

Abstract
Rat organic anion transporting protein 1a1 (oatp1a1), a hepatocyte basolateral plasma membrane protein, mediates transport of various amphipathic compounds. Our previous studies indicated that serine phosphorylation of a single tryptic peptide inhibits its transport activity without changing its cell surface content. The site of phosphorylation is unknown and was the subject of the present study. Following immunoaffinity chromatographic purification from rat liver, oatp1a1 was subjected to trypsin digestion and MALDI-TOF. Except for predicted N-glycosylated peptides, 97% of oatp1a1 tryptic peptides were observed. A single tryptic phosphopeptide was found in the C-terminus (aa 626-647), existing in unphosphorylated, singly, or doubly phosphorylated forms, and sensitive to alkaline phosphatase treatment. β-elimination reaction resulted in mass loss of 98 or 196 Da from this peptide, and subsequent Michael addition with cysteamine increased masses by the predicated 77 and 154 Da, indicating that oatp1a1 can be singly or doubly phosphorylated at serine or threonine residues in the C-terminal sequence SSATDHT (aa 634-640). Subsequent tandem MS/MS analysis revealed that phosphorylation at S634 accounted for all singly phosphorylated peptide, while phosphorylation at S634 and S635 accounted for all doubly phosphorylated peptide. These findings identify the site of oatp1a1 phosphorylation and demonstrate that it is an ordered process, in which phosphorylation at S634 precedes that at S635. The mechanism by which phosphorylation results in loss of transport activity in hepatocytes remains to be established. Whether phosphorylation near the C-terminus inhibits C-terminal oligomerization of oatp1a1, required for normal transport function, can be speculated upon, but is as yet unknown.
The rat organic anion transporting protein 1a1 (oatp1a1) is expressed on the basolateral plasma membrane of hepatocytes, on the apical plasma membrane of the S3 segment of the proximal tubule, and on the apical plasma membrane of the choroid plexus epithelial cell (1;2). This protein, formerly known as oatp1, has recently been renamed oatp1a1 in a proposal for standardization of nomenclature (3). It, as well as other members of the oatp family, have been shown to transport a wide variety of amphipathic organic compounds (3;4) and are thought to be involved in a broad range of physiological, pathophysiological, and pharmacological processes (3;4). Although function of the oatp’s has been studied extensively, their structure and regulation remain relatively unknown.
In earlier studies, we demonstrated that hepatocyte uptake of the oatp1a1 substrate sulfobromophthalein (5) is down-regulated rapidly, specifically, and reversibly by extracellular ATP (6), an event that coincides with serine phosphorylation of oatp1a1 at a single tryptic phosphopeptide(7). The location of this peptide within the oatp1a1 sequence is unknown and is the subject of the present study. Unlike the case for other cell surface proteins (8;9), phosphorylation did not alter the distribution of oatp1a1 on the cell surface, suggesting that the inhibitory effect might be due to conformational change of the transporter as has been described for aquaporin-4 (10) or possibly due to interference with oligomerization of oatp1a1, recently described as being required for optimal transport function (5). Extracellular ATP does not result in down regulation of transport function or phosphorylation of oatp1a1 in stably transfected HeLa cells (7), suggesting that the effectors that mediate this signal transduction pathway are lacking in these cells. However, modulation of transport function has also been shown in oatp1a1 expressing Xenopus oocytes in which PKC but not PKA activators suppressed transport activity, presumably as a result of oatp1a1 phosphorylation (11). All of these studies point out the significant role that phosphorylation of oatp1a1 can play in rapid regulation of its function. The importance of elucidating the effectors such as the kinases, phosphatases and other regulators involved in this process is clear (12). Identification of the phosphorylation site(s) is an important first step in achieving this goal.
Identification of phosphorylation sites on a hydrophobic, low abundance protein such as oatp1a1 (Figure 1) can be a challenging undertaking (13;14). As noted in a recent review, many methods are available to identify phosphorylation sites on proteins, but it is difficult to ascertain from the literature which of these methods are most useful in practice (14). In the present study, we adapted appropriate methods to purify oatp1a1 from rat liver by immunoaffinity chromatography (15), and to identify sites of posttranslational modification using matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). Aside from its physiologic importance as a transport protein of the hepatocyte plasma membrane, oatp1a1 is also a typical multitransdomain spanning integral plasma membrane protein. The methods used in this study may serve as a useful initial approach for studies of other integral membrane proteins.
Figure 1.
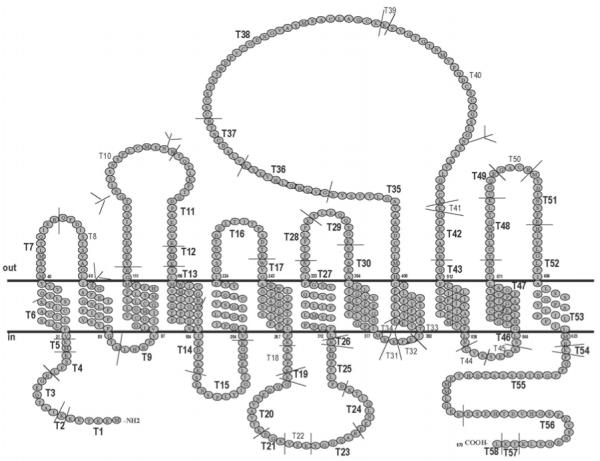
Predicted structure of oatp1a1, showing a 12 transmembrane domain model. Predicted tryptic peptides are indicated by T followed by an Arabic numeral and sites of cleavage are indicated by a short thin line C-terminal to the residue K or R. Tryptic peptides that are indicated in bold large font have been identified in the present study. The four potential N-linked glycosylation sites are designated by the branched structures at the appropriate N residues. The scheme was drawn according to the membrane protein secondary structure prediction program TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/).
MATERIALS AND METHODS
Tissues, enzymes and chemicals
Fresh frozen livers from adult male Sprague-Dawley rats were purchased from Pel-Freez Biologicals (Rogers, AZ) and stored at -80°C. Trypsin, endopeptidase Glu-C, α-cyano-4-hydroxycinnamic acid (α-CHCA), protease inhibitor cocktail and iodoacetamide were from Sigma-Aldrich (St. Louis, MO). Alkaline phosphatase was from Roche Diagnostics (Indianapolis, IN). Nonylphenyl-polyethylene glycol (NP-40), was from Fluka (Milwaukee, WI). Tris (2-carboxyethyl)-phosphine hydrochloride (TCEP) and dimethyl pimelimidate 2HCl (DMP) were from Pierce (Rockford, IL). Trifluoroacetic acid (TFA, protein sequencer grade) was from Applied Biosystems (Foster City, CA).
Sodium carbonate extraction of liver membranes
Livers were thawed in ice-cold 1 mM NaHCO3, homogenized, and extracted with 100 mM Na2CO3 as previously described (2). In brief, three rat livers, approximately 18 g total, were minced into small pieces and homogenized in 100 ml of 1 mM ice-cold NaHCO3 in a loose Dounce homogenizer. The homogenate was filtered through cheesecloth to remove debris, added to 300 ml of ice-cold 130 mM Na2CO3, and incubated with rotation for 15 min at 4°C. The mixture was centrifuged at 100,000 × g for 1 hr at 4°C, and the pellet in which oatp1a1 was highly enriched (16) was used for further analysis.
Antibody production and purification
Antibody was produced in rabbits against a KLH-linked synthetic peptide corresponding to the near C-terminal oatp1a1 sequence at aa 646-658, as described previously (1). The antibody was purified on a column containing Sulfo-Link gel (Pierce, Rockford, IL) to which the synthetic peptide was immobilized, following the manufacturer’s instructions. Bound antibody was eluted with 0.2 M glycine/HCl, pH 2.3 and the pH was neutralized immediately with Tris base. Following SDS-PAGE, fractions were analyzed by Coomassie blue stain to assess the presence and abundance of IgG.
Immunoaffinity purification of oatp1a1
Oatp1a1 antibody affinity gel was made according to Schneider et al (17). Briefly, 0.5 mg of affinity purified oatp1a1 antibody in 0.1 M borate buffer was rotated overnight at 4°C with 1 ml of protein A-agarose (Sigma-Aldrich), following which was added 20 mM (final concentration) dimethyl pimelimidate 2HCl in 100 mM triethylamine in borate buffer. The reaction was terminated after 30 min by addition of 40 mM ethanolamine. The gel was washed with 0.2 M glycine/HCl at pH 2.3 followed by PBS, and equilibrated with PBS containing 1% NP-40.
Na2CO3 extracted liver membrane pellets were resuspended to a protein concentration of 5-10 mg/ml in 1% NP-40 in PBS containing protease inhibitors. The pH was adjusted to 7.2 with HCl, and the mixture was gently stirred at room temperature for 1 hr following which it was centrifuged at 100,000 × g for 1 hr at 4°C. Solubilized liver membrane extract was loaded onto a mini-column containing the oatp1a1 immunoaffinity gel. After loading, the column was washed extensively with 1% NP-40 in PBS. Bound oatp1a1 was eluted with 0.2 M glycine/HCl, pH 2.3, into fractions of 400 μl and immediately neutralized with 1M Tris-base. Proteins in the eluent were analyzed by SDS-PAGE followed by Western blotting and silver staining of the gels. Silver stain was performed using the Silver Stain Plus kit (Bio-Rad, Hercules, CA). After elution, the column was regenerated by further washing with 1% NP-40 in PBS and stored in this buffer with 0.01% sodium azide. The column could be reutilized for purification multiple times.
Concentration of purified oatp1a1
Oatp1a1-containing fractions (approximately 3 ml) were pooled into one tube, ice-cold TCA was added to a final concentration of 10%, and the mixture was incubated for 30 min on ice. The resulting precipitate was centrifuged at 3,000 × g for 20 min at 4°C, and the pellet was washed three times with acetone to remove the residual TCA and NP-40. The pellet was dried at room temperature and incubated with SDS-PAGE sample buffer for 30 min at 37°C. Following SDS-PAGE, proteins were visualized by brief (about 10 min) staining with a low concentration of Coomassie blue (0.006% Coomassie blue in 50% methanol and 10% acetic acid).
Preparation of oatp1a1 tryptic peptides for MALDI-TOF MS analysis
Following SDS-PAGE, the Coomassie stained oatp1a1 band was excised, cut into 1×1 mm pieces, and washed with water. The gel pieces were destained three times by rotating at 37°C for 10 min with 50% acetonitrile in 50 mM ammonium bicarbonate (ABC). Subsequently, disulfide bonds were reduced by incubation in 20 mM TCEP at 50°C for 1 hr. Alkylation of reduced sulfhydryl groups was accomplished by incubation in an equal volume of 40 mM iodoacetamide at room temperature in the dark for 45 min. The gel pieces were then washed three times in water, another three times in 100% acetonitrile, and dried in a SpeedVac (Heto, Laurel, MD). The dried gel pieces were incubated overnight in 100 μl of 50 mM ABC containing 200 ng of proteomic grade trypsin (Sigma-Aldrich). Trifluoroacetic acid (TFA, 0.1% final) was added to stop the digestion. The supernatant was saved separately from the gel pieces that were then extracted with 50% acetonitrile, 0.1% TFA twice by bath sonicating for 10 min each time. For MALDI-TOF MS analysis, 1 μl of the supernatant or gel extract was mixed with 9 μl of 5% acetonitrile, 0.1% TFA and applied to a micro C18 Ziptip (0.6 μl bed volume, Millipore, Billerica, MA) following the manufacturer’s instructions. The bound peptides were step-eluted with 1.25 μl each of 25, 50, 75 and 95% acetonitrile containing 0.1% TFA and eluents were mixed with 1.25 μl of α-cyano-4-hydroxycinnamic acid matrix (α-CHCA, saturated solution in 50% acetonitrile, 0.1% TFA). The mixture was applied to a MALDI-TOF target plate and the matrix was allowed to crystallize at room temperature.
Methionine sulfoxidation of peptides
Because in the present study different levels of methionine oxidation complicated the peptide signals, in indicated studies we oxidized all methionine residues using H2O2 (18). One μl of the peptide solution was incubated with 10 μl of 30 mM H2O2, 0.1% acetic acid at 37°C for 30 min. After incubation, the sample was subjected to C18 Ziptip processing and then MALDI-TOF analysis.
Alkaline phosphatase treatment
In some experiments, samples were treated with alkaline phosphatase using the on-target protocol as described (19). In brief, 1.5 μl of a solution containing 0.075 U of alkaline phosphatase in 50 mM ABC was added onto a previously loaded spot on a MALDI target. The target was incubated at 37°C for 30 min following which 0.5 μl of 1% acetic acid was added to acidify the matrix for recrystallization and subsequent MALDI analysis.
β-elimination and Michael addition procedures
To confirm the presence of phosphorylation in candidate peptides, β-elimination and Michael addition were performed using a method described for cysteamine addition (20). In brief, 10 μl of the digest was dried and dissolved in 25 μl of a solution containing H2O, DMSO and ethanol at a ratio of 4:3:1. β-elimination was performed by adding 11.5 μl of saturated Ba(OH)2 and 0.5 μl of 5 M NaOH and incubating for 1 hr at 37°C. For Michael addition, 25 μl of 1 M cysteamine-HCl was subsequently added and incubated for up to 5 hr at 37°C. For identification of the phosphorylation sites, the β-eliminated peptides were modified with DTT(21). After extraction with activated thiol-sepharose 4B gel (Sigma) (21), the DTT modified peptide was subjected to MS/MS sequencing.
MALDI/TOF MS analysis
Spectra were recorded in positive or negative modes on a Voyager-DE STR MALDI-TOF mass spectrometer (Applied Biosystems, Foster City, CA), equipped with a 2.0 m flight tube and a 337 nm nitrogen laser. For MS/MS sequencing, the ABI 4700 TOF/TOF mass spectrometer (Applied Biosystems, Foster City, CA) with high energy collision-induced dissociation was used. Protein identification was accomplished through database searching (Swiss-Prot and NCBI) using MS-Fit and ProFound programs (Rodentia/Rattus, mass tolerance of 1Da, partially oxidized methionine, average and/or monoisotopic masses, and a maximum of two missed cleavages). Adrenocorticotropic hormone 18-39 (ACTH), bradykinin, and insulin were used for external mass calibration. Some frequently observed oatp1a1 tryptic peptides were used for internal mass calibration after they were confirmed as oatp1a1 peptides with ESI-MS (data not shown).
RESULTS
Immunoaffinity purification of oatp1a1
Sodium carbonate treatment is a simple method that can enrich integral membrane proteins by removing proteins loosely associated with the membrane (22;23) and is used here as the first step in oatp1a1 purification. Western blot analysis (Figure 2A) of oatp1a1 in the solubilized liver membrane extract (lane 1) and in the run-through (lane 2) indicates that the majority of oatp1a1 was retained on the antibody column. Silver staining of a replicate gel shows a high degree of purification of oatp1a1 following elution (Figure 2B). The amount of oatp1a1 in eluted fraction 3 (lane 5) is more than 50 ng as compared to a BSA standard (Figure 2B). In the silver stained gel, a minor band of approximately 50 kDa is seen in this fraction. Following trypsin digestion and MALDI-TOF MS analysis it was found to be rat IgG heavy chain (data not shown). It likely originated in the rat liver, and bound to unoccupied sites on the protein A column, eluting with the oatp1a1. To further concentrate and purify oatp1a1, the eluent was subjected to TCA precipitation and SDS-PAGE. Mild Coomassie blue staining of the gel reveals a broad band of oatp1a1 containing approximately 8 μg as compared to a BSA standard (Figure 2C). This final SDS-PAGE separated the approximately 80 kDa oatp1a1 from the contaminating IgG heavy chain.
Figure 2.
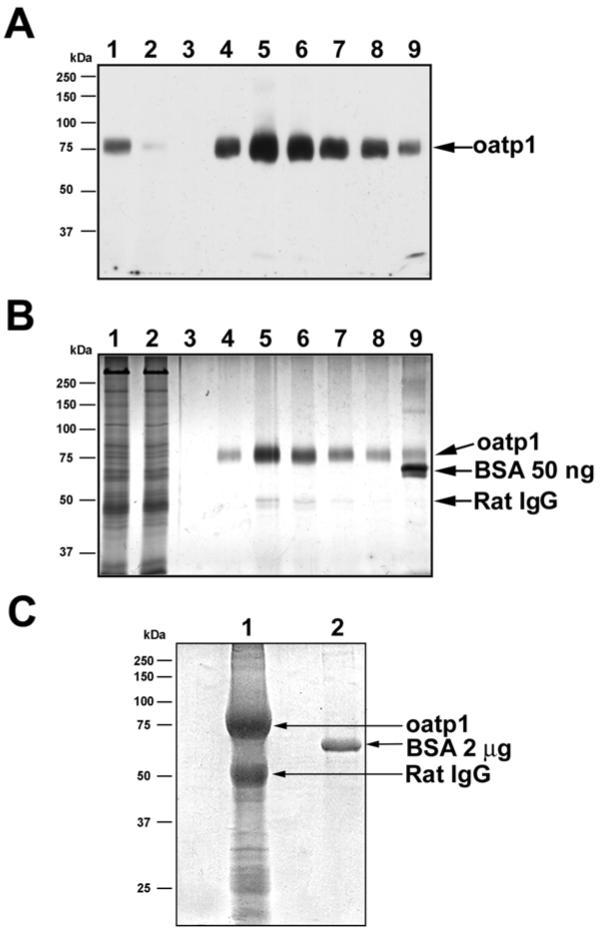
Immunoaffinity purification of oatp1a1 from rat liver. A. Western blot analysis of oatp1a1 in solubilized liver membrane (lane 1), run-through (lane 2), and different elution fractions (fractions 1 to 7 loaded in lanes 3 to 9, respectively). 20 μg of protein from the starting material was loaded onto a 10% gel. The same volume of sample from the column flow-through was loaded. One out of 400 μl of each eluent fraction was loaded. B. Silver stain analysis of proteins in solubilized liver membrane, flow-through, and different elution fractions of affinity chromatography. Samples were loaded in the same order as in panel A. 10 μg of protein from the starting material was loaded. The same volume of sample from the flow-through was loaded. For the eluted fractions, 2 μl of sample was loaded in each lane. 50 ng of BSA was also loaded in lane 9 for estimation of protein content. A minor amount of rat IgG heavy chain contamination was seen as indicated. C. Coomassie blue stain of a preparative 10% SDS-PAGE gel for in-gel oatp1a1 enzymatic digestion. Oatp1a1-containing fractions of the eluent were pooled and subjected to TCA precipitation and acetone wash. The pellet containing oatp1a1 was dissolved in SDS-PAGE buffer and loaded on the gel. 2 μg of BSA was also loaded as a control. The gel was weakly stained with 0.006% Coomassie blue in 50% methanol and 10% acetic acid for 10 min at room temperature by gentle shaking. The stained band was later excised, destained and subjected to in-gel trypsin digestion. Lane 1, TCA precipitated oatp1a1 eluent. Lane 2, 2 μg of BSA.
MALDI-TOF MS analysis of oatp1a1 tryptic digest
Following excision from the gel, the oatp1a1 band was subjected to proteolytic digestion with trypsin. Figure 3 shows a spectrum of the digest acquired in reflector mode. The majority of the peaks were identified as oatp1a1 tryptic peptides as predicted in Figure 1. Protein matching of masses from this spectrum with the ProFound program resulted in 37% sequence coverage of oatp1a1 with a probability of 1.0 and a Z score of 1.36. When detected in linear mode (Figure 4), peptides with larger masses were observed, including the peptides T55 and its incompletely digested form T54T55. By examining spectra acquired in different conditions, such as in reflector/linear, positive/negative modes, as well as in digests extracted with different concentrations of acetonitrile (data not shown), sequence coverage of 80% was obtained, as illustrated in Figure 1. In fact, when the predicted N-glycosylated peptides are excluded, nearly all the peptides with four or more amino acid residues were observed. The identified oatp1a1 peptides and their theoretical and observed masses are summarized in Table 1.
Figure 3.
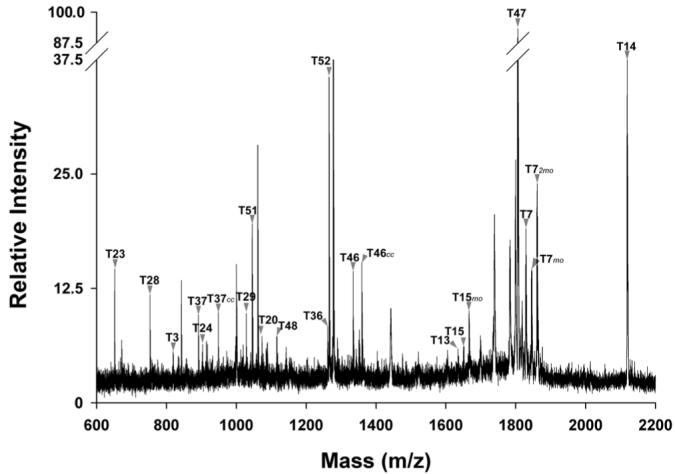
MALDI-TOF MS spectrum of oatp1a1 tryptic digest. The peptide digest was mixed with α-CHCA matrix saturated in 50% acetonitrile and 0.1% TFA. This representative spectrum was acquired in positive reflector mode. Tryptic peptides of oatp1a1 as illustrated in Figure 1 are indicated. The peaks were assigned either manually or automatically using peptide match software in ProFound. cc: carbamidomethylation; mo: methionine sulfoxidation.
Figure 4.
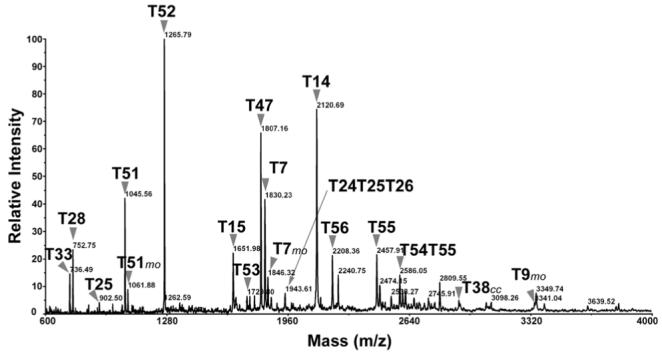
MALDI-TOF MS spectrum of oatp1a1 tryptic digest, acquired in linear mode. Tryptic peptides of oatp1a1 as illustrated in Figure 1 are indicated. The peaks were assigned either manually or automatically using peptide match software in ProFound. cc: carbamidomethylation; mo: methionine sulfoxidation.
Table 1. Summary of identified oatp1a1 tryptic peptides.
| Theoretical mass (MH+) |
Observed Mass (MH+) | Start | End | Sequence | ||
|---|---|---|---|---|---|---|
| Monoisotopic | Average | |||||
| T1 | 766.3293 | 766.8506 | 781.4 (mo) | 1 | 6 | (-) MEETEK (K) |
| T3 | 774.4110 | 774.8574 | 774.42, 902.51 (T2T3, ic) | 8 | 14 | (K) IATQEGR (L) |
| T4 | 494.2979 | 494.6155 | T4T5 (ic) | 15 | 18 | (R) LFSK (M) |
| T6 | 1411.7693 | 1412.8084 | 1525.76 (2cc), 1743.93 (T5T6, ic) | 21 | 33 | (K) VFLLSLTCACLTK (S) |
| T7 | 1828.8937 | 1830.1458 | 1828.89, 1844.89 (mo), 1860.89(2mo) | 34 | 49 | (K) SLSGVYMNSMLTQIER (Q) |
| T8 | 3617.8572 | 3620.1661 | nd (N -Glycosylated?) | 50 | 82 | (R) QFDISTSVAGLINGSFEIGNLFFIVFVSYFGTK (L) |
| T9 | 3266.7377 | 3269.1799 | 3382.53 (2cc) | 83 | 112 | (K) LHRPVVIGIGCVIMGLGCLLMSLPHFFMGR (Y) |
| T10 | 2756.2184 | 2758.0307 | nd (N-Glycosylated) | 113 | 136 | (R) YEYETTISPTGNLSSNSFLCMENR (T) |
| T11 | 1658.8423 | 1659.9111 | 1715.75 (cc) | 137 | 151 | (R) TQTLKPTQDPAECVK (E) |
| T12 | 407.1964 | 407.5125 | 2112.04 (T12T13, ic, 2mo, cc) | 152 | 154 | (K) EMK (S) |
| T13 | 1634.8585 | 1636.1101 | 1634.92, 1666.82 (mo), 1707.87, (mo, cc), 1723.87 (2mo, cc), 2112.04 (T12T13, ic, 2mo, cc) | 155 | 168 | (K) SLMWICVMVGNIIR (G) |
| T14 | 2119.1327 | 2120.4662 | 2119.12 | 169 | 188 | (R) GIGETPIVPLGISYIEDFAK (S) |
| T15 | 1650.8413 | 1651.9316 | 1650.82, 1666.82 (mo) | 189 | 203 | (K) SENSPLYIGILEMGK (V) |
| T16 | 3884.9632 | 3887.4260 | 3944.4 (cc) | 204 | 240 | (K) VAGPIFGLLLGSYCAQIYVDIGSVNTDDLTITPSDTR (W) |
| T17 | 3181.7105 | 3183.9000 | 3186.06 | 241 | 268 | (R) WVGAWWIGFLVCAGVNILTSIPFFFLPK (A) |
| T18 | 428.2873 | 428.5557 | nd | 269 | 272 | (K) ALPK (K) |
| T20 | 1073.5591 | 1074.1863 | 1073.48, 1201.63 (T19T20, ic) | 274 | 283 | (K) GQQENVAVTK (D) |
| T21 | 319.1618 | 319.3399 | T20T21 (ic) | 284 | 286 | (K) DGK (V) |
| T22 | 375.2244 | 375.4481 | nd | 287 | 289 | (K) VEK (Y) |
| T23 | 651.3214 | 651.7045 | 651.32 | 290 | 295 | (K) YGGQAR (E) |
| T24 | 903.4787 | 904.0143 | 903.49 | 296 | 303 | (R) EENLGITK (D) |
| T25 | 901.4494 | 902.1073 | 901.45, | 304 | 310 | (K) DFLTFMK (R) |
| T27 | 2787.4982 | 2789.4400 | 2846.24 (cc), 2862.81 (mo, cc) | 312 | 335 | (R) LFCNPIYMLFILTSVLQVNGFINK (F) |
| T28 | 752.4347 | 752.9378 | 752.45 | 336 | 341 | (K) FTFLPK (Y) |
| T29 | 1028.5053 | 1029.1449 | 1028.51 | 342 | 349 | (K) YLEQQYGK (S) |
| T30 | 3086.6714 | 3088.8095 | 3163.44 (mo, cc) | 350 | 378 | (K) STAEAIFLIGVYSLPPICLGYLIGGFIMK (K) |
| T33 | 460.3135 | 460.5981 | ic with T32 | 382 | 385 | (K) ITVK (K) |
| T35 | 4109.9929 | 4112.9149 | 4283.68 (3cc) | 387 | 423 | (K) AAYLAFCLSVFEYLLFLCHFMLTCDNAAVAGLTTSYK (G) |
| T36 | 1261.6653 | 1262.4200 | 1261.67 | 424 | 434 | (K) GVQHQLHVESK (V) |
| T37 | 891.4358 | 892.0282 | 891.44, 948,49 (cc) | 435 | 442 | (K) VLADCNTR (C) |
| T38 | 2856.1230 | 2858.2808 | 2915.28 (cc), 2932.68 (mo, cc) | 443 | 470 | (R) CSCSTNTWDPVCGDNGVAYMSACLAGCK (K) |
| T40 | 3079.4361 | 3081.6007 | nd (N-glycosylated) | 472 | 501 | (K) FVGTGTNMVFQDCSCIQSLGNSSAVLGLCK (K) |
| T42 | 746.3255 | 746.8253 | 803.34 (cc) | 503 | 509 | (K) GPECANR (L) |
| T43 | 3308.8777 | 3311.1229 | 3311.84 | 510 | 537 | (R) LQYFLILTIIISFIYSLTAIPGYMVFLR (C) |
| T44 | 349.1910 | 349.4756 | nd | 538 | 540 | (R) CVK (S) |
| T45 | 492.2306 | 492.5096 | nd | 541 | 544 | (K) SEEK (S) |
| T46 | 1302.6993 | 1303.5792 | 1302.70, 1359.73 (cc) | 545 | 556 | (K) SLGVGLHTFCIR (V) |
| T47 | 1805.9954 | 1807.1595 | 1805.98 | 557 | 573 | (R) VFAGIPAPVYFGALIDR (T) |
| T48 | 1058.5457 | 1059.2836 | 1058.52, 1115.52 (cc) | 574 | 582 | (R) TCLHWGTLK (C) |
| T49 | 463.2087 | 463.5394 | 520.15 (cc) | 583 | 586 | (K) CGQR (G) |
| T50 | 406.1873 | 406.4872 | nd | 587 | 590 | (R) GACR (M) |
| T51 | 1045.4777 | 1046.1976 | 1045.50, 1061.47 (mo) | 591 | 598 | (R) MYDINSFR (H) |
| T52 | 1265.7734 | 1266.5825 | 1265.79 | 599 | 609 | (R) HIYLGLPIALR (G) |
| T53 | 1727.9559 | 1729.1514 | 1745.05 (mo) | 610 | 624 | (R) GSSYLPAFFILILMR (K) |
| T55 | 2456.0750 | 2457.7142 | 2457.83, 2473.92 (mo), 2489.98 (2mo), 2538.00 (p), 2554.07 (mo, p), 2569.59 (2mo, p), 2618.09 (2p), 2633.88 (2p, mo) | 626 | 647 | (K) FQFPGDIDSSATDHTEMMLGEK (E) |
| T56 | 2206.9853 | 2208.2740 | 2208.21 | 648 | 667 | (K) ESEHTDVHGSPQVENDGELK (T) |
Immunoaffinity purified oatp1a1 was subjected to in-gel trypsin digestion. The spectra were acquired under various conditions, such as linear/reflector, negative/positive mode. Observed peptides with masses lower than 2,200 Da are shown in monoisotopic masses, those that are greater than 2,200 are shown in average masses. cc: cysteine carbamidomethylation, increases mass by 57 Da; ic: incomplete cleavage; mo: methionine sulfoxidation, increases mass by 16 Da; nd: not detected; p: phosphorylation, increases mass by 80 Da.
Identification of phosphopeptides
MALDI-TOF MS in reflector mode provides highly accurate monoisotopic masses. However, its sensitivity is relatively low, especially for detection of peptides with masses greater than 2000 Da. These peptides are more easily detected in linear mode as shown in Figure 4 where several signals with masses (MH+, average) over 2000 were apparent. A cluster of peaks was found in the range of 2400 to 2800, in which two major peaks could be assigned as T55 (2457.91 Da) and its incompletely cleaved form T54T55 (2586.05 Da) where the N-terminal lysine residue was not removed by trypsin. Even when the digestion time was extended to greater than 24 hours with addition of more trypsin, this missed cleavage still remained (data not shown). This cluster of peaks is shown in detail in Figure 5A. The two peaks at 2473.86 and 2489.95, 16 and 32 Da higher than T55, could be assigned as mono- and di-oxidized forms of T55. This is consistent with the fact that there are two methionine residues in T55. Similarly, the two peaks at 2602.06 and 2618.05 represent mono- and di-oxidized forms of T54T55. Each of these peaks is followed by clusters of peaks that are 80 Da higher, as indicated as pT55 and pT54T55, consistent with mono phosphorylated forms of these peptides (Figure 5A). Clusters of peaks that are 160 Da higher also exist, as indicated as ppT54 and ppT54T55, consistent with diphosphorylated forms of these peptides. This is in accordance with the fact that there are four potential phosphorylation sites on T55 at S635, S636, T638 and T641.
Figure 5.
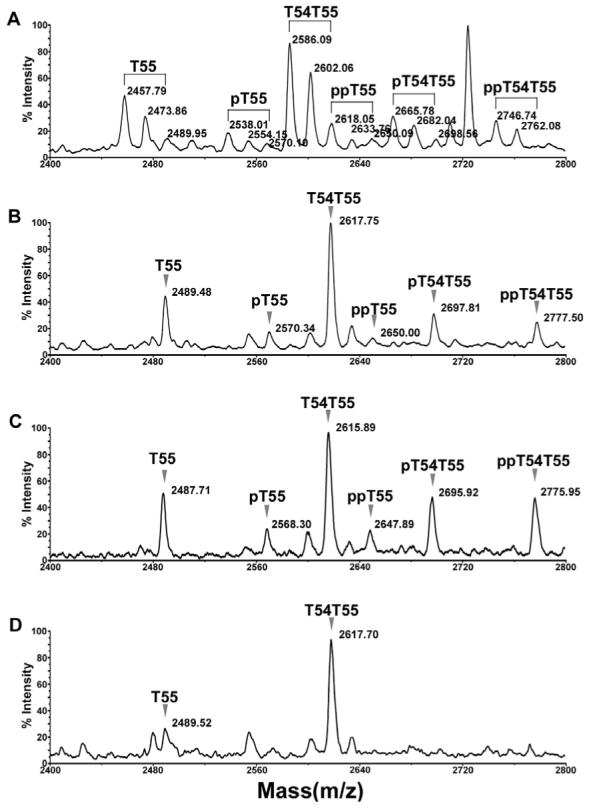
Confirmation of phosphorylation on oatp1a1 peptide T55. A. Spectrum of oatp1a1 tryptic digest showing the clusters of T55 peaks with different levels of methionine oxidation and phosphorylation. The spectrum was acquired in positive linear mode. B. Improvement of spectral resolution following methionine oxidation. The oatp1a1 digest was incubated with 30 mM H2O2 in 0.1% acetic acid at 37°C for 30 min to fully oxidize methionine residues. The sample was then processed with a C18 Ziptip and analyzed by MALDI-TOF in positive linear mode. C. Spectrum acquired in negative linear mode from the same MALDI target spot as in panel B, showing the increased relative intensity of phosphorylated peptides. D. On-target alkaline phosphatase treatment of the sample loaded on the same spot as in panel B. The sample was dissolved in 1.5 μl of a solution containing 0.075 U of alkaline phosphatase in 50 mM ammonium bicarbonate. The target was incubated at 37°C for 30 min. 0.5 μl of 1% acetic acid was added to acidify the matrix for recrystallization and subsequent MALDI analysis. pT55, pT54T55: the singly phosphorylated forms of T55 and T54T55, respectively. ppT55, ppT54T55: the doubly phosphorylated forms of T55 and T54T55, respectively.
Methionine oxidation and alkaline phosphatase treatment of phosphopeptide
As described above, the signals of candidate phosphopeptides are confounded by different levels of methionine oxidation. To obviate this problem, the digest was treated with H2O2 to fully oxidize methionine residues. The effect of H2O2 treatment is apparent; the resulting fully oxidized peaks are higher in intensity and more accurate in mass (Figure 5B). When detected in negative mode, the intensity of the candidate phosphopeptide peaks, relative to their unphosphorylated forms, is further improved (Figure 5C). To confirm the presence of phosphopeptides the digest was pretreated with alkaline phosphatase prior to MALDI analysis. As seen in Fig 5D, each of the candidate phosphopeptides essentially disappeared after the treatment.
Glu-C secondary digestion of the tryptic peptides
The tryptic digest of oatp1a1 was subjected to a secondary digestion with the Endoproteinase Glu-C that cleaves peptide bonds C-terminal to glutamate. The digest was analyzed with MALDI-TOF in positive/negative and linear/reflector modes. As shown in Figure 6 A (positive linear mode) and B (negative linear mode), both the Glu-C secondary digestion products of T55/T54T55 (peptides FQFPGDIDSSATDHTE and its incompletely digested form KFQFPGDIDSSATDHTE) and T55/T54T55 itself were observed due to incomplete digestion. The phosphorylated peptides are easily recognizable. Reflector mode allowed for more accurate detection of the masses of the Glu-C digestion products of T55 (Figure 6 E and F). Again, negative mode detection leads to higher signals of phosphorylated peptide (Figure 6 B and F) than those of positive mode (Figure 6 A and E). The effect of alkaline phosphatase is also apparent (Figure 6 C, D, G, and H).
Figure 6.
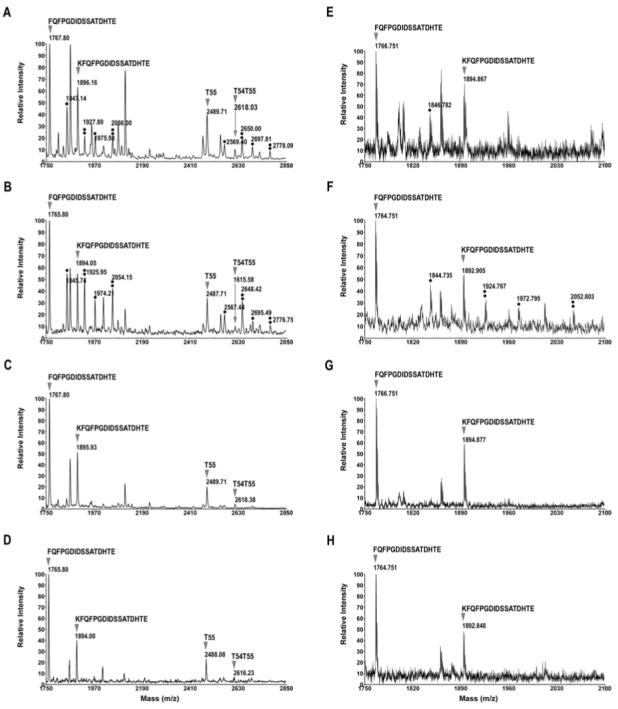
Secondary digestion of the oatp1a1 tryptic digest with endoproteinase Glu-C. The oatp1a1 tryptic digest was further digested with Glu-C in 100 mM ammonium bicarbonate at 37°C overnight. Under this condition, digestion did not go to completion. A. Spectrum acquired in positive linear mode. B. Spectrum acquired in negative linear mode. C. On-target alkaline phosphatase treatment of the same spot as in panel B. This spectrum was acquired in linear, positive mode. D. On-target alkaline phosphatase treatment. This spectrum was acquired in negative linear mode. E, F, G and H, same as A, B, C and D respectively, except that the spectra were acquired in reflector mode. Only mass ranges from 1750 to 2100 are shown for reflector mode, because the T55 and T54T55 peaks could not be detected clearly in this mode. A single asterisk represents singly phosphorylated peptides, a double asterisk represents doubly phosphorylated peptides. Sequences of oatp1a1 T55 or T54T55 Glu-C products are also shown on top of each corresponding peak.
β-elimination and Michael addition of the phosphopeptides
To further confirm their identity, the phosphopeptides were subjected to β-elimination and Michael addition. β-elimination occurs when the phosphoryl group of phosphoserine or phosphothreonine residues is exposed to base, leading to a 98 Da decrease in mass (24). The β-eliminated serine and threonine residues undergo Michael addition in the presence of nucleophiles such as alkanethiols and cysteamine (20;24). The combination of β-elimination and Michael addition provides signature signals in MALDI-TOF spectra that can be used to confirm the existence of phosphopeptides in a mixture of protease digested peptides (24). Figure 7 compares MALDI spectra of T55 peptides before and after β-elimination and Michael addition with cysteamine. As demonstrated in the figure, after β-elimination, the phosphorylated peaks of T54T55 disappear and two new peaks at 2581.75 and 2599.75 are observed (Panel B). These peaks, 196 and 98 Da lower than the phosphorylated ones, represent the β-eliminated doubly and singly phosphorylated peaks respectively. Michael addition with cysteamine adds 154 and 77 Da to the two new peaks, respectively, resulting in the detection of peaks at 2676.95 and 2736.13 (Panel C).
Figure 7.
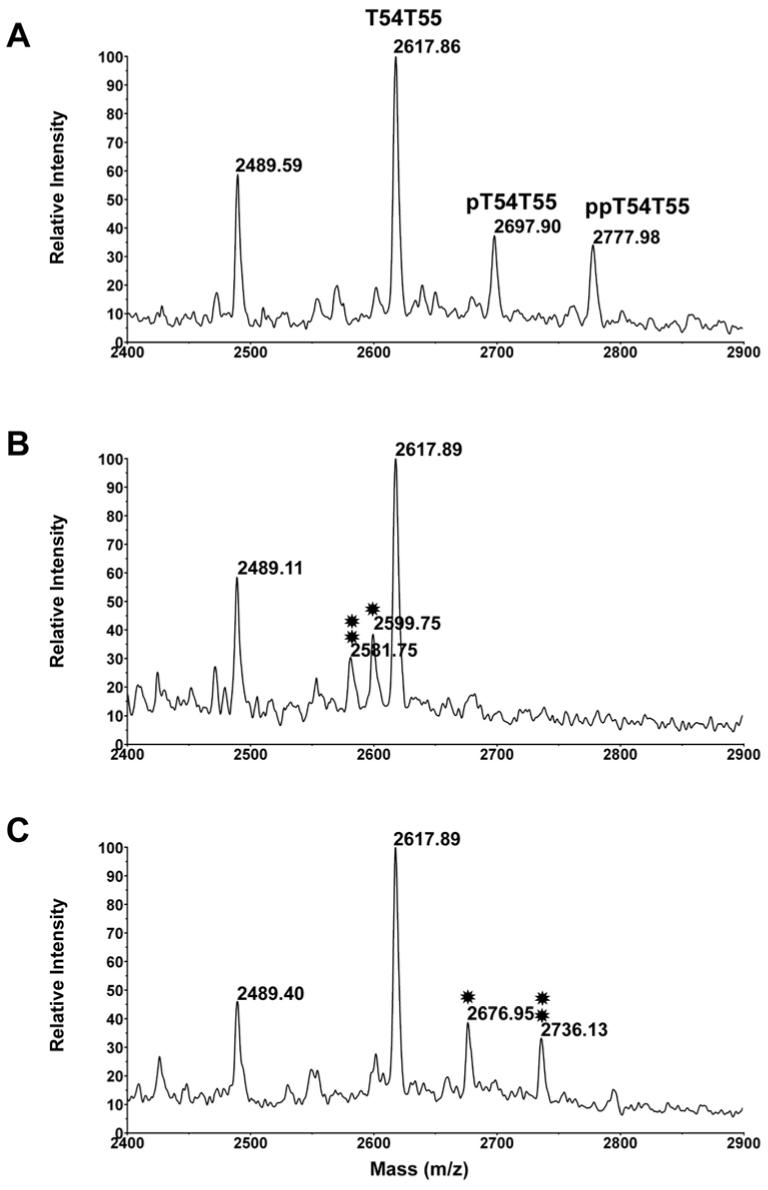
Effect of β-elimination and Michael addition with cysteamine on T55 and related peaks. A. The oatp1a1 tryptic digest was preoxidized with H2O2 prior to obtaining this spectrum. B. The preoxidized peptides were subjected to β-elimination with barium and sodium hydroxide. The single asterisk indicates the β-eliminated pT54T55, while the double asterisk indicates the β-eliminated ppT54T55. C. The β-eliminated peptides were subjected to Michael addition with cysteamine. The single asterisk indicates the cysteamine modified product of pT54T55, while the double asterisk indicates the cysteamine modified product of ppT54T55.
TOF/TOF MS/MS sequencing of DTT modified phosphopeptides
The phosphopeptides were subjected to β-elimination and Michael addition with DTT using the BEMAD method(21) that allows the specific extraction of the modified peptide with thiol gel. The results of DTT modification and thiol gel extraction are shown in Fig. 8. Modification is observed only for T55 and T54T55 (panels A and D), at masses 2625.99 (T55+DTT) and 2754.92 (T54T55+DTT), reflecting a 136 Da increase in mass due to a single DTT addition (panel D). After thiol gel extraction, these two modified peptides were enriched in the eluent (panels C and F). However, we didn’t find the di-DTT modified forms that would have been expected to result from the doubly phosphorylated peptide. Instead, two unexpected peaks at 2667.85 and 2735.90 Da were observed (panel D). These two peptides were not extracted with thiol gel and remained in the flow through (panels B and E). By their sizes, these peptides could well fit into a novel type of modification (named here as DTT bridge and illustrated as DTT) in which the two thiol groups of a DTT molecule bind to the two β-eliminated sites within the same peptide of T55 or T54T55, leading to mass increase of 118, as illustrated in panel G. The modified peptides were easily detected in reflector mode (data not shown), allowing MALDI TOF/TOF MS/MS analysis to locate the modification sites. Such direct analysis is usually not possible for phosphopeptides due to the loss of the phosphate group during CID.
Figure 8.
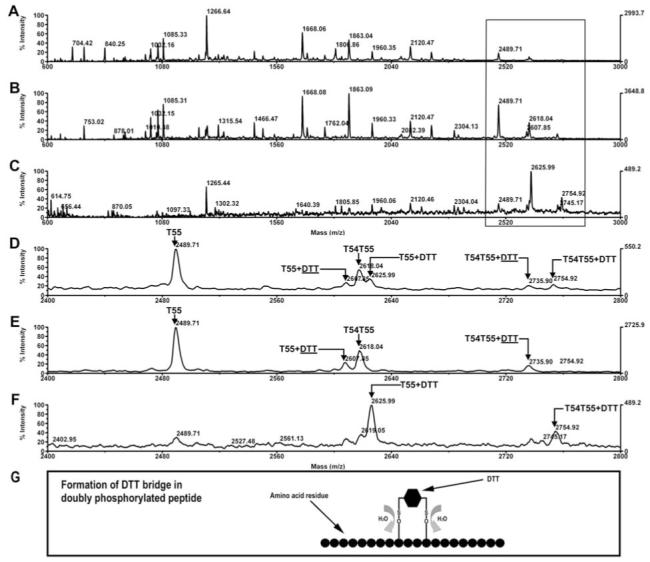
Modification of the phosphopeptides with DTT after ß-elimination. A. The full spectrum of oatp1a1 tryptic digest after ß-elimination and Michael addition with DTT. B. The full spectrum of the peptides in the flow through of the thiol gel. C. The full spectrum of the peptides released from the thiol gel with DTT. D, E, and F display detailed views in the mass range of 2400 to 2800 Da of panels A, B, and C respectively. G. Illustration of the formation of DTT bridge in doubly phosphorylated peptide. DTT: DTT bridge, formed by binding of both thiol groups in DTT to two ß-eliminated residues on the same peptide, resulting in mass increase by 118.2 Da
The thiol extracted T55+DTT (2625.12 Da) was then subjected to sequencing with MALDI TOF/TOF CID (Fig 9, Panel D). Other peptides, including the unphosphorylated T54T55 (2616.21 Da, panel B), the singly phosphorylated T54T55 (2696.02, panel C), the DTT modified T54T55 (2752.17 Da, panel E) and the DTT bridge modified T54T55 (2735.14, panel F) were also subjected to CID analysis. All spectra showed a clear y9 signal as well as y9-64 Da, resulting from loss of methane sulfenic acid (SO2, 64 Da) from sulfoxidized methionine residues during CID. This indicates that the threonine residue T640 is not a candidate for phosphorylation. The unphosphorylated T54T55 produced y ions from y9 to y16, except for y10 and y15 (panel B). The b ions from b14 to b17 are also apparent. Two a ions, a15 and a17, are also found. Although only weakly observed in reflector mode, the singly phosphorylated T54T55 (panel C) produced a clear y13 ion, indicating that S635, T637 and T640 are not phosphorylated in this peptide species. The only candidate site left is thus S634. In fact, the absence of y14 and y14+80 Da, coincided with the appearance of y14-18Da, clearly indicating phosphorylation at S634. Therefore, single phosphorylation of oatp1a1 is due exclusively to phosphorylation at S634. The CID spectra (panel D and E) of DTT modified single phosphopeptides T55+DTT and T54T55+DTT generated y9, y10, y12, y13, b14+DTT, y14+DTT, b15+DTT, and y16+DTT ions. This set of data indicates that the two threonine residues are not used for phosphorylation. Observation of y13 indicates that S635 is not phosphorylated, while the observation of b14+DTT, y14+DTT, b15+DTT and y16+DTT strongly suggest phosphorylation at S634. The CID spectrum (panel F) of T54T54+DTT generated y9 and y11, again indicating that the two threonine residues are not phosphorylated in doubly phosphorylated T55. The observation of y14+DTT, b15+DTT and y16+DTT indicates that S634 and S635 are the two sites of DTT bridge formation. This implies that S634 and S635 are the residues that are phosphorylated in the doubly phosphorylated T55 species.
Figure 9.
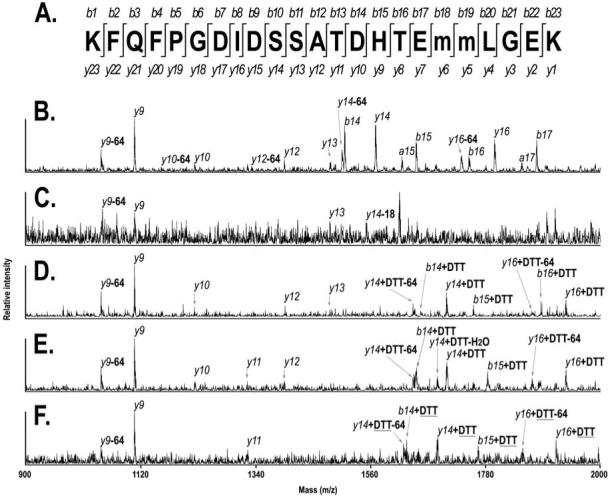
CID spectra of T55 and its derived peptides. A. Illustration of the sequences of b ions and y ions generated from T54T55 during MALDI-TOF/TOF CID. The lower case “m” indicates sulfoxidized methionine residue. B. CID spectrum of unmodified T54T55. Parent ion mass is 2616.02 Da. C. CID spectrum of singly phosphorylated T54T55. Parent ion mass is 2696.02. D. CID spectrum of DTT modified T55 (not T54T55). Parent ion mass is 2625.12. E. CID spectrum of DTT modified T54T55. Parent ion mass is 2752.17 Da. F. CID spectrum of DTT modified T54T55. Parent ion mass is 2735.14 Da. DTT: dithiothreitol. DTT: DTT bridge. -64: less 64 Da in mass, due to loss of methane sulfoneic acid (SO2). -18: less 18 Da in mass.
DISCUSSION
Removal of xenobiotics and endogenous organic anionic compounds from the circulation represents a major function of the hepatocyte (25). Initial studies that were performed by expressing rat liver mRNA in a Xenopus laevis oocyte expression system identified a candidate transporter, originally named organic anion transport protein 1 (oatp1), and now named oatp1a1 (5;26). Subsequently, over 20 additional members of the oatp family have been described, with widespread tissue distributions and overlapping and broad substrate specificities (3). The amino acid sequences of all of these proteins have high levels of homology, and for the most part they have similar predicted membrane topologies and biochemical characteristics (3;5). Expression of these proteins can be regulated at the transcriptional level, and a role for specific transcription factors and nuclear hormone receptors has been demonstrated (27-29). However, this is a relatively slow process and cannot explain the rapid modulation of transport function that has been described following exposure of rat hepatocytes to extracellular ATP or PKC activators, conditions that result in phosphorylation of oatp1a1 (6;7;11). Although previous studies showed that a single tryptic peptide was phosphorylated on a serine residue (7), there was no information as to where this peptide was located in the oatp1a1 sequence. This information is essential for the design of future studies to examine the mechanism by which phosphorylation regulates transport function. Other studies indicate that oatp1a4 (formerly known as oatp2) is regulated similarly by phosphorylation (11) and it is likely that methods and findings for oatp1a1 will serve as a prototype for elucidating regulation of other oatp family members as well.
Immunoaffinity purification of oatp1a1 was essential for the analytical procedures that were employed in the present study. This single step procedure resulted in a high degree of purification (Figure 2), with approximately 8 μg (100 pmole) of oatp1a1 protein obtained from three rat livers. This is an amount sufficient for multiple mass spectrometric analyses, as these generally require only subpicomole to picomole amounts of sample per assay (30). Identity of the purified oatp1a1 was confirmed by MALDI-TOF MS analysis of tryptic digests. By examining spectra acquired under different conditions, such as from linear/reflector, positive/negative settings of the MALDI-TOF instrument, from the original tryptic digest, the secondary digest with Glu-C or ASP-N (data not shown), and the gel extract, peptides covering 84% of the oatp1a1 sequence could be identified. When the predicted N-glycosylated peptides (T8, T10 and T40, occupying 13% of the whole sequence) and some peptides with masses lower than 600 Da (occupying 3% of the whole sequence) are excluded, all the remaining peptides were identified. Of the predicted N-glycosylated peptides, T10 and T40 were confirmed to be glycosylated by mutagenesis and PNGase F deglycosylation studies (unpublished data). N-glycosylation adds approximately 5 kDa to each peptide, making them unsuitable for efficient detection by MALDI-TOF using the present experimental conditions. Mutation at Asn62 in peptide T8 did not change the apparent molecular weight of oatp1a1 on Western blot (unpublished data), suggesting that it is not glycosylated. However, in the present study we could not detect T8. One possibility is that it may be glycosylated with minor glycans. A number of signals in the spectra could not be identified, even when common modifications such as oxidation, cysteine alkylation, phosphorylation and O-glycosylation were considered. This might be due to unusual modification or peptide truncations (31;32).
Methionine oxidation in the tryptic peptides of oatp1a1 made analysis difficult by reducing the signals of otherwise homogeneous peptides. For example, it divides the T55 signal into three peaks, each of relatively low intensity (Figure 5A). In addition, the doubly phosphorylated T55 (ppT55) overlaps with the di-oxidized T54T55, making it difficult to identify the individual species. This problem was overcome by utilizing a relatively simple sulfoxidation procedure with H2O2 prior to MALDI analysis. Sulfoxidation of methionine residues was confirmed from loss of methane sulfenic acid (-64 Da) in the CID spectra. Although methionine oxidation may be physiological (33-35), it occurs commonly in vitro during sample preparation. The extent of oatp1a1 oxidation in vivo remains to be determined.
T55 was the only phosphopeptide that was detected in these studies. The mass differences of 80 and 160 Da that were found in the cluster of T55-related peaks strongly indicate existence of single and double phosphorylation. However, this mass difference could also represent sulfation, a protein modification that also adds 80 Da to the mass of a peptide (30;36). The finding that pretreatment with alkaline phosphatase results in disappearance of these peaks effectively obviates this possibility. A higher ratio of the intensities of phosphorylated to unphosphorylated peaks in negative as compared to positive mode, as we saw in our spectra, is characteristic of phosphopeptides (37;38). Identical results were found in both the tryptic digest and the secondary digest with Glu-C. In the latter case we were able to detect the phosphopeptide in reflector mode that allows accurate detection of the monoisotopic masses of shorter peptides. However, cleavage with Glu-C could not be carried to completion, possibly due to the existence of phosphorylation in this peptide. Secondary digestion with the endoproteinase ASP-N also gave similar results (data not shown).
Immobilized metal affinity chromatography (IMAC) has been suggested as a good method to enrich phosphopeptides from tryptic digests (14). However, using the Millipore ZipTip MC system, the yield of oatp1a1-derived phosphopeptide was poor (data not shown), and we used β-elimination and Michael addition reactions as an alternative approach. These reactions are characteristic of serine and threonine phosphorylation (20). Using cysteamine for Michael addition, all expected modification products including the β-eliminated peaks were observed (Figure 7). Michael addition with other nucleophiles such as butanethiol and dithiothreitol also resulted in all expected products (data not shown). Therefore, these results not only confirm T55 phosphorylation, but also indicate that the phosphorylation occurs at serine or threonine residues. These residues are located in the short sequence SSATDHT. Unfortunately, no proteolytic enzyme is available for further efficient cleavage within that sequence that can lead to the ultimate identification of the phosphorylation sites. Direct fragmentation of the peptide under varied conditions during MS/MS analysis caused loss of the phosphate groups in most cases, precluding assignment of the phosphorylated residues (data not shown).
MALDI TOF/TOF CID was used to identify phosphorylated amino acid residues in T55 and T54T55 after modification with DTT. As presented in the results section, the monophosphorylated peptide derives from phosphorylation at S634 only, while S634 and S635 are phosphorylated in the diphosphorylated peptide. These findings not only indicate the sites of phosphorylation, but also demonstrate that oatp1a1 phosphorylation occurs in an ordered process, i.e., phosphorylation at S634 precedes that at S635. The BEMAD method (21) is efficient for extracting and concentrating modified peptides for subsequent CID analysis, as was done in the present study. However, the formation of DTT bridges prevented the extraction of these peptides, and could result in failure to identify doubly phosphorylated peptides.
The present study identified T55 (Figure 1) as the only phosphopeptide in the oatp1a1 sequence. Under the same conditions of MALDI detection, e.g., linear positive mode, the ratios of areas of phosphorylated and unphosphorylated peaks of T55 in the tryptic digest from different batches of purification remain relatively stable. We could therefore estimate that about 20% of the T55 peptide is singly phosphorylated and another 20% is doubly phosphorylated. Since the protein is purified from the livers of normal rats, this amount should represent the minimum level of oatp1a1 phosphorylation in vivo. As noted above, multispanning membrane proteins represent challenging subjects for mass spectrometric analysis (39). Some abundant membrane proteins such as aquaporin and rhodopsin can be isolated relatively easily in amounts adequate for subsequent mass spectrometric analysis (32;40-42). However, we are not aware of any other low abundance membrane protein purified from native tissue that has been subjected to similar analysis, and we propose that the methods utilized in this study may help to provide a new strategy for efficient structural analysis of such proteins.
The mechanism by which phosphorylation of oatp1a1 results in loss of transport activity in hepatocytes remains to be established. The C-terminus of oatp1a1 has been shown recently to bind to two of the four PDZ domains on PDZK1 (5). This interaction is required for expression of the transporter on the cell surface and also suggests the potential presence of other proteins in an oligomeric complex (5). Phosphorylation of oatp1a1 occurs 32 amino acids upstream of its PDZ consensus binding site at the C-terminus. It is appealing to consider the hypothesis that phosphorylation of the transporter at this site modulates interaction with PDZK1 or other members of the PDZK1 complex. Such a situation exists for interaction of the cystic fibrosis transmembrane conductance regulator (CFTR) with ezrin/radixin/moesin-binding protein 50 (EBP50), a PDZ-domain binding protein (43;44). CFTR binds to EBP50 through its C-terminus (44), forming a functionally active complex with the β2 adrenergic receptor which also binds to EBP50 (43). Phosphorylation of CFTR at its R domain, located far upstream, inhibits its activity (45) by preventing its binding to EBP50 (43). Whether a similar mechanism exists for oatp1a1 complex formation is not known. Future studies regarding the tertiary structure of oatp1a1 as well as elucidation of its binding partners will be required to answer this important question.
ABBREVIATIONS
- oatp1a1
rat organic anion transporting protein 1a1
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- MALDI-TOF MS
matrix assisted laser desorption/ionization time-of-flight mass spectrometry
- DTT
Dithiothreitol
- α-CHCA
α-cyano-4-hydroxycinnamic acid
- TCEP
tris (2-carboxyethyl)-phosphine hydrochloride
- TFA
trifluoroacetic acid
- NP-40
nonylphenyl-polyethylene glycol
- PBS
phosphate buffered saline
- ABC
ammonium bicarbonate
- DMSO
dimethyl sulfoxide
- Glu-C
endopeptidase specific C-terminal to glutamate residue
- CID
collision induced dissociation
Footnotes
This work was supported by NIH grants DK23026 and CA101150.
References
- 1.Bergwerk AJ, Shi X, Ford AC, Kanai N, Jacquemin E, Burk RD, Bai S, Novikoff PM, Stieger B, Meier PJ, Schuster VL, Wolkoff AW. Immunologic distribution of an organic anion transport protein in rat liver and kidney. Am. J. Physiol. 1996;271:G231–G238. doi: 10.1152/ajpgi.1996.271.2.G231. [DOI] [PubMed] [Google Scholar]
- 2.Angeletti RH, Novikoff PM, Juvvadi S, Fritschy J-M, Meier PJ, Wolkoff AW. The choroid plexus epithelium is the site of the organic anion transport protein in the brain. Proc. Natl. Acad. Sci. USA. 1997;94:283–286. doi: 10.1073/pnas.94.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 4.Pang KS, Wang PJ, Chung AY, Wolkoff AW. The modified dipeptide, enalapril, an angiotensin-converting enzyme inhibitor, is transported by the rat liver organic anion transport protein. Hepatology. 1998;28:1341–1346. doi: 10.1002/hep.510280524. [DOI] [PubMed] [Google Scholar]
- 5.Wang P, Wang JJ, Xiao Y, Murray JW, Novikoff PM, Angeletti RH, Orr GA, Lan D, Silver DL, Wolkoff AW. Interaction with PDZK1 is required for expression of organic anion transporting protein 1A1 on the hepatocyte surface. J. Biol. Chem. 2005;280:30143–30149. doi: 10.1074/jbc.M503969200. [DOI] [PubMed] [Google Scholar]
- 6.Campbell CG, Spray DC, Wolkoff AW. Extracellular ATP4- modulates organic anion transport by rat hepatocytes. J. Biol. Chem. 1993;268:15399–15404. [PubMed] [Google Scholar]
- 7.Glavy JS, Wu SM, Wang PJ, Orr GA, Wolkoff AW. Down-regulation by extracellular ATP of rat hepatocyte organic anion transport is mediated by serine phosphorylation of oatp1. J. Biol. Chem. 2000;275:1479–1484. doi: 10.1074/jbc.275.2.1479. [DOI] [PubMed] [Google Scholar]
- 8.Anwer MS, Gillin H, Mukhopadhyay S, Balasubramaniyan N, Suchy FJ, Ananthanarayanan M. Dephosphorylation of Ser-226 facilitates plasma membrane retention of Ntcp. J Biol Chem. 2005;280:33687–33692. doi: 10.1074/jbc.M502151200. [DOI] [PubMed] [Google Scholar]
- 9.Zmuda-Trzebiatowska E, Oknianska A, Manganiello V, Degerman E. Role of PDE3B in insulin-induced glucose uptake, GLUT-4 translocation and lipogenesis in primary rat adipocytes. Cell Signal. 2006;18:382–390. doi: 10.1016/j.cellsig.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Zelenina M, Zelenin S, Bondar AA, Brismar H, Aperia A. Water permeability of aquaporin-4 is decreased by protein kinase C and dopamine. Am. J. Physiol Renal Physiol. 2002;283:F309–F318. doi: 10.1152/ajprenal.00260.2001. [DOI] [PubMed] [Google Scholar]
- 11.Guo GL, Klaassen CD. Protein kinase C suppresses rat organic anion transporting polypeptide 1- and 2-mediated uptake. J. Pharmacol. Exp. Ther. 2001;299:551–557. [PubMed] [Google Scholar]
- 12.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 13.Arnott D, Gawinowicz MA, Grant RA, Neubert TA, Packman LC, Speicher KD, Stone K, Turck CW. ABRF-PRG03: phosphorylation site determination. J. Biomol. Tech. 2003;14:205–215. [PMC free article] [PubMed] [Google Scholar]
- 14.Loyet KM, Stults JT, Arnott D. Mass spectrometric contributions to the practice of phosphorylation site mapping through 2003: a literature review. Mol Cell Proteomics. 2005;4:235–245. doi: 10.1074/mcp.R400011-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Burgess RR, Thompson NE. Advances in gentle immunoaffinity chromatography. Curr. Opin. Biotechnol. 2002;13:304–308. doi: 10.1016/s0958-1669(02)00340-3. [DOI] [PubMed] [Google Scholar]
- 16.Angeletti RH, Bergwerk AJ, Novikoff PM, Wolkoff AW. Dichotomous development of the organic anion transport protein in liver and choroid plexus. Am. J. Physiol. 1998;275:C882–C887. doi: 10.1152/ajpcell.1998.275.3.C882. [DOI] [PubMed] [Google Scholar]
- 17.Schneider C, Newman RA, Sutherland DR, Asser U, Greaves MF. A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem. 1982;257:10766–10769. [PubMed] [Google Scholar]
- 18.Neumann NP. Methods in Enzymology. Vol. 25. Academic Press; 1972. [31] Oxidation with hydrogen peroxide; pp. 393–400. [DOI] [PubMed] [Google Scholar]
- 19.Larsen MR. Mass spectrometric characterization of posttranslationally modified proteins--phosphorylation. Methods Mol Biol. 2004;251:245–262. doi: 10.1385/1-59259-742-4:245. [DOI] [PubMed] [Google Scholar]
- 20.Knight ZA, Schilling B, Row RH, Kenski DM, Gibson BW, Shokat KM. Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat. Biotechnol. 2003;21:1047–1054. doi: 10.1038/nbt863. [DOI] [PubMed] [Google Scholar]
- 21.Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics. 2002;1:791–804. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi X, Bai S, Ford AC, Burk RD, Jacquemin E, Hagenbuch B, Meier PJ, Wolkoff AW. Stable inducible expression of a functional rat liver organic anion transport protein in HeLa Cells. J. Biol. Chem. 1995;270:25591–25595. doi: 10.1074/jbc.270.43.25591. [DOI] [PubMed] [Google Scholar]
- 24.Molloy MP, Andrews PC. Phosphopeptide derivatization signatures to identify serine and threonine phosphorylated peptides by mass spectrometry. Anal. Chem. 2001;73:5387–5394. doi: 10.1021/ac0104227. [DOI] [PubMed] [Google Scholar]
- 25.Hata S, Wang P, Eftychiou N, Ananthanarayanan M, Batta A, Salen G, Pang KS, Wolkoff AW. Substrate specificities of rat oatp1 and ntcp: implications for hepatic organic anion uptake. Am J Physiol Gastrointest Liver Physiol. 2003;285:G829–G839. doi: 10.1152/ajpgi.00352.2002. [DOI] [PubMed] [Google Scholar]
- 26.Jacquemin E, Hagenbuch B, Stieger B, Wolkoff AW, Meier PJ. Expression cloning of a rat liver Na+-independent organic anion transporter. Proc. Natl. Acad. Sci. USA. 1994;91:133–137. doi: 10.1073/pnas.91.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 28.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322–342. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Larsen MR, Sorensen GL, Fey SJ, Larsen PM, Roepstorff P. Phospho-proteomics: evaluation of the use of enzymatic de-phosphorylation and differential mass spectrometric peptide mass mapping for site specific phosphorylation assignment in proteins separated by gel electrophoresis. Proteomics. 2001;1:223–238. doi: 10.1002/1615-9861(200102)1:2<223::AID-PROT223>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 31.Schey KL, Little M, Fowler JG, Crouch RK. Characterization of human lens major intrinsic protein structure. Invest Ophthalmol. Vis. Sci. 2000;41:175–182. [PubMed] [Google Scholar]
- 32.Ball LE, Garland DL, Crouch RK, Schey KL. Post-translational modifications of aquaporin 0 (AQP0) in the normal human lens: spatial and temporal occurrence. Biochemistry. 2004;43:9856–9865. doi: 10.1021/bi0496034. [DOI] [PubMed] [Google Scholar]
- 33.Bigelow DJ, Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta. 2005;1703:121–134. doi: 10.1016/j.bbapap.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Davies MJ. The oxidative environment and protein damage. Biochim. Biophys. Acta. 2005;1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Merker K, Grune T. Proteolysis of oxidised proteins and cellular senescence. Exp. Gerontol. 2000;35:779–786. doi: 10.1016/s0531-5565(00)00140-6. [DOI] [PubMed] [Google Scholar]
- 36.Seibert C, Cadene M, Sanfiz A, Chait BT, Sakmar TP. Tyrosine sulfation of CCR5 N-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11031–11036. doi: 10.1073/pnas.172380899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma Y, Lu Y, Zeng H, Ron D, Mo W, Neubert TA. Characterization of phosphopeptides from protein digests using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and nanoelectrospray quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2001;15:1693–1700. doi: 10.1002/rcm.426. [DOI] [PubMed] [Google Scholar]
- 38.Xu CF, Lu Y, Ma J, Mohammadi M, Neubert TA. Identification of Phosphopeptides by MALDI Q-TOF MS in Positive and Negative Ion Modes after Methyl Esterification. Mol Cell Proteomics. 2005;4:809–818. doi: 10.1074/mcp.T400019-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.van Montfort BA, Canas B, Duurkens R, Godovac-Zimmermann J, Robillard GT. Improved in-gel approaches to generate peptide maps of integral membrane proteins with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Mass Spectrom. 2002;37:322–330. doi: 10.1002/jms.288. [DOI] [PubMed] [Google Scholar]
- 40.Han J, Schey KL. Proteolysis and mass spectrometric analysis of an integral membrane: aquaporin 0. J. Proteome. Res. 2004;3:807–812. doi: 10.1021/pr049945w. [DOI] [PubMed] [Google Scholar]
- 41.Ervin LA, Ball LE, Crouch RK, Schey KL. Phosphorylation and glycosylation of bovine lens MP20. Invest Ophthalmol. Vis. Sci. 2005;46:627–635. doi: 10.1167/iovs.04-0894. [DOI] [PubMed] [Google Scholar]
- 42.Kraft P, Mills J, Dratz E. Mass spectrometric analysis of cyanogen bromide fragments of integral membrane proteins at the picomole level: application to rhodopsin. Anal. Biochem. 2001;292:76–86. doi: 10.1006/abio.2001.5072. [DOI] [PubMed] [Google Scholar]
- 43.Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci U S A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brone B, Eggermont J. PDZ proteins retain and regulate membrane transporters in polarized epithelial cell membranes. Am J Physiol Cell Physiol. 2005;288:C20–C29. doi: 10.1152/ajpcell.00368.2004. [DOI] [PubMed] [Google Scholar]
- 45.Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]