Y, Fylaktakidou KC, Mitchell HJ, Wei HX, Weyershausen B. Angew Chem Int Ed. 2001;40:3849–3854. [PubMed] [Google Scholar]
- 14.Schwartz C, Raible J, Mott K, Dussault PH. Org Lett. 2006;8:3199–3201. doi: 10.1021/ol061001k. [DOI] [PubMed] [Google Scholar]
- 15.a) Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49–56. [Google Scholar]; b) Molander GA, Ellis NM. Acc Chem Res, ASAP. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, compound characterization data, and NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.