

Abstract
A total synthesis of (+)-bullatacin has been accomplished via a diastereoselective [3+2] annulation reaction of the highly enantiomerically enriched allylsilane 3 and racemic aldehyde 4, which provides the key bis-tetrahydrofuran fragment 15 with ≥ 20 : 1 ds.
(+)-Bullatacin (1) is one of more than 350 Annonaceous acetogenins isolated from the tropical plant family Annonaceae (Figure 1). Many members of this structurally diverse family of natural products exhibit impressive antitumor activity in human tumor cell lines.1 The acetogenins contain a long aliphatic backbone bearing a terminal butenolide unit and one or more tetrahydrofuran rings and hydroxyl groups at internal positions of the aliphatic chain. These compounds are intriguing synthetic targets owing to the variation of stereochemistry around the tetrahydrofuran rings and at the sites bearing additional hydroxyl groups.2, 3
Figure 1.
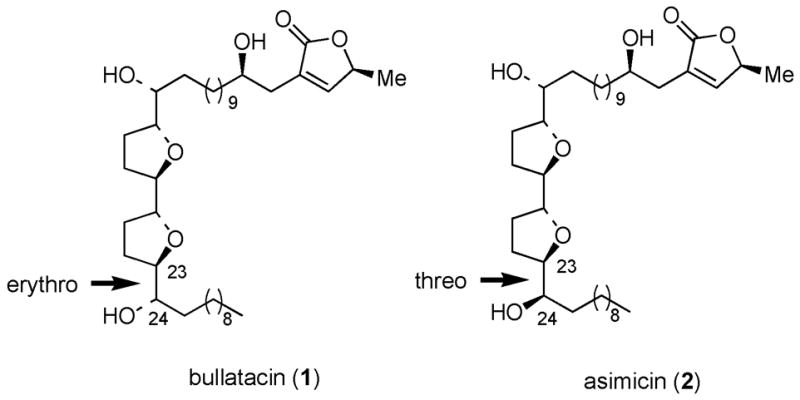
Structures of Bullatacin and Asimicin.
The [3+2]-annulation reaction of aldehydes and chiral allylsilanes is an important method for the stereocontrolled synthesis of substituted tetrahydrofurans.4, 5 β-Silyloxy-substituted allylsilanes undergo [3+2] annulation reactions with aldehydes and certain electrophilic ketones to give either 2, 5-trans or 2, 5-cis substituted tetrahydrofurans with excellent selectivity, depending on the use of chelating or non-chelating Lewis acids, respectively.6 We have recently utilized this methodology in a highly stereoselective total synthesis of asimicin (2).7
As part of ongoing studies focusing on the development of a stereochemically general synthesis of members of the acetogenin family, we have developed and report herein a highly stereoselective synthesis of bullatacin (1),8 which differs from asimicin (2) at a single stereocenter (C-24). We envisaged that the bis-tetrahydrofuran core unit of bullatacin could be synthesized from sequential chelate-controlled [3+2] annulation reactions of allylsilanes 3 and 6 (Figure 2). The proposed [3+2] annulation of 3 and 4 is expected to be a stereochemically matched double asymmetric reaction under chelate-controlled conditions, by analogy with the corresponding reaction in our asimicin synthesis that proceeded with ≥20 : 1 ds.7
Figure 2.
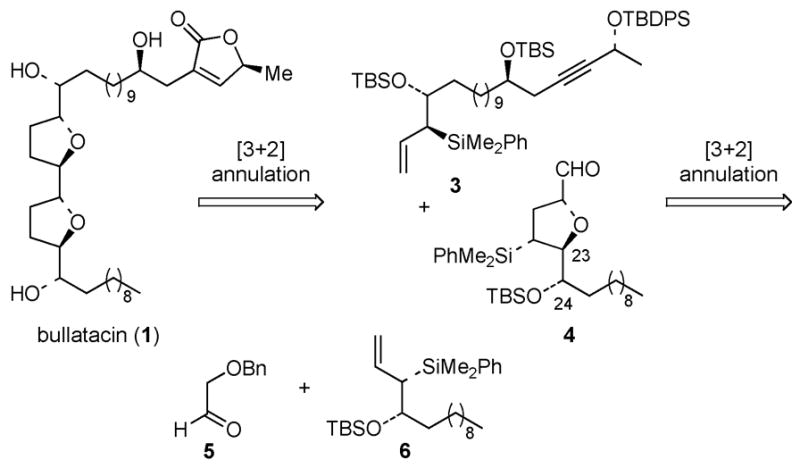
Bullatacin, Retrosynthetic Analysis
The erythro stereochemistry of C(23)–C(24) of aldehyde 4 requires that a syn-β-silyloxy allylsilane 6 be used in a chelate-controlled [3+2] annulation reaction with α-benzyloxy acetaldehyde (5). Initial attempts to develop an enantioselective synthesis of syn-β-silyloxy allylsilanes related to 6 focused on asymmetric allylboration reactions using (Z)-γ-dimethylphenylsilyallylboronate 7 (Scheme 1). Thus, silylcupration9 of acetylene, addition of the intermediate vinylcopper species to diisopropyl iodomethylboronate,10 hydrolysis of the crude alkylation product and then esterification of the intermediate allylic boronic acid with diisopropyl (R, R)-tartrate provided (R, R)-7 in 57–60% yield. This reagent underwent the expected11 syn-selective allylboration reaction of achiral aldehydes in 74–95% yield. However, the syn-β-hydroxyallylsilanes 8 were obtained with only 50–64% ee. Because the synthesis of 7 proved difficult to scale up, alternative strategies for synthesis of the targeted allylsilanes was pursued.
Scheme 1.
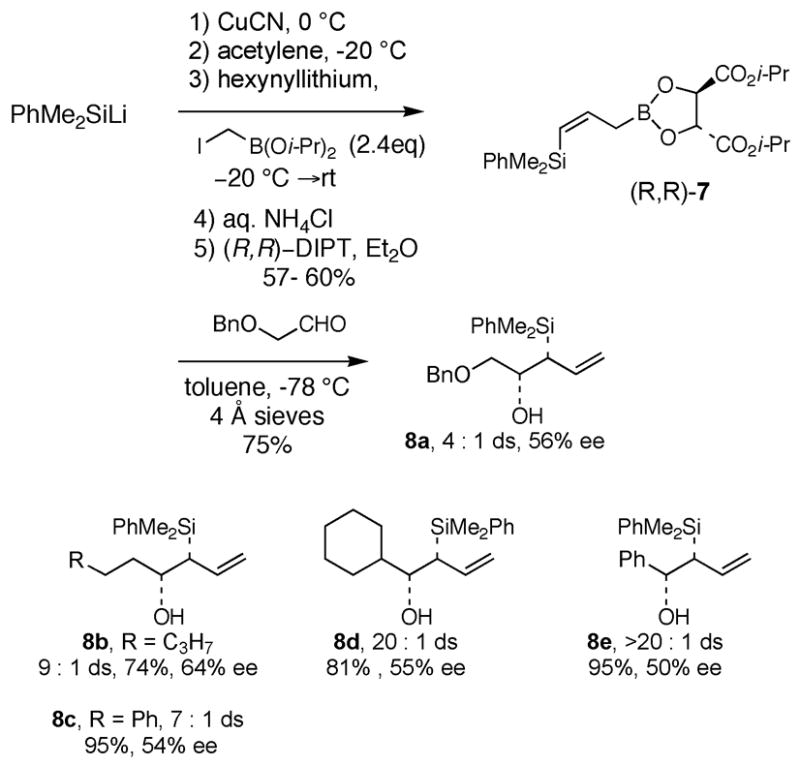
Synthesis of syn-β-Hydroxyallylsilanes via Asymmetric Allylboration
A more convenient route to (racemic) syn-β-hydroxyallylsilanes 8 involves the γ-silylallylstannation of aldehydes using γ-(dimethylphenylsilyl)allylstannane 9.12 Treatment of cyclohexanecarboxaldehyde with 9 at −78 °C in CH2Cl2 in the presence of BF3·OEt2 provided 8d in 51% yield, along with 11% of homoallylic alcohol 10. The latter compound presumably arises from a secondary reaction of 8d with cyclohexanecarboxaldehyde via an oxonia-Cope process.13 Production of the vinylsilane byproducts can be minimized by using an excess of 9 (tyically 2 equiv) in the allylstannation reaction, and by maintaining the reaction temperature at −78 °C. This method consistently provides the targeted (racemic) syn-β-hydroxyallylsilanes 8 in 51–68% yield with ≥20 : 1 ds (Scheme 2). Efforts to accomplish these reactions by using a chiral Lewis acid catalyst are in progress.
Scheme 2.
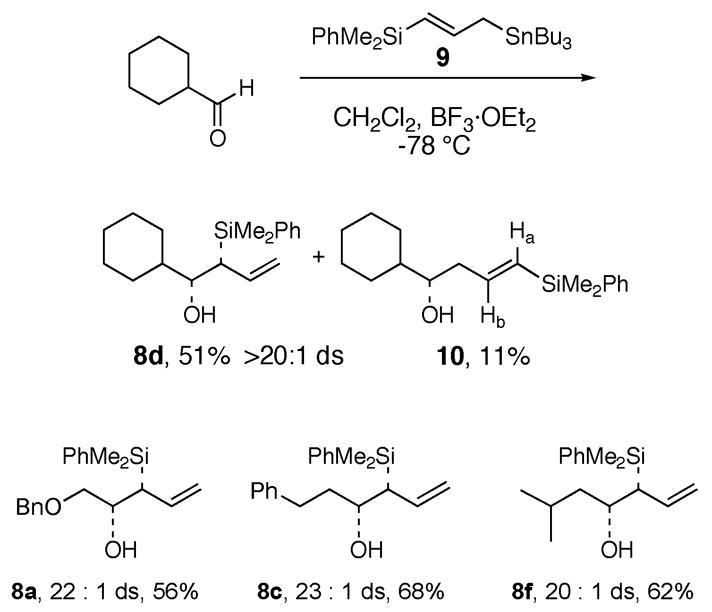
Synthesis of Racemic syn-β-Hydroxyallylsilanes via γ-Silylallylstannation
We decided to pursue a kinetic resolution strategy in the bullatacin synthesis based on observations made in our asimicin synthesis.7, 14 Specifically, initial batches of allylsilane 3 were prepared that contained significant amounts of the diastereomer 11. When these mixtures were subjected to the [3+2]-annulation reaction with transtetrahydrofuran aldehyde 12 (>95% ee), only one diastereomeric bis-tetrahydrofuran 13 was obtained. This result implied that the reaction of the (R)-allylsilane 3 with 12 is stereochemically matched and that the reaction of these two components is substantially faster and/or chemically more efficient than the mismatched reaction of the diastereomeric (S)-allylsilane 11 with 12.15 This conclusion is supported by studies in our laboratory on double asymmetric reactions of chiral tetrahydrofuran aldehydes and chiral allylsilanes.16
The synthesis of bullatacin commenced with the synthesis of racemic tetrahydrofuran carboxaldehyde 4 (Scheme 4). Allylsilane 6 was readily prepared from the BF3·OEt2 catalyzed reaction of silylallylstannane 9 and undecanal, followed by protection of the resulting secondary alcohol as a TBS ether.17 The chelate controlled [3+2]-annulation reaction of allylsilane (±)-6 and α-benzyloxy acetaldehyde (5) in the presence of SnCl4 at −45 ºC afforded the 2, 5-trans tetrahydrofuran (±)-14 in 80% yield and ≥15:1 diastereoselectivity. Hydrogenolysis of the benzyl ether using Pd(OH)2 and subsequent oxidation of the alcohol with SO3-pyridine and DMSO provided (±)-4. 18
Scheme 4.
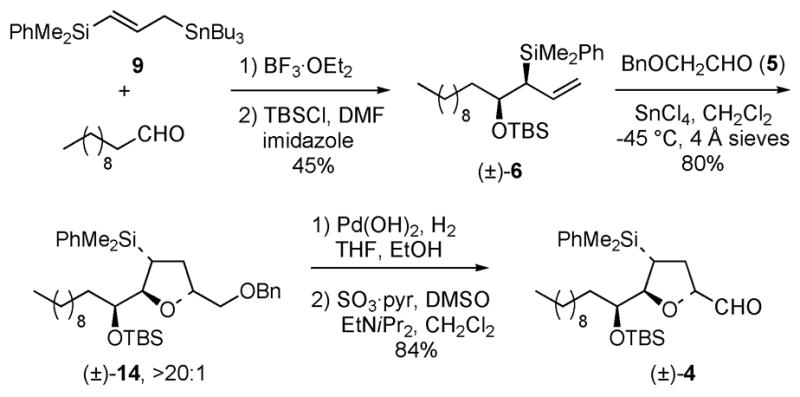
Synthesis of erythro (±)-Tetrahydrofuryl Carboxaldehyde 4
Treatment of highly enantiomerically enriched allylsilane 37 with 1 equiv of racemic aldehyde 4 in the presence of SnCl4 (1 equiv) at 0 ºC provided the bis-tetrahydrofuran 15 as a single diastereomer in 25% yield (Scheme 2). 19 When 2 equiv of the aldehyde were used in the annulation reaction, the yield of 15 increased to 50%, and a 76% yield was obtained when 3 equiv of (±)-4 was employed. In each case, bis-tetrahydrofuran 15 was obtained as a single diastereomer. The high selectivity of this reaction is attributed to the matched facial selectivity of the chiral allylsilane and the (2R, 4R, 5S)-enantiomer of the SnCl4-chelated chiral aldehyde in the favored syn-synclinal transition state 16.6, 16, 20
Woerpel and coworkers have previously described a kinetic resolution in a double asymmetric [3+2] annulation reaction of a racemic allylsilane and a chiral auxiliary-bearing α-keto ester to prepare highly-substituted tetrahydrofurans as single enantiomers.21 Our methodology is complementary in that it takes advantage of the stereochemistry inherent in the annulation substrates. In principle, our approach may be expanded to other reaction partners.
Protiodesilylation of the bis-tetrahydrofuran 15 was accomplished by treatment with TBAF in a 1 : 1 mixture of THF and DMF at 90 ºC; 22 this provided tetraol 17 in 49% yield (Scheme 6). The butenolide ring was then installed by using a procedure developed by Marshall and coworkers.7, 23 Thus, per-trifluoroacetylation of 17 followed by Pd(0)-catalyzed hydroxycarbonylation and Ag(I)-promoted cyclization of the intermediate allenyl carboxylic acid gave the tris-trifluoroacetate ester of bullatacin. Deprotection of the three trifluoroacetates by treatment of the tri-ester with KCN in MeOH then provided synthetic (+)-bullatacin (1) in 60% yield for this four step sequence. The spectroscopic properties of synthetic (+)-bullatacin were in excellent agreement with literature data (see Supporting Information).
Scheme 6.
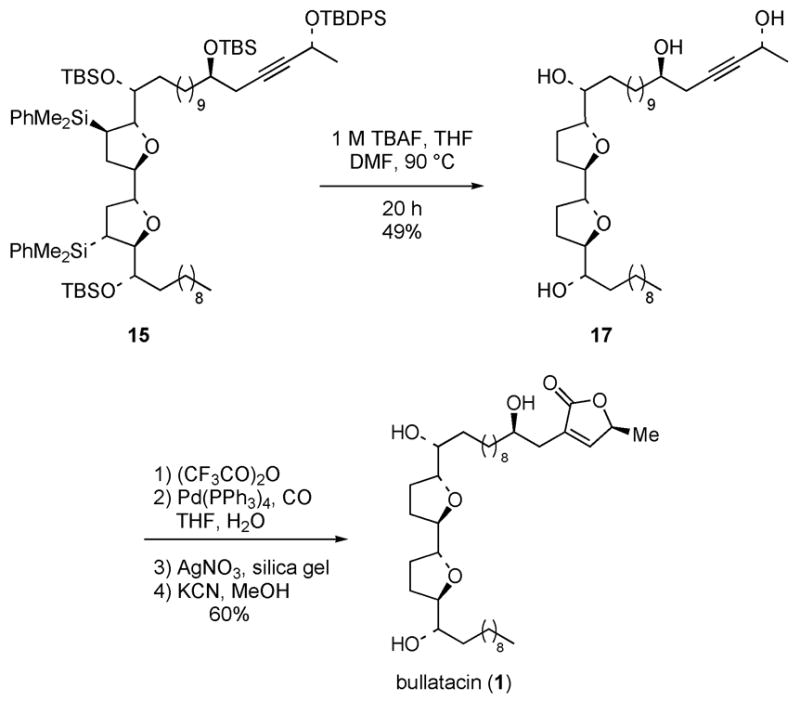
Completion of Total Synthesis of Bullatacin
In summary, the total synthesis of (+)-bullatacin has been accomplished via a highly diastereoselective [3+2] annulation reaction of allylsilane 3 and racemic aldehyde 4 in the kinetic resolution manifold. Applications of the [3+2]-annulation sequence to the synthesis of other members of the Annonaceous acetogin family will be reported in due course.
Supplementary Material
Experimental procedures and spectroscopic data for all intermediates in the bullatacin synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.
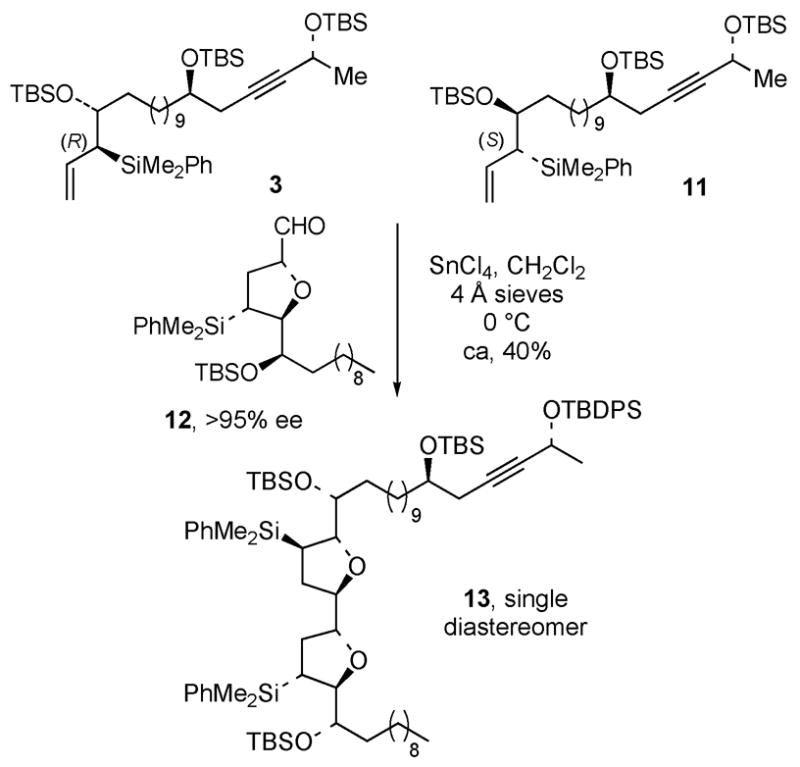
Kinetic Diastereomer Resolution in the Asimicin Synthesis.
Scheme 5.
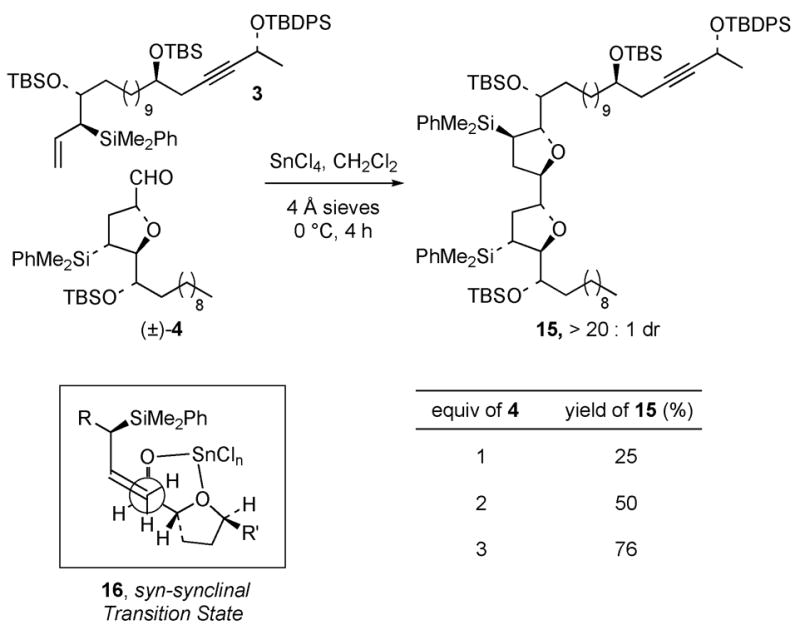
Kinetic Resolution in the [3+2]-Annulation Reaction of 3 and (±)-4
Acknowledgments
This work is supported by a grant from the National Institutes of Health (GM 38907). E.M. is grateful for fellowship support from the National Cancer Institute (CA 103507).
References
- 1.Alali FQ, Liu XX, McLaughlin JL. J Nat Prod. 1999;62:504. doi: 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]
- 2.Representative acetogenin total syntheses: Marshall JA, Jiang H. J Org Chem. 1999;64:971. doi: 10.1021/jo982057y.Kuriyama W, Ishigami K, Kitahara T. Heterocycles. 1999;50:981.Marshall JA, Jiang H. J Nat Prod. 1999;62:1123. doi: 10.1021/np990132+.Hu TS, Wu YL, Wu YK. Org Lett. 2000;2:887. doi: 10.1021/ol005504g.Emde U, Koert U. Eur J Org Chem. 2000:1889.Hoppen S, Baurle S, Koert U. Chem - Eur J. 2000;6:2382.Dixon DJ, Ley SV, Reynolds DJ. Angew Chem, Int Ed. 2000;39:3622. doi: 10.1002/1521-3773(20001016)39:20<3622::aid-anie3622>3.0.co;2-h.Harcken C, Bruckner R. New J Chem. 2001;25:40.D’Souza LJ, Sinha SC, Lu SF, Keinan E, Sinha SC. Tetrahedron. 2001;57:5255.Makabe H, Hattori Y, Tanaka A, Oritani T. Org Lett. 2002;4:1083. doi: 10.1021/ol0102803.Crimmins MT, She J. J Am Chem Soc. 2004;126:12790. doi: 10.1021/ja0455852.Zhang Q, Lu H, Richard C, Curran DP. J Am Chem Soc. 2004;126:36. doi: 10.1021/ja038542e.Nattrass GL, Diez E, McLachlan MM, Dixon DJ, Ley SV. Angew Chem, Int Ed. 2005;44:580. doi: 10.1002/anie.200462264.
- 3.Previous syntheses of bullatacin: Hoye TR, Hanson PR. Tetrahedron Lett. 1993;34:5043.Hoye TR, Tan L. Tetrahedron Lett. 1995;36:1981.Naito H, Kawahara E, Maruta K, Maeda M, Sasaki S. J Org Chem. 1995;60:4419. doi: 10.1248/cpb.46.154.Sinha SC, Sinha-Bagchi A, Yazbak A, Keinan E. Tetrahedron Lett. 1995;36:9257.Avedissian H, Sinha SC, Yazbak A, Sinha A, Neogi P, Sinha SC, Keinan E. J Org Chem. 2000;65:6035. doi: 10.1021/jo000500a.
- 4.For a review: Masse CE, Panek JS. Chem Rev. 1995;95:1293.
- 5.(a) Panek JS, Yang M. J Am Chem Soc. 1991;113:9868. [Google Scholar]; (b) Panek JS, Beresis R. J Org Chem. 1993;58:809. [Google Scholar]; (c) Beresis R, Panek JS. Bioorg Med Chem Lett. 1993;3:1609. [Google Scholar]; (d) Micalizio GC, Roush WR. Org Lett. 2001;3:1949. doi: 10.1021/ol0160250. [DOI] [PubMed] [Google Scholar]; (e) Smitrovich JH, Woerpel KA. Synthesis. 2002:2778. [Google Scholar]; (f) Heo JN, Micalizio GC, Roush WR. Org Lett. 2003;5:1693. doi: 10.1021/ol034347t. [DOI] [PubMed] [Google Scholar]
- 6.Micalizio GC, Roush WR. Org Lett. 2000;2:461. doi: 10.1021/ol9913082. [DOI] [PubMed] [Google Scholar]
- 7.Tinsley JM, Roush WR. J Am Chem Soc. 2005;127 doi: 10.1021/ja051986l. ASAP. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hui YH, Rupprecht JK, Liu YM, Anderson JE, Smith DL, Chang CJ, McLaughlin JL. J Nat Prod. 1989;52:463. doi: 10.1021/np50063a002. [DOI] [PubMed] [Google Scholar]; (b) Rieser MJ, Hui YH, Rupprecht JK, Kozlowski JF, Wood KV, McLaughlin JL, Hanson PR, Zhuang Z, Hoye TR. J Am Chem Soc. 1992;114:10203. [Google Scholar]; (c) Ahammadsahib KI, Hollingworth RM, McGovren JP, Hui YH, McLaughlin JL. Life Sci. 1993;53:1113. doi: 10.1016/0024-3205(93)90547-g. [DOI] [PubMed] [Google Scholar]; (d) Morré DJ, de Cabo R, Farley C, Oberlies NH, McLaughlin JL. Life Sci. 1994;56:343. doi: 10.1016/0024-3205(94)00957-0. [DOI] [PubMed] [Google Scholar]; (e) Oberlies NH, Croy VL, Harrison ML, McLaughlin JL. Cancer Lett. 1997;115:73. doi: 10.1016/s0304-3835(97)04716-2. [DOI] [PubMed] [Google Scholar]
- 9.Fleming I, Newton TW, Roessler F. J Chem Soc, Perkin Trans 1. 1981:2527. [Google Scholar]
- 10.(a) Matteson DS. Tetrahedron. 1998;54:10555. [Google Scholar]; (b) Soundararajan R, Li G, Brown HC. J Org Chem. 1996;61:100. [Google Scholar]
- 11.Roush WR. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Pergamon Press; Oxford: 1991. p. 1. [Google Scholar]
- 12.(a) Keck GE, Romer D. J Org Chem. 1993;58:6083. [Google Scholar]; (b) Yamamoto Y, Saito Y, Maruyama K. J Organomet Chem. 1985;292:311. [Google Scholar]
- 13.(a) Roush WR, Dilley GJ. Synlett. 2001:955. [Google Scholar]; (b) Sumida S, Ohga M, Mitani J, Nokami J. J Am Chem Soc. 2000;122:1310. [Google Scholar]
- 14.Tinsley JM. PhD Thesis. University of Michigan; 2005. [Google Scholar]
- 15.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem, Int Ed Engl. 1985;24:1. [Google Scholar]
- 16.Mertz E, Tinsley JM, Roush WR. J Org Chem. 2005 doi: 10.1021/jo0511290. submittted. [DOI] [PubMed] [Google Scholar]
- 17.Corey EJ, Venkateswarlu A. J Am Chem Soc. 1972;94:6190. [Google Scholar]
- 18.Parikh JR, von Doering EW. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 19.Aldehyde 4 recovered from this reaction was reduced with NaBH4 to give the corresponding primary alcohol. Moster ester analysis of this intermediate indicated that the recovered 4 had an enantiomeric purity of ca. 20% ee.
- 20.Keck GE, Savin KA, Cressman ENK, Abbott DE. J Org Chem. 1994;59:7889. [Google Scholar]
- 21.Peng ZH, Woerpel KA. Org Lett. 2002;4:2945. doi: 10.1021/ol026343e. [DOI] [PubMed] [Google Scholar]
- 22.Heitzman CL, Lambert WT, Mertz E, Shotwell JB, Tinsley JM, Va P, Roush WR. Org Lett. 2005;7:2405. doi: 10.1021/ol0506821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall JA, Hinkle KW. J Org Chem. 1997;62:5989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectroscopic data for all intermediates in the bullatacin synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.