

Abstract
Deoxysugars are critical structural elements for the bioactivity of many natural products. Ongoing work toward elucidating a variety of deoxysugar biosynthetic pathways has paved the way for manipulation of these pathways to generate structurally diverse glycosylated natural products. In the course of this work, the biosynthesis of D-mycaminose in the tylosin pathway of Streptomyces fradiae was investigated. Attempts to reconstitute the entire mycaminose biosynthetic machinery in a heterologous host led to the discovery of a previously overlooked gene, tyl1a, encoding an enzyme thought to convert TDP-4-keto-6-deoxy-D-glucose to TDP-3-keto-6-deoxy-D-glucose, a 3,4-ketoisomerization reaction in the pathway. Tyl1a has now been overexpressed, purified, assayed, and its activity verified by product analysis. Incubation of Tyl1a and the C-3 aminotransferase TylB, the next enzyme in the pathway, produced TDP-3-amino-3,6-dideoxy-D-glucose, confirming that these two enzymes act sequentially. Steady state kinetic parameters of the Tyl1a-catalyzed reaction were determined, and the ability of Tyl1a and TylB to process a C-2 deoxygenated substrate and a CDP-linked substrate was also demonstrated. Enzymes catalyzing 3,4-ketoisomerization of hexoses represent a new class of enzymes involved in unusual sugar biosynthesis. The fact that Tyl1a shows relaxed substrate specificity holds potential for future deoxysugar biosynthetic engineering endeavors.
Deoxysugars, such as D-mycaminose (1, Scheme 1), are present in the structures of many secondary metabolites possessing antigenic, antibiotic, and chemotherapeutic properties (1). These unusual sugars have been shown to be important for the biological activities of the parent compounds by functional studies of natural products with altered glycosylation patterns (2, 3), and by structural studies of glycosylated natural products complexed with their targets (4). Due to the direct role of deoxysugars in conferring natural product bioactivity, there has been much interest in elucidating deoxysugar biosynthetic pathways (5–10). Recently, the feasibility of manipulating the sugar biosynthetic machinery to generate new glycosylated natural products has been demonstrated (6, 8, 11–13). However, to further exploit this strategy, enzymes involved in the formation of a diverse set of deoxysugars must be identified, their activities demonstrated, preferred substrates identified, and tolerance for other substrates assessed. A detailed understanding of the biochemical properties of these enzymes is important because any rational attempt to effectively utilize a specific enzyme in biosynthetic applications requires an understanding of the details of its catalytic process.
Scheme 1.

The TDP-D-mycaminose pathway in the biosynthesis of tylosin in Streptomyces fradiae, and the tylosin gene cluster highlighting the positions of tyl1a and the mycaminose biosynthetic genes. The original assignments of enzymes catalyzing each step are shown in parentheses, and the revised assignments are shown in boldface. The reaction catalyzed by FdtA from Aneurinibacillus thermoaerophilus is shown in the inset.
The biosynthesis of mycaminose (1) has been studied for more than ten years. This 3-NN, -dimethylamino-3,6-dideoxyhexose is found as a substituent on a number of 16-membered ring macrolide antibiotics, including leucomycins, carbomycins, maridomycins, platenomycins, midecamycins, and spiramycins (14). It is also the first sugar attached to tylactone (2), a 16-membered macrolactone, in the formation of tylosin (3) in Streptomyces fradiae (Scheme 1). Extensive genetic and phenotypic complementation studies revealed the genetic organization of the tylosin (tyl) biosynthetic gene cluster in which the tylG region harbors the polyketide synthase (PKS) genes for making tylactone and the flanking tylLM, tylIBA and tylCK regions contain the genes for unusual sugar formation (15). The tylLM, tylIBA and tylCK regions were sequenced in previous studies, and 17 open reading frames (ORFs) were identified within these regions (16). Sequence similarities with other sugar biosynthetic genes, especially those reported by Cundliffe and coworkers who had also sequenced the tylIBA and tylLM regions of the tyl cluster (17), led to the assignment of tylA1, tylA2, tylB, tylM1, tylM2, and tylM3 as genes involved in mycaminose formation and attachment.
The tylA1, tylA2, and tylM2 genes all show high sequence identity with their well-characterized counterparts in other sugar biosynthetic pathways, and thus were assigned the following functions: tylA1 encodes an β-D-glucose-1-phosphate thymidylyltransferase responsible for conversion of 4 to 5, tylA2 encodes a TDP-D-glucose 4,6-dehydratase converting 5 to 6, and tylM2 encodes a glycosyltransferase responsible for the attachment of 1 to tylactone (2). The tylB and tylM1 genes encode a pyridoxal 5′-phosphate (PLP)-dependent aminotransferase and an S-adenosylmethionine (SAM)-dependent methyltransferase, respectively. As depicted in Scheme 1, TylB catalyzes the C-3 transamination of TDP-3-keto-6-deoxy-D-glucose (7) to form TDP-3-amino-3,6-dideoxy-D-glucose (8), and TylM1 catalyzes the N,N-dimethylation of 8 to give TDP-D-mycaminose (9) (16, 18).
While most of the steps in the proposed mycaminose biosynthetic pathway are supported by sequence alignment data or biochemical evidence, the process by which TDP-4-keto-6-deoxy-D-glucose (6) isomerizes to 7 remains unknown. On the basis of two early reports in which a portion of 6 was transformed to 7 during purification by Dowex-1 ion exchange chromatography, a reversible non-enzymatic ketoisomerization between 6 and 7 was thought to occur (19, 20). However, in our study of TylB, when the reaction was run in reverse using 8 and α-ketoglutarate as substrates, only 7 was produced and no trace of 6 was detected. Also, no product was formed upon incubation of 6 with TylB. These results indicated that, at least under the in vitro conditions used, there was no chemical isomerization between 6 and 7, suggesting that the 3,4-ketoisomerization is more likely enzyme-catalyzed (18). This activity was tentatively assigned to the tylM3 gene product, which displays low sequence similarity to P-450 enzymes but lacks the conserved cysteine residue that coordinates the heme iron.
Subsequent attempts to reconstitute the mycaminose biosynthetic pathway in a non-producing strain showed that expression of tylB, tylM1, tylM2, and tylM3 failed to convert 6 to TDP-D-mycaminose (9) and couple 9 to tylactone (2). These studies were performed by heterologous expression of tylB, tylM1, tylM2, and tylM3 in a mutant (KdesI/VII) of Streptomyces venezuelae, which is the producer of the D-desosamine (11) containing macrolide antibiotics methymycin (12), neomethymycin (13), pikromycin (14), and narbomycin (15) (Scheme 2A). With the desI and desVII genes disrupted, intermediate 6 was expected to accumulate in vivo. It was also expected that expression of tylB, tylM1, tylM2, and tylM3 would produce all the necessary enzymes to convert 6 to TDP-D-mycaminose (9), which could then be used by TylM2 to glycosylate appropriate aglycones. Surprisingly, feeding exogenous tylactone (2) to this recombinant strain led to quinovosyl tylactone (17) rather than the anticipated mycaminosyl tylactone (10, Scheme 2B) (8). Production of quinovosylated macrolides had previously been observed in an S. venezuelae mutant in which desI was disrupted. It was proposed that quinovose was generated by C-4 reduction of 6 by a non-specific reductase to give 16 in the KdesI mutant (21). A similar reduction of 6 to 16 likely occurs in the KdesI/VII mutant. Thus, the above results strongly suggested that conversion of 6 to 7 did not occur in the recombinant strain.
Scheme 2.

(A) Biosynthesis of desosamine (11) and its incorporation into methymycin (12), neomethymycin (13), pikromycin (14) and narbomycin (15) in Streptomyces venezuelae. (B) Pathway for the formation of quinovosyltylactone (17) in the KdesI/KdesVII/tylM3, tylM2, tylM1, tylB mutant. (C) Pathway for the formation of mycaminosyltylactone (10) in the KdesI/KdesVII/tyl1a, tylM3, tylM2, tylM1, tylB mutant.
The inability to reconstitute the mycaminose pathway with the tylB, tylM1, tylM2, and tylM3 genes in the above experiment prompted us to re-examine all unassigned open reading frames (ORFs) in the tylosin gene cluster. This effort led to the identification of an ORF, 1a, which is immediately downstream from and translationally coupled to tylB, and exhibits modest sequence homology (34% identity, 52% similarity) to a recently reported TDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase (FdtA, catalyzing 6 → 18, see Scheme 1, inset) from the thermophilic bacillus species Aneurinibacillus thermoaerophilus (22). The FdtA enzyme is involved in S-layer polysaccharide biosynthesis, and is the first hexose-3,4-ketoisomerase to be characterized biochemically. Subsequent heterologous expression of ORF 1a (hereafter referred to as tyl1a) together with tylB, tylM1, tylM2, and tylM3 in the KdesI/VII S. venezuelae mutant resulted in the quantitative conversion of exogenously fed tylactone (2) to 5-O-mycaminosyl-tylactone (10, Scheme 2C). These findings identified Tyl1a as the TDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase in the mycaminose pathway (Scheme 1) (8).
Herein, we report the overexpression, purification, and biochemical characterization of Tyl1a. We showed via in situ 1H NMR spectroscopic analysis that Tyl1a converts 6 to 7, which can then be converted to 8 by incubation with the next enzyme in the mycaminose pathway, TylB. These results firmly establish Tyl1a as the 3,4-ketoisomerase in the mycaminose pathway. We also explored the substrate specificity of this enzyme and demonstrated that Tyl1a processes the alternate substrate TDP-4-keto-2,6-dideoxy-D-glucose (22, Scheme 4), and can also act on CDP-4-keto-6-deoxy-D-glucose (26, Scheme 5), albeit at a much reduced rate. Additionally, we demonstrated that TylB is able to convert the Tyl1a products generated using 22 and 26 to TDP-3-amino-2,3,6-trideoxy-D-glucose (25, Scheme 4) and CDP-3-amino-3,6-dideoxy-D-glucose (28, Scheme 5), respectively. These findings have important implications for deoxysugar pathway engineering efforts and for the functional elucidation and characterization of other Tyl1a and FdtA homologues.
Scheme 4.

Enzymatic synthesis of 22 using 5, RfbB, TylX3, and SpnN; and conversion of 22 to 25 using Tyl1a and TylB. The Tyl1a reaction product 23 and the degradation product 24 are shown.
Scheme 5.

Enzymatic synthesis of 26, and its conversion to 28 by Tyl1a and TylB. The Tyl1a reaction product 27 and the degradation product 19 are shown.
Experimental Procedures
Materials
The tyl1a and tylB genes were amplified from the cosmid pSET552, generously provided by Dr. Eugene Seno of Eli Lilly Research Laboratories. Escherichia coli strain DH5α and Salmonella enterica serovar Typhimurium LT2 (ATCC 15277) were purchased from Bethesda Research Laboratories (Gaithersburg, MD) and the American Type Culture Collection (Manassas, VA), respectively. Vector pET28b(+) and the overexpression hosts E. coli BL21 and BL21(DE3) were purchased from Novagen (Madison, WI). Enzymes and molecular weight standard used for molecular cloning experiments were products of Invitrogen (Carlsbad, CA) or New England Biolabs (Ipswich, MA). Ni-NTA agarose and kits for DNA gel extraction and spin miniprep were obtained from Qiagen (Valencia, CA). pfu DNA polymerase was purchased from Stratagene (La Jolla, CA), and growth media components were acquired from Becton Dickinson (Sparks, MD). Antibiotics and chemicals were products of Sigma-Aldrich Chemical Co. (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). Bio-gel P2 resin and all reagents for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad (Hercules, CA), with the exception of the prestained protein molecular weight marker, which was obtained from New England Biolabs. Amicon YM-10 filtration products were purchased from Millipore (Billerica, MA). Sephadex G-10 resin was acquired from Amersham (Piscataway, NJ), CarboPac PA1 HPLC column was obtained from Dionex (Sunnyvale, CA), and Mono-Q H/R 16/10 and Superdex 200 HR 10/30 FPLC columns were obtained from Pharmacia (Uppsala, Sweden). Oligonucleotide primers for cloning of Tyl1a were prepared by Integrated DNA Technologies (Coralville, IA).
General
Protein concentrations were determined according to Bradford (23) using bovine serum albumin as the standard. The relative molecular mass and purity of enzyme samples were determined using SDS-PAGE as described by Laemmli (24). The native molecular mass of Tyl1a was determined by the gel filtration method of Andrews (25). NMR spectra were acquired on either a Varian Unity 300 or 500 MHz spectrometer, and chemical shifts (δ in ppm) are reported relative to that of dimethylsulfoxide (DMSO, δ 2.54 for 1H NMR). DNA sequencing was performed by the Core Facilities of the Institute of Cellular and Molecular Biology at the University of Texas at Austin. Mass spectra were obtained by the Mass Spectrometry Core Facility in the Department of Chemistry and Biochemistry at the University of Texas at Austin. The general methods and protocols for recombinant DNA manipulations followed those described by Sambrook et al (26). Kinetic data were analyzed by non-linear fit using Grafit5 (Erithacus Software Ltd., UK).
Gene Amplification and Cloning of tyl1a
Two oligonucleotide primers complementary to the sequence at each end of tyl1a were prepared to amplify the tyl1a gene from the cosmid pSET552. These two primers, Tylorf1a-24/28-N-up (5′-GGAATTCCATATGGCGGCGAGC-ACTACGACGGAGGG-3′) and Tylorf1a-28-H-down (5′-GCGCAAGCTTTCACGGGTGGCT-CCTGCC-3′), were designed to amplify tyl1a with engineered 5′ NdeI and 3′ HindIII restriction sites (shown in bold) to be cloned into pET28b(+), and thereby encode expression of Tyl1a with an N-terminal His6-tag. The PCR-amplified tyl1a gene was purified, digested with NdeI and HindIII restriction enzymes, and ligated into the NdeI/HindIII-digested vector, pET28b(+), to give the recombinant plasmid tyl1a/pET28b(+). This plasmid was used to transform E. coli BL21 for protein overexpression.
Growth of E. coli BL21-tyl1a/pET28b(+) Cells
An overnight culture of E. coli BL21-tyl1a/pET28b(+) grown in Luria-Bertani (LB) media containing 50 μg/mL kanamycin at 37 °C, was used (2 mL each) to inoculate 6 L (in 6 × 1 L aliquots) of LB culture containing 35 μg/mL kanamycin. These cultures were incubated at 37 °C until the OD600 reached 0.6. Protein expression was then induced with addition of isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 0.2 mM, and the cells were allowed to grow at 37 °C for an additional 5 h. After this time, OD600 had reached 1.5. The cells were harvested by centrifugation at 6000 g for 10 min and stored at −80 °C until lysis. A total of 18 g (wet weight) of cells was obtained.
Purification of N-His6-Tyl1a Protein
All purification steps were carried out at 4 °C, except the FPLC step, which was carried out at 25 °C. Thawed cells were suspended in lysis buffer containing 15% glycerol (37 mL). Two 1000× protease inhibitor cocktails were used in the preparation: one containing 16 mg/mL each of phenylmethylsulfonylfluoride (PMSF) and N-p-tosyl-L-phenylalanine (TPCK) in 1:1 DMSO/i-PrOH, and the other containing 2.4 mg/mL each of leupeptin and lima bean trypsin inhibitor in water. These two solutions were added to the cell suspension at 1× final concentration every 30 min prior to Ni-NTA chromatography. To this cell suspension were also added EDTA and lysozyme to 0.3 mM and 1 mg/mL final concentrations, respectively. The mixture was stirred gently for 30 min on ice. Next, 360 μg (360 μL of 1 mg/mL) of each DNase and RNase (20 μg/g of cells) were added to the mixture, and the resulting solution was incubated for an additional 15 min on ice with gentle stirring. Subsequent sonication was performed using 12 × 20 s pulses with 30 s pauses between each pulse. The lysate was centrifuged at 15,000 g for 30 min and the supernatant was subjected to Ni-NTA chromatography.
The general purification procedure is a modified version of that described in the QIAexpressionist handbook provided by Qiagen for use with Ni-NTA agarose resin. Specifically, Ni-NTA slurry (10 mL) was washed twice with lysis buffer before use. The soluble protein fraction was incubated with 10 mL of Ni-NTA beads, which had been washed with lysis buffer, on a rotator at 4 °C for 1 h. Lysate and beads were then loaded onto a column, which was allowed to drain, and then washed with wash buffer containing 15% glycerol (75 mL). Bound protein was eluted using elution buffer containing 15% glycerol (4 mL) and collected in 4 × 1 mL portions, which were pooled and dialyzed against 3 × 1 L of 50 mM NaH2PO4 buffer, 300 mM NaCl, 15% glycerol, pH 8.0. Tyl1a was greater than 90% pure after Ni-NTA chromatography, and the yield was 45 mg per liter of cell culture. Further purification of Tyl1a was performed on an FPLC Mono-Q 16/10 column using a linear gradient of 0 to 50% buffer B in buffer A as eluant. Buffer A was 20 mM Tris·HCl, pH 7.5, and buffer B was 20 mM Tris·HCl, 250 mM NaCl, pH 7.5. The detector was set at 280 nm and the flow rate was 3 mL/min. Collected fractions were analyzed by SDS-PAGE, the desired fractions pooled, mixed with an equal volume of 20 mM Tris·HCl buffer, 250 mM NaCl, 30% glycerol, pH 7.5, and concentrated to 1–2 mg/mL using an Amicon YM-10 filter. On the basis of SDS-PAGE analysis, the isolated Tyl1a was estimated to be greater than 95% pure. The purified Tyl1a was flash frozen and stored at −80 °C.
Cleavage of N-His6-tag from Tyl1a by Thrombin
Tyl1a expressed from pET28b(+) contains a thrombin cleavage site between the His6-tag and the first amino acid of Tyl1a. The Novagen Thrombin Cleavage Capture Kit containing biotinylated thrombin and straptavidin agarose for removal of thrombin was used for the preparation of N-His6-cleaved Tyl1a. Small-scale trials showed that Tyl1a-N-His6 could be efficiently cleaved after a 2 h incubation with 0.5 units of thrombin per mg Tyl1a. Accordingly, Tyl1a-N-His6 (5 mg) was incubated with 2.5 units of thrombin at 25 °C overnight. Thrombin was then removed using streptavidin agarose according to the manufacturer’s protocol. Cleaved Tyl1a with remaining non-native N-terminal sequence (GSH), was further purified by FPLC using a Mono Q column as described above. The desired fractions (> 95% pure) were pooled, mixed with an equal volume of 20 mM Tris·HCl buffer containing 250 mM NaCl, 30% glycerol, pH 7.5, and concentrated to 1–2 mg/mL using an Amicon YM-10 filter. The purified Tyl1a was flash frozen and stored at −80 °C.
Molecular Mass Determination
The native molecular mass of Tyl1a was determined by gel filtration performed on an FPLC equipped with a Superdex 200 HR 10/30 column. The proteins were eluted using 50 mM NaH2PO4 buffer, 150 mM NaCl, pH 7.0, at a flow rate of 0.5 mL/min. The system was calibrated with protein standards (Sigma), and the void volume (V0) was measured using blue dextran. The data were analyzed by the method of Andrews (25).
Preparation of Enzymes used to Synthesize TDP-4-keto-6-deoxy-D-glucose (6)
The TK, TMK, and NDK used in this synthesis were prepared as described by Takahashi et al (9). Rabbit muscle pyruvate kinase was purchased from Sigma as a 400–800 units/mg ammonium sulfate precipitate. This ammonium sulfate precipitate was dissolved in water to a concentration of 2500 units/mL, dialyzed against 50 mM NaH2PO4 buffer, 300 mM NaCl, pH 8.0, to remove ammonium sulfate, and stored at −80 °C. RfbA was prepared as follows. The rfbA gene was amplified from Salmonella enterica serovar Typhimurium LT2 genomic DNA using PCR. The start primer contained an engineered BamHI restriction site (in italics), ribosomal binding sequence (underlined), and AT rich region upstream of the native start codon (shown in bold), with sequence 5′-CGGGATCCGAAGGAGATATATAATGAAAACGCGTA-AGGGC-3′; and the halt primer contained an engineered PstI restriction site (in italics) and a C-terminal His5-tag (underlined) immediately downstream of the stop codon (in bold), with sequence 5′-CTTGCATGCCTGCAGTTAATGATGATGATGATGTAAACCTTTCACCATC-3′. The PCR-amplified gene was purified, digested with BamHI and PstI, and ligated into BamHI/PstI digested pUC18. The resulting construct was used to transform E. coli BL21(DE3). Overexpression was achieved by growth of the transformed host in LB media in the presence of 100 μg/mL ampicillin at 37 °C overnight. Protein was purified from the harvested cells by Ni-NTA affinity chromatography in an identical manner to that used for purification of Tyl1a. RfbB used in the synthesis of 6 was prepared in the following manner. The rfbB gene was amplified from Salmonella enterica serovar Typhimurium LT2 genomic DNA by PCR. The start primer contained an engineered EcoRI restriction site (in italics), ribosomal binding sequence (underlined), and AT rich region upstream of the native start codon (shown in bold), with sequence 5′-GGAATTCGAAGGAGATATATAATGGTGAAGATACTTATTACTGG-3′. The halt primer contained an engineered BamHI restriction site (in italics) and a His5-tag sequence (underlined) immediately downstream of the stop codon (in bold), with sequence 5′-CGGGATCCTTAATGATGATGATGATGCTGGCGTCCTTCATAGTTC-3′. The PCR-amplified gene was purified, digested with EcoRI and BamHI, and ligated into EcoRI/BamHI digested pUC18. The resulting construct was used to transform E. coli BL21(DE3). Overexpression was achieved by growth of the transformed host in LB medium supplemented with 100 μg/mL ampicillin at 37 °C overnight. Protein was purified from the harvested cells by Ni-NTA affinity chromatography in an identical manner to that used for the purification of Tyl1a.
Enzymatic Synthesis of TDP-4-keto-6-deoxy-D-glucose (6)
The large-scale enzymatic preparation of Tyl1a substrate was initiated by coupling thymidine with glucose-1-phosphate (4) to make TDP-D-glucose (5), which was then converted to TDP-4-keto-6-deoxy-D-glucose (6). Preparation of TDP-D-glucose from thymidine and glucose-1-phosphate (4) was conducted in a two-stage, “one-pot” reaction (9). In the first stage, a mixture containing 76.2 mM phosphoenolpyruvate (PEP), 24 mM thymidine, 1.6 mM ATP, 27 mM MgCl2, 25 μM thymidine kinase (TK), 25 μM thymidylate kinase (TMK), 25 μM nucleoside diphosphate kinase (NDK), and 1000 units of rabbit muscle pyruvate kinase (PK) in 17 mL of 45 mM Tris·HCl buffer, pH 7.5, was incubated at 37 °C for 4 h, generating thymidine triphosphate (TTP). The enzymes were removed by filtration through an Amicon YM-10 membrane, and glucose-1-phospate (4), MgCl2, and α-D-glucose-1-phosphate thymidylyltransferase (RfbA) from S. enterica LT2 were added to the filtrate to give final concentrations of 28 mM, 50 mM, and 36 μM, respectively. The mixture was incubated for 16 h at 37 °C, centrifuged at 5,000 g for 10 min to remove precipitate, and filtered through an Amicon YM-10 membrane to remove enzymes. The crude product (5), with a theoretical yield of 228 mg, was stored at 4 °C.
The enzyme-free filtrate from the previous step was loaded onto a Bio-gel P2 column (25 mm × 100 cm) pre-washed with water, and run at a flow rate of 12 mL/h with water as the eluant, with 8 mL fractions collected. Fractions showing UV absorption at 267 nm were lyophilized, and the identity and purity of the compounds in each fraction assessed by 1H and 31P NMR spectroscopy. TDP-D-glucose (5) containing fractions (total weight 170 mg), which varied in purity from 25–70%, were pooled according to their purities. Further purification of TDP-D-glucose was performed by FPLC equipped with a Mono Q 16/10 column. A linear gradient of 0 to 40% of a solution of 400 mM NH4HCO3 in water was used as the eluant. The detector was set at 280 nm, and the flow rate was 5 mL/min. Fractions containing the major peak were lyophilized individually, redissolved in water, and lyophilized again to remove NH4HCO3. The purities of these fractions (total weight 123 mg), which ranged from 50–90%, were assessed by 1H and 31P NMR spectroscopy. From these fractions, 23 mg of 90% pure TDP-D-glucose (5) was obtained. 1H NMR (300 MHz, D2O) δ 1.78 (3H, s, 5″-Me), 2.22 (2H, m, 2′-H), 3.28–3.40 (2H, m, 2-H, 3-H), 3.59–3.78 (2H, m, 4-H, 5-H), 3.95–4.06 (3H, m, 4′-H, 5′-H), 4.45 (1H, m, 3′-H), 5.44 (1H, dd, J = 6.9, 3.3, 1-H), 6.20 (1H, t, J = 6.9, 1′-H), 7.59 (1H, s, 6″-H).
TDP-D-glucose (5, 23 mg) obtained from the previous step was dissolved in 47 mM KH2PO4 buffer, pH 7.5, to give a final concentration of 29 μM. This solution was incubated with TDP-D-glucose 4,6-dehydratase (RfbB) from S. enterica LT2 (18 μM) at 37 °C for 2 h, after which RfbB was removed by filtration through an Amicon YM-10 membrane. The filtrate was loaded onto a Sephadex G-10 column pre-washed with water and run at a flow rate of 1 mL/min using water as the eluant, with 10 mL fractions collected. Those fractions exhibiting absorption at 267 nm were lyophilized and their purities assessed by 1H and 31P NMR spectroscopy. Those fractions containing pure TDP-4-keto-6-deoxy-D-glucose (6, 12.3 mg, >90% purity) were combined and the concentration of 6 in the solution was determined spectrophotometrically at 267 nm, (ε =9600 M−1cm−1). 1H NMR (300 MHz D2O) of 6 (a mixture of hydrate and keto forms): δ 1.08 (3H, d, J = 6.5, 5-Me of hydrate form), 1.12 (3H, d, J = 6.5, 5-Me of keto form), 1.79 (3H, s, 5″-Me), 2.09–2.26 (2H, m, 2′-H), 3.48 (1H, m, 2-H of hydrate form), 3.64 (1H, d, J = 10.0, 3-H of hydrate form), 3.68 (1H, m, 2-H of keto form), 3.96 (1H, q, J = 6.5, 5-H of hydrate form), 4.01–4.07 (3H, m, 4′-H, 5′-H), 4.48 (1H, m, 3′-H), 5.41 (1H, dd, J = 7.3, 3.8, 1-H of hydrate form), 5.59 (1H, dd, J = 7.0, 3.0, 1-H of keto form), 6.20 (1H, t, J = 6.9, 1′-H), 7.60 (1H, s, 6″-H).
Enzymatic Preparation of TDP-4-keto-2,6-dideoxy-D-glucose (22)
This compound was prepared enzymatically from TDP-D-glucose (5) using purified enzymes RfbB from S. enterica LT2, TylX3 from Streptomyces fradiae (27), and SpnN, the TDP-3,4-diketo-2,6-dideoxy-D-glucose 3-ketoreductase from the spinosyn biosynthetic pathway of Saccharopolyspora spinosa (28) (Scheme 4). A typical reaction mixture containing 28 mM TDP-D-glucose (5), 34 μM RfbB, 17 mM NADPH, 10 μM TylX3, 17 μM SpnN, in 50 mM Tris·HCl buffer, pH 7.5, 10% glycerol, was incubated at 25 °C for 4 h. Enzymes were removed using a Centricon YM-10 microconcentrator, and the filtrate was separated on a Bio-gel P2 gel filtration column (25 mm × 100 cm) pre-washed with 25 mM NH4HCO3, and run at a flow rate of 12 mL/h with 25 mM NH4HCO3 as the eluant. Fractions (8 mL each) showing UV absorbance at 267 nm were lyophilized, and the identity and purity of the compounds in each fraction assessed by 1H and 31P NMR spectroscopy. The 1H-NMR spectrum of the purified TDP-4-keto-2,6-dideoxy-D-glucose (22) was identical to that previously reported (29).
HPLC Activity Assay for Tyl1a
A reaction mixture (35 μL) containing 2.85 μM Tyl1a (with or without N-His6-tag) and 1 mM TDP-4-keto-6-deoxy-D-glucose (6) in 50 mM KH2PO4 buffer, pH 7.5, was incubated at 25 °C. Aliquots of 5 μL were removed at various time points, quenched by flash freezing in liquid nitrogen, thawed at 4 °C, diluted by addition of 20 μL of 50 mM KH2PO4 buffer, pH 7.5, filtered through a Microcon YM-10 membrane to remove enzyme, and the filtrate flash frozen until HPLC analysis. HPLC analysis was performed using a Dionex Carbopac PA1 column with 9.5 μL of sample for each injection. The sample was eluted by a gradient of water as solvent A and 500 mM NH4OCOCH3 (adjusted to pH 7.0 with aqueous NH3) as solvent B where the gradient ran from 5 to 20% B over 15 min, then 20 to 60% B over 20 min, 60 to 100% B over 2 min, 3 min wash at 100% B, 100–5% B over 5 min, and re-equilibration at 5% B for 15 min. The flow rate was 1 mL/min, and the detector was set at 267 nm. The retention times were 35.3 min for TDP-4-keto-6-deoxy-D-glucose (6), 39.0 min for TDP-3-keto-6-deoxy-D-glucose (7), 41.9 min for TDP, and 1.8 min for the degradation product, (2R,3R)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran (19). The substrate and product ratios were calculated from the integration of the corresponding peaks on the HPLC chromatogram.
In situ 1H NMR Assay for Tyl1a and Characterization of Products
A reaction mixture (600 μL) containing 10 mM TDP-4-keto-6-deoxy-D-glucose (6), 50 mM KH2PO4 buffer, pH 7.5, 5% (v/v) DMSO-d6 (as a reference) was prepared in an NMR tube. After shimming and initial peak integration in a 500 MHz NMR spectrometer, the sample was removed from the tube, mixed thoroughly with glycerol-free His6-tagged Tyl1a (6 or 10 μM final concentration), and returned to the NMR tube. Data were acquired at 5-min intervals for 150 min. Spectral data for TDP-3-keto-6-deoxy-D-glucose (7) and the degradation product (2R,3R)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran (19) were assigned from the spectra generated during the in situ assay. 1H NMR (DMSO-d6) of 7: δ 1.23 (3H, d, J =5.5, 5-Me), 1.73 (3-H, s, 5″-Me), 2.15–2.20 (2H, m, 2′-H), 3.92–4.01 (5H, m, 4-H, 5-H, 4′-H, 5′-H), 4.00–4.05 (1H, m, 2-H), 4.45–4.50 (1H, m, 3′-H), 5.68 (1H, dd, J =7.0, 4.5, 1-H), 6.15 (1H, t, J = 7.0, 1′-H), 7.55 (1H, s, 6″-H). 1H NMR (DMSO-d6) of 19: δ 1.28 (3H, d, J = 6.0, 5-Me), 4.03 (1H, d, J = 12.3, 4-H), 4.10 (1H, dq, J = 12.3, 6.0, 5-H), 7.32 (1H, s, 1-H).
Determination of Kinetic Parameters for Tyl1a
The steady state kinetic parameters of the Tyl1a-catalyzed reaction were determined by the HPLC activity assay as described above. Reactions containing 100 nM N-His6-tagged Tyl1a and varied amounts of TDP-4-keto-6-deoxy-D-glucose (6) (3 μM to 1 mM) in 50 mM KH2PO4 buffer, pH 7.5, were incubated at 25 °C. Larger reaction volumes were used for the lower substrate concentrations to facilitate HPLC analysis. Aliquots were taken at four different time points for each of 16 substrate concentrations. Incubation mixtures with lower substrate concentrations were monitored for shorter periods of time (2 to 3 min), and those with higher substrate concentrations were monitored for periods as long as 7 min. Since the concentration of substrate in each sample was known, the percent conversion determined by HPLC could be used to calculate μmoles of product formed for each time point. The amount of product formed at the four time points for a given substrate concentration were plotted against time, and the slope of each line was plotted versus substrate concentration. The resulting data were fit to the Michaelis–Menten equation by non-linear regression using Grafit 5 to determine the kcat and Km values.
As a comparison, data obtained from the HPLC time course study and those obtained from the in situ NMR assay by following changes in integration of individual proton signals over time (for example, the 5-methyl signal of 6, 7, and the 2-methyl signal of 19), were also used to calculate the apparent kcat. In each case, peak integrations were normalized to initial substrate concentration and data sets were individually fit to either single or double exponential equations by non-linear regression analysis using Grafit 5. The rate constant for the disappearance of substrate from these data was used to calculate the apparent kcat in each experiment.
HPLC Assay of Coupled Tyl1a-TylB Reaction
The C-3 aminotransferase TylB (18) used in this coupled assay and the TylB product standard, TDP-3-amino-3,6-dideoxy-D-glucose (8) (16), were prepared according to published procedures. The reaction mixture (100 μL) contained 28.5 μM Tyl1a, 10 μM TylB, 1 mM TDP-4-keto-6-deoxy-D-glucose (6), 10 mM L-glutamate, 50 μM pyridoxal-5′-phosphate (PLP), in 50 mM KH2PO4 buffer, pH 7.5. Aliquots were removed at various time points and analyzed by HPLC in the same manner as used for Tyl1a activity assays. The HPLC retention time for TDP-3-amino-3,6-dideoxy-D-glucose (8) was 13.7 min. For isolation and MS characterization of the TylB product (8) and Tyl1a degradation product (19), 300 μg of TDP-4-keto-6-deoxy-D-glucose (6) at a concentration of 1 mM was incubated with 3 μM Tyl1a, 10 μM TylB, 50 μM PLP, and 10 mM L-glutamate in 50 mM KH2PO4 buffer, pH 7.5. The total reaction volume was 550 μL. The reaction was incubated at 25 °C for 70 min, quenched by flash freezing in liquid nitrogen, thawed on ice, and enzymes removed by filtration through Microcon YM-10 at 4 °C. The sample was then separated using a semi-preparative Dionex CarboPac PA-1 column and a gradient elution program identical to that used for the analytical Dionex CarboPac PA-1 column, with a flow rate of 5 mL/min. Compounds 8 and 19, which eluted at 12.5 and 1.8 min, respectively, were collected manually and lyophilized to dryness. HPLC analysis showed 35% conversion of 6 to 8 and 65% conversion of 6 to 19 (the degradation product) and TDP. Compound 8 was resuspended in water to a concentration of 0.1 mg/mL and 19 was resuspended in methanol to a concentration of 1 mg/mL. High resolution ESI-MS of 8: calculated for C16H27N3O14P2 (M − H) 546.0883, found 546.0885; 19: calculated for C6H4O4 (M + H) 145.0501, found 145.0500.
HPLC Analysis of Incubation Mixture Containing Tyl1a with TDP-4-keto-2,6-dideoxy-D-glucose (22)
The reaction mixture (70 μL) contained 2.85 μM Tyl1a and 1 mM 22 in 50 mM KH2PO4 buffer, pH 7.5, and was incubated at 25 °C. Aliquots were removed at various time points, and analyzed by HPLC in the same manner as used for other Tyl1a activity assays. The retention time of the substrate 22 was 33.4 min, that of the product, TDP-3-keto-2,6-dideoxy-D-glucose (23), was 37.0 min, and that of the degradation product, (2R,3R)-2-methyl-3-hydroxy-4-keto-2,3-dihydropyran (24), was 1.7 min. Peak integrations were normalized to initial substrate concentration and data sets were individually fit to either single or double exponential equations by non-linear regression analysis using Grafit 5. The rate constant for the disappearance of substrate obtained from these data was used to calculate the apparent kcat in each experiment.
Coupled Assay of Tyl1a-TylB with TDP-4-keto-2,6-dideoxy-D-glucose (22)
The reaction mixture (50 μL), which was incubated at 25 °C, contained 2.85 μM Tyl1a, 28.5 μM TylB, 1 mM 22, 28.5 mM L-glutamate, and 142.5 μM PLP in 50 mM KH2PO4 buffer, pH 7.5. Aliquots were removed at various time points and subjected to HPLC analysis as described for the Tyl1a activity assays. The retention time of the product, TDP-3-amino-2,3,6-trideoxy-D-glucose (25), was 9.5 min. For isolation and MS characterization of the TylB product (25) and Tyl1a degradation product (24), 1.0 mg of the SpnN product TDP-4-keto-2,6-dideoxy-D-glucose (22) at a concentration of 2 mM was incubated with 1 μM Tyl1a, 30 μM TylB, 250 μM PLP, and 50 mM L-glutamate in 50 mM KH2PO4 buffer, pH 7.5. The total reaction volume was 940 μL. The reaction was incubated at 25 °C for 12 h and enzymes removed by filtration through Microcon YM-10 at 4 °C. The sample was then separated using a semi-preparative Dionex CarboPac PA-1 column and a gradient elution program identical to that used for the analytical Dionex CarboPac PA-1 column, with a flow rate of 5 mL/min. Compounds 25 and 24, which eluted at 8.9 and 1.8 min, respectively, were collected. HPLC analysis showed 15% conversion of 22 to 25 and 85% conversion of 22 to 24 (the degradation product) and TDP. Compound 24 was lyophilized to dryness and resuspended in methanol to a concentration of 1 mg/mL. Compound 25, which is unstable when lyophilized to dryness, or when stored at 25 °C, was lyophilized to reduce its volume tenfold, and stored at −80 °C. High resolution EI-MS of 24: calculated for C6H4O3 (M+) 128.0473, found 128.0486. High resolution ESI-MS 25: calculated for C16H27N3O13P2 (M − H) 530.0941, found 530.0941.
HPLC Analysis of Incubation Mixture Containing Tyl1a and CDP-4-keto-6-deoxy-D-glucose (26)
Preparation and quantitation of CDP-4-keto-6-deoxy-D-glucose (26) was conducted as described by Chen et al (30). Preparation of enzymes used to make 26 was performed as described by Thorson et al. (31, 32). The reaction mixture (100 μL) contained 100 μM Tyl1a and 1 mM 26 in 50 mM KH2PO4 buffer, pH 7.5, and the incubation was carried out at 25 °C. Aliquots were removed at various time points and analyzed by HPLC in the same manner as used for other Tyl1a activity assays. The retention times of 26, the product CDP-3-keto-6-deoxy-D-glucose (27), and CDP were 33.1, 36.6, and 42.3 min, respectively. Peak integrations were normalized to initial substrate concentration and data sets were individually fit to either single or double exponential equations by non-linear regression analysis using Grafit 5. The rate constant for the disappearance of substrate obtained from these data was used to calculate the apparent kcat in each experiment.
Coupled Assay of Tyl1a, TylB with CDP-4-keto-6-deoxy-D-glucose (26)
The reaction mixture (100 μL), which was incubated at 25 °C, contained 35 μM Tyl1a, 35 μM TylB, 1 mM 26, 35 mM L-glutamate, and 175 μM PLP in 50 mM KH2PO4 buffer, pH 7.5. Aliquots were removed at various time points and subjected to HPLC analysis as described for the Tyl1a activity assays. The retention time of the product, CDP-3-amino-3,6-dideoxy-D-glucose (28), was 11.4 min. For isolation and MS characterization of the TylB product (28), 1.8 mM CDP-4-keto-6-deoxy-D-glucose (26) was incubated with 35 μM Tyl1a, 35 μM TylB, 175 μM PLP, and 35 mM L-glutamate in 100 μL of 50 mM KH2PO4 buffer, pH 7.5. The reaction mixture was incubated at 25 °C for 48 h and enzymes were removed by filtration using a Microcon YM-10 at 4 °C. The sample was then further purified using a semi-preparative Dionex CarboPac PA-1 column and a gradient elution program identical to that used for the analytical Dionex CarboPac PA-1 column, with a flow rate of 5 mL/min. Compound 28, which was eluted at 11.1 min, was collected manually, lyophilized to dryness and resuspended in water to a concentration of 0.1 mg/mL. High resolution ESI-MS of 28: calculated for C15H25N4O14P2 (M − H) 547.0843, found 547.0841. HPLC analysis showed 46% conversion of 26 to 28, with the remaining 54% consisting of 26 and 27.
Results
Purification and Characterization of Tyl1a
The tyl1a gene (33) was amplified, cloned, and overexpressed to give N-terminal His6-tagged Tyl1a. Production of soluble protein was quite efficient, with 275 mg of Tyl1a obtained from a 6 L culture after the Ni-NTA chromatographic step. Further purification by FPLC using a Mono Q column improved the purity to >95% as assessed by SDS-PAGE. Subunit molecular mass was estimated to be 19 kDa based on SDS-PAGE analysis (Figure 1), which agrees well with the calculated mass of 18,806 Da for the N-terminal methionine-cleaved His6-tagged Tyl1a. Thrombin cleavage was also carried out to generate Tyl1a without the N-terminal His6-tag for comparative kinetic studies. Gel filtration analysis revealed a native molecular mass of 31.0 kDa for Tyl1a, suggesting a homodimeric structure in solution.
Figure 1.
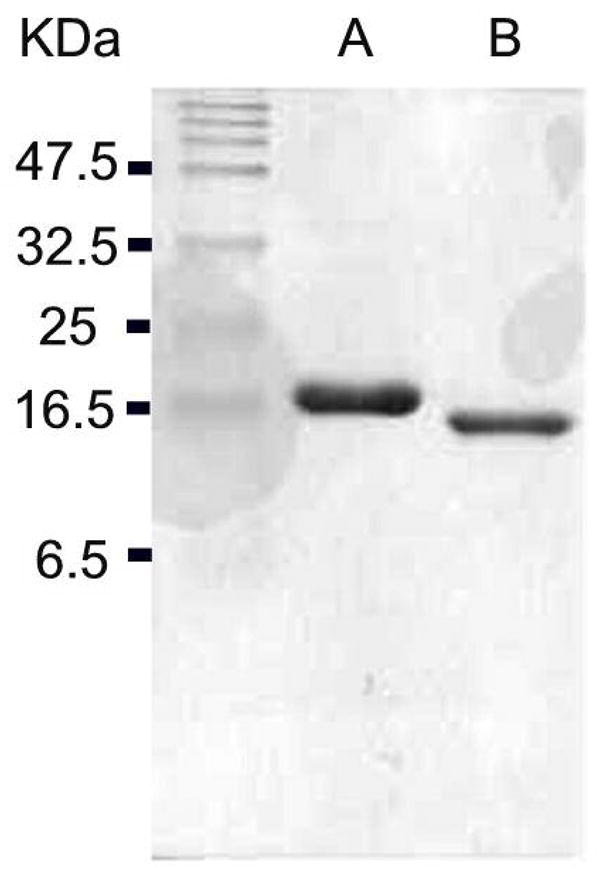
SDS-PAGE gel (18%) of (A) purified N-His6-tagged Tyl1a, and (B) purified Tyl1a with His6-tag removed by thrombin cleavage.
Catalytic Properties of Tyl1a
Because Tyl1a exhibits modest sequence identity (34% identity, 52% similarity) with FdtA, which catalyzes the conversion of TDP-4-keto-6-deoxy-D-glucose (6) to TDP-3-keto-6-deoxy-D-galactose (18) (22), we hypothesized that Tyl1a might also be a hexose 3,4-ketoisomerase catalyzing a similar reaction in the mycaminose pathway. The predicted substrate of Tyl1a, TDP-4-keto-6-deoxy-D-glucose (6), was therefore prepared enzymatically from thymidine and glucose-1-phosphate (4) using six enzymes in a two-stage, one-pot reaction (9). Subsequent HPLC analysis of an incubation mixture containing 6 and N-His6-tagged Tyl1a showed time-dependent depletion of substrate and the appearance of three new products (Figure 2A, traces a, b). Identical results were also observed using thrombin-cleaved Tyl1a under the same reaction conditions, indicating that the N-terminal His6-tag has no effect on Tyl1a activity. Thus, the N-His6-tagged Tyl1a was used for all subsequent work. Analysis of the full reaction time course revealed that 6 (retention time = 35.3 min) was first converted to an intermediate (retention time = 39.0 min), which was swiftly depleted with concomitant formation of TDP (retention time = 41.9 min) and a new product (retention time =1.8 min) (Figure 2B). Integration of the peak corresponding to the substrate and each of the three new peaks over time gave the traces shown in Figure 2C. Evidently, the immediate product having a retention time of 39.0 min is unstable, degrading to TDP and a new species with a retention time of 1.8 min. In an attempt to divert the transient intermediate to a more stable product, TylB, which catalyzes the subsequent step in the mycaminose pathway, was added to the incubation mixture. As expected, the addition of TylB along with PLP and L-glutamate to the reaction mixture led to a new product (retention time = 13.7 min), which co-eluted with the chemically synthesized TDP-3-amino-3,6-dideoxy-D-glucose (8) (Figure 2A, traces c, d). The identity of this new product was further confirmed to be 8 by high resolution ESI mass spectrometry. Clearly, the unstable intermediate generated in the Tyl1a reaction is TDP-3-keto-6-deoxy-D-glucose (7), which is converted to 8 in the presence of TylB.
Figure 2.

(A) HPLC traces showing product formation in the Tyl1a and TylB reactions: (a) incubation mixture containing TDP-4-keto-6-deoxy-D-glucose (6, 1 mM) in 50 mM KH2PO4 buffer (pH 7.5) without Tyl1a; (b) incubation mixture containing 6 (1 mM) and Tyl1a (2.85 μM) in the same phosphate buffer as in (a); (c) incubation mixture containing 6 (1 mM) and Tyl1a (28.5 μM) in the presence of TylB (10 μM), PLP (50 μM), and L-glutamate (10 mM), in the same phosphate buffer as in (a). Note that TMP and TDP peaks are not visible due to the adjustment of the scaling to keep the strong peaks of 8 and 19 in scale; (d) chemically synthesized TDP-3-amino-3,6-dideoxy-D-glucose (8). (B) HPLC traces of Tyl1a reaction time course study (2 – 120 min) using 1 mM 6 and 2.85 μM Tyl1a in 50 mM KH2PO4 buffer (pH 7.5). (C) Time course of Tyl1a (2.85 μM)-catalyzed reaction using 6 (1 mM) as substrate. Integrations of the HPLC peaks of substrate, product, and degradation products were plotted versus time. (○) represents 6, (●) 7, (■) 19, and (□) TDP. (See Experimental Procedures for details).
In situ 1H NMR Analysis of Tyl1a Reaction and Identification of Reaction Products
In order to directly characterize the unstable Tyl1a reaction product, in situ 1H NMR analysis was performed. In this experiment, TDP-4-keto-6-deoxy-D-glucose (6) was incubated with glycerol-free Tyl1a in an NMR tube, and the reaction was monitored at 5-min intervals for 150 min. A stack plot of the resulting spectra in which 6 μM Tyl1a was used is shown in Figure 3. Disappearance of the proton signals for 6 is seen along with the appearance of a new set of signals for the Tyl1a product. These signals reached a maximum intensity at about 50 min, and then diminished. A third set of signals corresponding to the degradation product also appeared after a short lag. Comparison of these spectra to those of 6 and the chemoenzymatically synthesized 7 (18) enabled assignment of the 1H NMR signals for 7. Thus, the Tyl1a product is indeed TDP-3-keto-6-deoxy-D-glucose (7) which confirms that Tyl1a is a TDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase. Signals corresponding to the degradation product were also identified. The chemical shifts and coupling constants of these signals are consistent with the structure of (2R,3R)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran (19), which is likely formed by C-2 deprotonation of 7 followed by elimination of TDP to give the 1,2-unsaturated ketone (Scheme 3). High resolution CI-MS analysis of the 1.8 min peak collected from the HPLC assay confirms the assigned structure.
Figure 3.
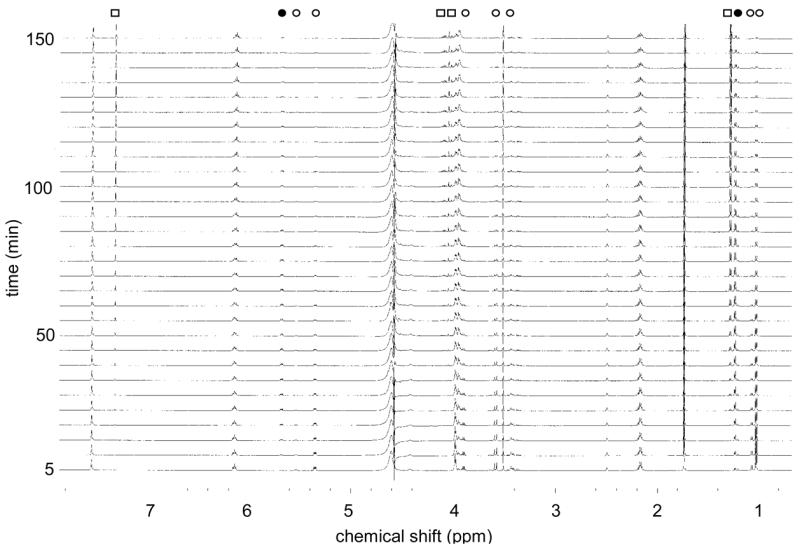
1H NMR stack plot of Tyl1a reaction (10 mM 6, 6 μM Tyl1a in 50 mM KH2PO4 buffer, pH 7.5) monitored over 150 min. The signals correspond to each compound are labeled as (○) from 6, (●) from 7, and (□) from 19 (See Experimental Procedures for details).
Scheme 3.

Conversion of 6 to 8 by Tyl1a and TylB as part of the biosynthesis of TDP-D-mycaminose. The proposed Tyl1a reaction intermediate 20, the degradation product 19, and the Tyl1a reaction product 7 are shown.
Determination of Kinetic Parameters for Tyl1a-Catalyzed Reaction
To determine the steady state kinetic parameters for the Tyl1a-catalyzed conversion of 6 to 7, a discontinuous HPLC assay was developed and performed. The plot of v0 (initial velocity) versus [S] (3 μM -1 mM) (Figure 4A) was fitted to the Michaelis-Menton equation by non-linear regression to yield the values of kcat of 6.1 min−1 and Km of 27 μM for 6. Data obtained by fitting the experimental data from the HPLC time course study (Figure 2C) and those obtained from the in situ NMR assay (Figure 4B) by non-linear regression using single or double exponential equations, were also used to calculate apparent kcat values. A kcat of ~ 7.0 min−1 was obtained from the HPLC data, and an apparent kcat of 2.4 ± 0.6 min−1 was obtained from the in situ 1H NMR assays. These values are in good agreement with the kcat value determined by the HPLC initial velocity assays.
Figure 4.
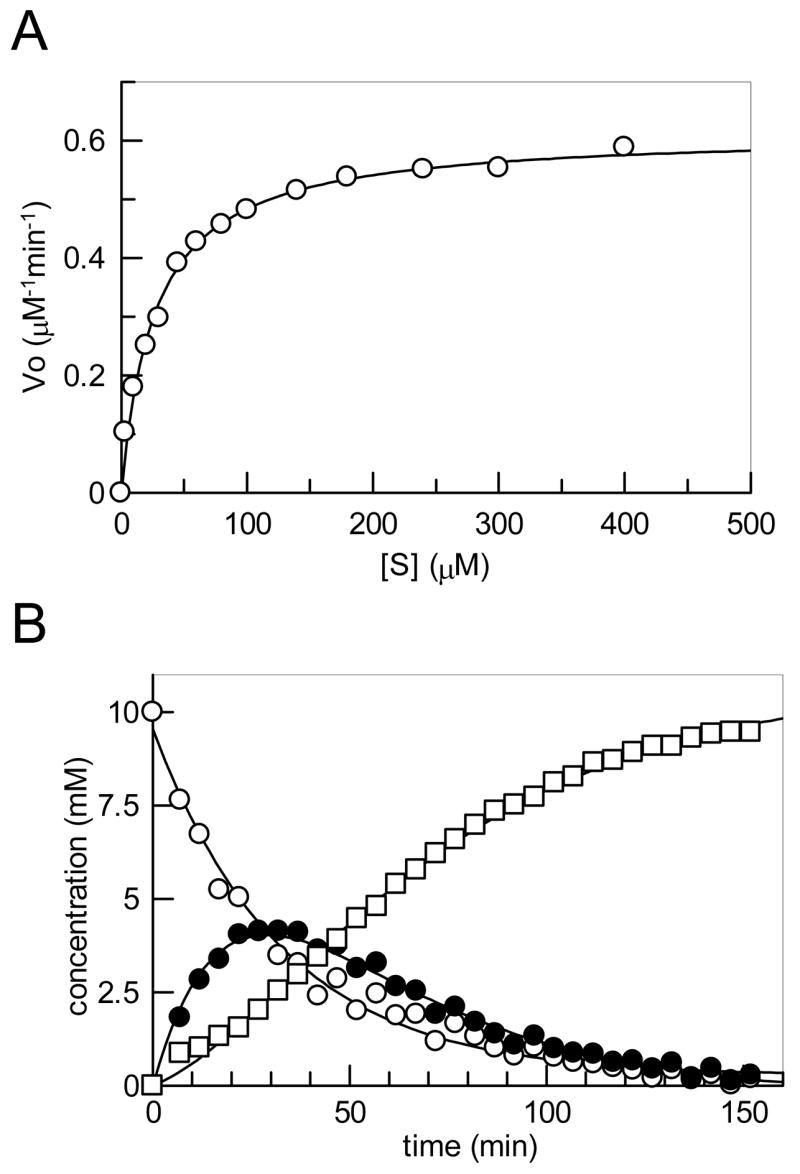
(A) Plot of v0 versus [S] determined via HPLC assay from which the steady state kinetic constants for the Tyl1a reaction were determined. (B) Plot of the integration of the 1H NMR signals (see Figure 3) of the 5-methyl group of 6 (○) and 7 (●), and that of 2-methyl group of 19 (□) during the in situ 1H NMR assay (10 mM 6 and 10 μM Tyl1a in 50 mM KH2PO4 buffer, pH 7.5) (See Experimental Procedures for details).
Tyl1a Substrate Specificity
To determine the substrate specificity of Tyl1a, the 2-deoxy analogue of 6, TDP-4-keto-2,6-dideoxy-D-glucose (22), and the CDP version of 6, CDP-4-keto-6-deoxy-D-glucose (26), were tested as possible substrates. Compound 22 was prepared enzymatically from TDP-4-keto-6-deoxy-D-glucose (6) using TylX3 (27) from the mycarose biosynthetic pathway of S. fradiae, and SpnN (28) from the forosamine biosynthetic pathway of S. spinosa, as the catalysts (Scheme 4). HPLC analysis of an incubation mixture containing 22 (1 mM) and Tyl1a (2.85 μM) showed time-dependent disappearance of the substrate peak at 33.4 min, accumulation of a small amount of a new peak at 37.0 min, and the tandem formation of TDP and another new peak at 1.7 min (Figure 5, trace a, b). Based on the retention times and confirmed identities of products from the assay of Tyl1a with 6, the peaks at 37.0 and 1.7 min likely correspond to TDP-3-keto-2,6-dideoxy-D-glucose (23) and its degradation product (2R,3R)-2-methyl-3-hydroxy-4-keto-2,3-dihydropyran (24), respectively (Scheme 4). While no spectral data was collected for 23 due to its low yield and instability, high resolution CI-MS data of the 1.7 min peak is consistent with the assigned structure of 24. By plotting the peak integration of substrate, product, and degradation products of this reaction versus time (Figure 6), it was found that 23 is more prone to degradation than 7, as less of it accumulates during the course of the reaction, and TDP and 24 are formed significantly more rapidly. By fitting the substrate depletion data from this time course to a single exponential equation, the apparent kcat for the conversion of 22 to 23 by Tyl1a was estimated to be 7.9 min−1 (Figure 6). These analyses indicate that 22 and 6 are comparable substrates for Tyl1a.
Figure 5.
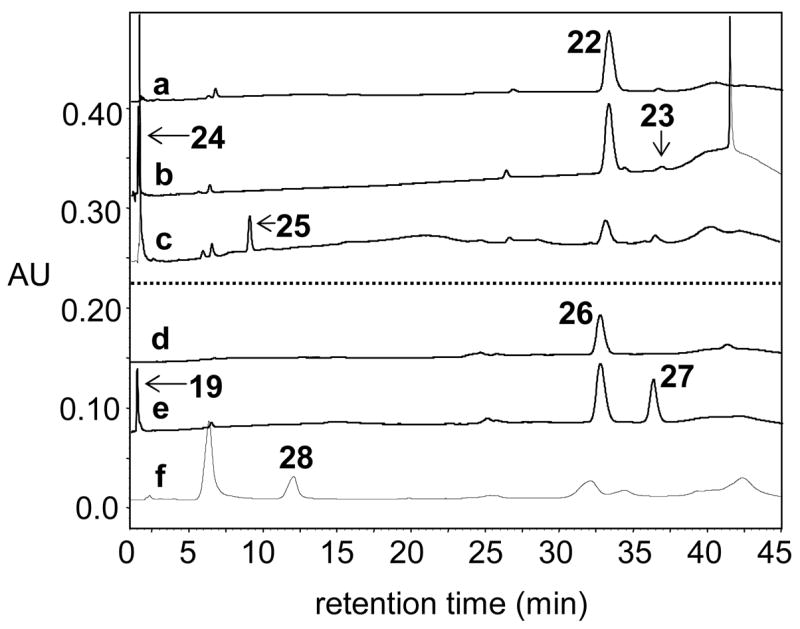
HPLC traces showing conversion of alternate substrates 22 and 26 by Tyl1a and TylB: (a) 1 mM 22 without Tyl1a, (b) 1 mM 22 + 2.85 μM Tyl1a, (c) 1 mM 22 + 2.85 μM Tyl1a, 28.5 μM TylB, 142.5 μM PLP, and 28.5 mM L-glutamate, (d) 1 mM 26 without Tyl1a, (e) 1 mM 26 + 100 μM Tyl1a, (f) 1 mM 26 + 35 μM Tyl1a, 35 μM TylB, 175 μM PLP, and 35 mM L-glutamate.
Figure 6.
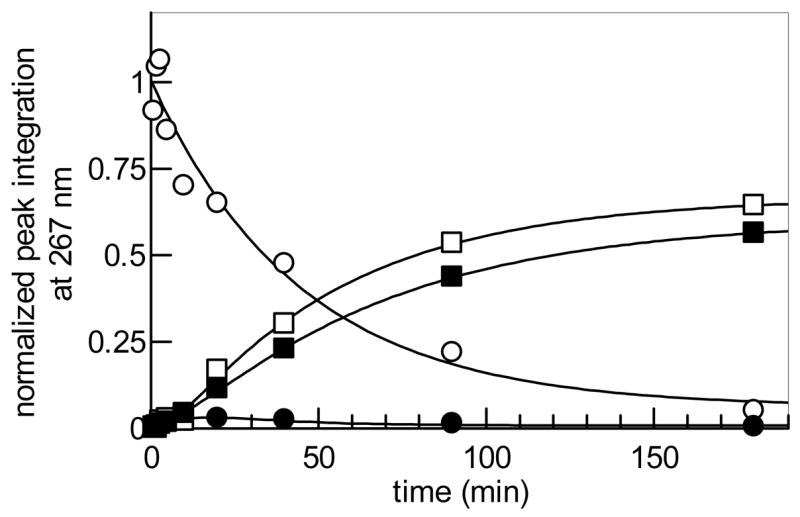
Time course of Tyl1a (2.85 μM)-catalyzed reaction using 22 (1 mM) as substrate. Integrations of the HPLC peaks of substrate, product, and degradation products were plotted versus time. (○) represents 22, (●) 23, (■) 24, and (□) TDP.
We next examined whether Tyl1a can accept CDP-4-keto-6-deoxy-D-glucose (26) as a substrate. This compound was generated enzymatically from CTP and glucose-1-phosphate (4) by action of α-D-glucose cytidylyltransferase (Ep) and CDP-D-glucose 4,6-dehydratase (Eod) from Yersinia pseudotuberculosis as described previously (Scheme 5) (30). The assay was initially performed using 1 mM 26 and 1 μM Tyl1a, and monitored by HPLC as before. Although formation of a new peak at 36.6 min was discernible, its formation was very slow, with only 2% conversion observed after a 24 h incubation. The assay was repeated with an increased amount of Tyl1a (100 μM), and the time-dependent disappearance of substrate at 33.1 min, the formation of product at 36.6 min, and the degradation of the product to TDP and a new product at 1.8 min were clearly noted, with product levels reaching a maximum of 30% conversion after 8 h (Figure 5, traces d, e). The pattern of product formation and degradation is similar to that observed for the Tyl1a reaction with its natural substrate, suggesting that the product in this case is likely CDP-3-keto-6-deoxy-D-glucose (27) and the degradation product seen at 1.8 min is the same pyran (19) formed in the reaction of Tyl1a with 6. By fitting the substrate depletion data from this time course to a single exponential, the apparent kobs value for the conversion of 26 to 27 by Tyl1a under these conditions was calculated to be 0.015 min−1 (Figure 7). Thus, Tyl1a appears to also be able to catalyze a 3,4-ketoisomerization of 26, although the efficiency of conversion is low, requiring a high concentration of enzyme and a long incubation time to achieve significant turnover.
Figure 7.
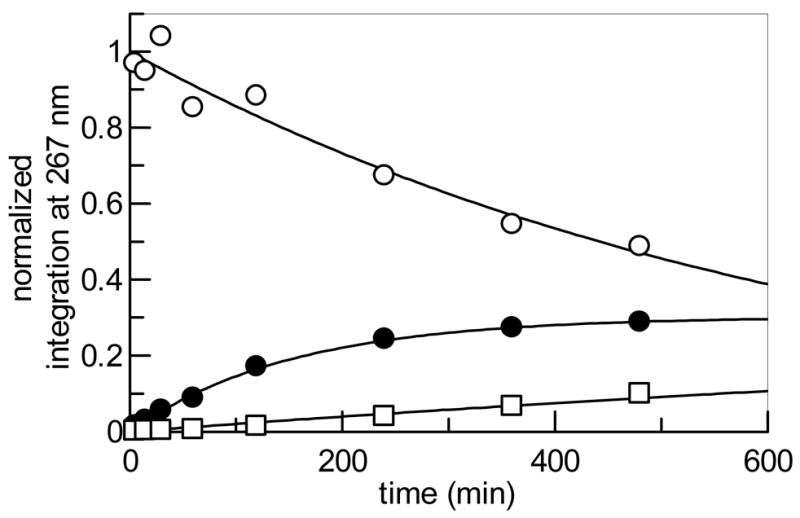
Time course of Tyl1a (100 μM)-catalyzed reaction using 26 (1 mM) as substrate. Integrations of the HPLC peaks of substrate, product, and degradation products were plotted versus time. (○) represents 26, (●) 27, and (□) 19.
TylB Substrate Specificity
Incubation of 22 with Tyl1a (2.85 μM) and TylB (28.5 μM) was also carried out to test whether TylB could accept 23 as a substrate. As expected, HPLC analysis of the incubation mixture showed substrate depletion as well as formation of the degradation products, TDP and 24. Interestingly, the time-dependent formation of a new peak at 9.5 min was also observed. The conversion of 22 to this new species was less than 2% as estimated from peak integration. Inclusion of less Tyl1a (1 μM) and more TylB (50 μM) along with a longer incubation time (12 h) resulted in a 15% overall conversion (Figure 5, trace c). In view of the effect of the increased TylB concentration, this new product is likely TDP-3-amino-2,3,6-trideoxy-D-glucose (25). High resolution ESI-MS data of this product purified by HPLC is consistent with the assigned structure (25). These results strongly suggest that TylB is capable of converting 23 to 25 in spite of the inherent instability of 23.
Encouraged by the observation that Tyl1a was able to convert 23 to 25, we subsequently incubated CDP-4-keto-6-deoxy-D-glucose (26) (1 mM) with Tyl1a (35 μM) and TylB (35 μM) to test the ability of TylB to accept 27 as a substrate. As expected, we observed slow substrate depletion and concomitant formation of Tyl1a product 27. However, we also observed formation of a new peak at 11.4 min. Incubation of the reaction mixture for 32 h led to 34% conversion of the starting material to this new product (Figure 5, trace f). Based on the dependence of product formation on the presence of TylB, this compound is likely to be CDP-3-amino-3,6-dideoxy-D-glucose (28). High resolution ESI-MS data of this product purified by HPLC is consistent with the assigned structure (28). These results strongly suggest that TylB is capable of converting 27 to 28.
Discussion
At the time when the function of FdtA from A. thermoaerophilus was first verified, less than 10 ORFs encoding homologous proteins had been identified. There are currently at least 65 ORFs exhibiting homology to fdtA in the NCBI database, the vast majority of which were uncovered as part of whole genome sequencing projects. These ORFs exist exclusively in bacteria, and are often clustered with genes proposed to be involved in outer membrane polysaccharide biosynthesis. A significant portion (18%) of these homologues are found to encode the C-terminal domains of putative bifunctional sugar ketoisomerase/N-acetyltransferases. One example of this type of bifunctional enzyme, WxcM from Xanthomonas campestris pv. campestris, has been shown through genetic studies to be involved in lipopolysaccharide (LPS) biosynthesis (34). Interestingly, tyl1a and the Spi seq 25 gene from the spiramycin biosynthetic cluster of Streptomyces ambofaciens are the only two homologues found in natural product biosynthetic gene clusters. The structure of spiramycin and the organization of its biosynthetic cluster bear significant similarity to those of tylosin. Since a mycaminose moiety is also present in the structure of spiramycin, the protein encoded by Spi seq 25 likely serves an analogous role to Tyl1a in mycaminose formation in S. ambofaciens. It is worth noting that most fdtA/tyl1a homologous are found in close proximity to genes encoding sugar aminotransferases, with the two often being co-transcribed. In view of the instability of the Tyl1a product, this close linkage between 3,4-ketoisomerase and aminotransferase may be advantageous, allowing coordinate expression of these two genes in order to minimize degradation of the unstable 3-keto sugar product.
The mechanism for the 3,4-ketoisomerization catalyzed by both Tyl1a and FdtA could proceed with deprotonation at C-3 of 6 to form an enediol (or enediolate) intermediate (i.e., 20 in Scheme 3) followed by reprotonation at C-4 to give the 3-keto product (7 or 18). Protein fold and structure analysis of Tyl1a and FdtA using the Phyre program predicts that these ketoisomerases belong to the RmlC-like cupin superfamily (35). Interestingly, RmlC, the TDP-4-keto-6-deoxy-D-glucose-3,5-epimerase involved in L-rhamnose biosynthesis, is a dimeric protein which also processes 6. In fact, many members in this superfamily are NDP-4-ketohexose epimerases responsible for inversion of the C-3 and/or C-5 centers of their substrates. The epimerization catalyzed by these enzymes is thought to involve two sequential cycles of abstraction of the proton α to the keto group followed by reprotonation at the opposite face of the enolate intermediate (36). Thus, there are apparent parallels between the mechanism of RmlC and that proposed for Tyl1a. It is also interesting to note that Tyl1a and FdtA use the same substrate, TDP-4-keto-6-deoxy-D-glucose (6), yet form products 7 and 18 (Scheme 1), respectively, which are C-4 epimers of each other. This observation suggests that deoxysugar products with opposite C-4 stereochemistry can be produced by replacing Tyl1a with FdtA (or vice versa). Mechanistically, the stereospecificity of Tyl1a- and FdtA-catalyzed reactions may be determined by the position of the proton donating residue in the active site relative to C-4 of the sugar substrate. Further studies are required to examine the predicted mechanistic and structural similarities between Tyl1a and RmlC-like epimerases, and to decipher the molecular basis for the difference in stereospecificity of Tyl1a- and FdtA-catalyzed reactions.
The identification and in vitro characterization of Tyl1a and the successful reconstitution of the reactions by Tyl1a and TylB to convert 6 to 8 make possible the enzymatic preparation of TDP-3-amino-3,6-dideoxyhexoses, such as 8 and TDP-D-mycaminose (9). These sugars can be used in enzymatic synthesis of glycosylated natural products. The presence of 3-amino-2,3,6-trideoxyhexoses in a number of bioactive natural products prompted us to investigate the ability of Tyl1a and TylB to process 2-deoxysugar substrates. The only example of an NDP-3-amino-2,3,6-trideoxyhexose whose biosynthesis has been fully characterized is L-eremosamine (29) from the chloroeremomycin pathway of Amycolatopsis orientalis (37). This pathway contains a specific aminotransferase, EvaB, capable of acting on C-3 of the highly unstable intermediate TDP-3,4-diketo-2,6-dideoxy-D-glucose (21) to give TDP-3-amino-4-keto-2,3,6-trideoxy-D-glucose (30), which may serve as a general precursor for 3-amino-2,3,6-trdeoxyhexoses (Scheme 6, route A). In the case of L-eremosamine (29), compound 30 is further modified by a methyltransferase and an epimerase before reduction of the C-4 keto group.
Scheme 6.

Formation of TDP-L-eremosamine (29) by Route A and an alternative route to 3-amino-2,3,6-trideoxy sugars via Route B (21 → 22 → 23 → 25).
Having verified the function of Tyl1a, we envisioned that a pathway including a 3,4-ketoisomerization step may be an alternative biosynthetic route to 3-amino-2,3,6-trideoxyhexoses. This pathway, starting from 6, involves C-2 deoxygenation and subsequent C-3 ketoreduction to give 22, followed by 3,4-ketoisomerization and C-3 transamination catalyzed by homologues of Tyl1a and TylB, respectively, to give 25 (Scheme 6, route B). The feasibility of such a pathway could be determine by examining the ability of Tyl1a and TylB to act on 2-deoxysugar substrates. The fact that we were able to demonstrate the formation of 25 enzymatically from 6 via a 6 → 21 → 22 → 23 → 25 route in vitro suggests that this pathway is plausible. However, due to the instability of the 3-keto-2,6-dideoxy sugar 23, an excess of TylB relative to Tyl1a is required to yield appreciable amounts of 25. Since each gene cluster encoding production of 3-amino-2,3,6-trideoxyhexoses discovered thus far possesses an aminotransferase gene closely related to that encoding EvaB, it seems that nature has adopted a more efficient route to make 3-amino-2,3,6-trideoxyhexoses by evolving an aminotransferase that captures the unstable intermediate 21 (route A) rather than one that captures 23 (route B).
While most deoxysugars used in the biosynthesis of secondary metabolites are TDP-derivatives, deoxysugar structures bearing CDP-, GDP-, and UDP- groups are not uncommon. Although some studies have demonstrated the ability of the sugar thymidylyltransferase RmlA (Ep) from Salmonella enterica to use UTP (38), there are few studies that have assessed the ability of deoxysugar biosynthetic enzymes to use substrates with alternate nucleotides. Identification of deoxysugar biosynthetic enzymes having relaxed specificity with respect to the nucleoside portion of their NDP-sugar substrates would be useful for preparing NDP-activated sugars. Here, we demonstrated that Tyl1a is capable of turnover of 26, the CDP version of its natural substrate, but at a significantly reduced rate (about 400-fold slower than 6), requiring high concentrations of enzyme for significant product formation. However, large amounts of Tyl1a are readily obtainable so that in vitro synthesis of CDP-sugars involving a 3,4-ketoisomerization reaction in their biosynthesis may be feasible using Tyl1a. More extensive testing of deoxysugar biosynthetic enzymes for NDP promiscuity will be necessary to assess the synthetic feasibility of this approach. As the number of X-ray crystal structures of deoxysugar biosynthetic enzymes increases, more sophisticated approaches involving mutagenesis may be employed to generate NDP-promiscuous enzymes for use in construction of a variety of NDP-activated deoxysugars.
The work described herein has established the function of Tyl1a as the TDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase in the mycaminose biosynthetic pathway in S. fradiae. Tyl1a is only the second example of an enzyme from this newly discovered class to be characterized in vitro. The biochemical characterization of Tyl1a has significant implications for the correct functional assignment of its many uncharacterized homologues, and for future investigation of the structure and mechanism of this group of enzymes. The availability of Tyl1a and its demonstrated substrate flexibility are also important for the in vitro and in vivo preparation of a variety of NDP-deoxysugars and thus, the glycodiversification of natural products.
Acknowledgments
We thank Dr. Eugene Seno at Eli Lilly Research Laboratories for providing us with pSET552, from which Tyl1a and TylB were amplified. We also thank Steve Sorey at the NMR Core Facility in the Department of Chemistry and Biochemistry at The University of Texas at Austin for his help and expertise in conducting the in situ 1H NMR assays of Tyl1a, and Dr. Mehdi Moini and coworkers at the Mass Spectrometry Core Facility in the Department of Chemistry and Biochemistry at the University of Texas at Austin for mass analysis of samples.
This work was supported in part by the National Institutes of Health Grants (GM35906, 54346).
Abbreviations
- CDP
cytidine 5′-diphosphate
- CI-MS
chemical ionization mass spectrometry
- CTP
cytidine 5′-triphosphate
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediamine tetraacetic acid
- ESI-MS
electrospray ionization mass spectrometry
- FPLC
fast protein liquid chromatography
- GDP
guanidine 5′-diphosphate
- HPLC
high performance liquid chromatography
- IPTG
isopropyl β-D-thiogalactoside
- LB
Luria-Bertani
- NCBI
National Center for Biotechnology Information
- NDP
nucleotide 5′-diphosphate
- NMR
nuclear magnetic resonance
- ORF
open reading frame
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- PEP
phosphoenolpyruvate
- PKS
polyketide synthase
- PLP
pyridoxal 5′-phosphate
- PMSF
phenylmethylsulfonyl fluoride
- SAM
S-adenosylmethionine
- SDS
sodium dodecyl sulfate
- TDP
thymidine 5′-diphosphate
- TPCK
N-p-tosyl-L-phenylalanine
- TTP
thymidine 5′-triphosphate
- Tyl1a
TDP-4-keto-6-deoxy-D-glucose 3,4-ketoisomerase
- TylB
TDP-3-keto-6-deoxy-D-glucose 3-aminotransferase
- UDP
uridine 5′-diphosphate
- UTP
uridine 5′-triphosphate
References
- 1.Johnson DA, Liu H-w. Deoxysugars: occurrence, genetics, and mechanisms of biosynthesis. In: Barton DHR, Nakanishi K, editors. Comprehensive Natural Products Chemistry. Vol. 3. Elsevier; Amsterdam, New York: 1999. pp. 311–365. [Google Scholar]
- 2.Kren V, Martinkova L. Glycosides in medicine: “the role of glycosidic residue in biological activity”. Curr Med Chem. 2001;8:1303–1328. doi: 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- 3.Weymouth-Wilson AC. The role of carbohydrates in biologically active natural products. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 4.Hermann T. Drugs targeting the ribosome. Curr Opin Struct Biol. 2005;15:355–366. doi: 10.1016/j.sbi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Trefzer A, Salas JA, Bechthold A. Genes and enzymes involved in deoxysugar biosynthesis in bacteria. Nat Prod Rep. 1999;16:283–299. doi: 10.1039/a804431g. [DOI] [PubMed] [Google Scholar]
- 6.He X, Liu H-w. Formation of unusual sugars: mechanistic and biosynthetic applications. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 7.Szu P-h, He X, Zhao L, Liu H-w. Biosynthesis of TDP-D-desosamine: identification of a strategy for C4 deoxygenation. Angew Chem. 2005;44:6742–6746. doi: 10.1002/anie.200501998. [DOI] [PubMed] [Google Scholar]
- 8.Melançon CEI, Yu W-l, Liu H-w. TDP-mycaminose biosynthetic pathway revised and conversion of desosamine pathway to mycaminose pathway with one gene. J Am Chem Soc. 2005;127:12240–12241. doi: 10.1021/ja053835o. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi H, Liu Y-n, Liu H-w. A two-stage one-pot enzymatic synthesis of TDP-L-mycarose from thymidine and glucose-1-phosphate. J Am Chem Soc. 2006;128:1432–1433. doi: 10.1021/ja0562144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z, Hong L, Liu H-w. Characterization of the protein encoded by spnR from the spinosyn gene cluster of Saccharopolyspora spinosa: mechanistic implications for forosamine biosynthesis. J Am Chem Soc. 2005;127:7692–7693. doi: 10.1021/ja042702k. [DOI] [PubMed] [Google Scholar]
- 11.Blanchard S, Thorson JS. Enzymatic tools for engineering natural product glycosylation. Curr Opin Chem Biol. 2006;10:263–271. doi: 10.1016/j.cbpa.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Salas JA, Mendez C. Biosynthesis pathways for deoxysugars in antibiotic-producing Actinomycetes: isolation, characterization and generation of novel glycosylated derivatives. J Mol Microbiol Biotechnol. 2005;9:77–85. doi: 10.1159/000088838. [DOI] [PubMed] [Google Scholar]
- 13.Hong JSJ, Park SH, Choi CY, Sohng JK, Yoon YJ. New olivosyl derivatives of methymycin/pikromycin from an engineered strain of Streptomyces venezuelae. FEMS Microbiol Lett. 2004;238:391–399. doi: 10.1016/j.femsle.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Bryskier AJ. In: Macrolides: chemistry, pharmacology, and clinical uses. Bryskier AJ, Butzler J-P, Neu HC, Tulkens PM, editors. Arnette Blackwell; Paris, Boston: 1993. [Google Scholar]
- 15.Baltz RH, Seno ET. Genetics of Streptomyces fradiae and tylosin biosynthesis. Annu Rev Microbiol. 1988;42:547–574. doi: 10.1146/annurev.mi.42.100188.002555. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Guo Z, Liu H-w. Expression, purification, and characterization of TylM1, an N, N-dimethyltransferase involved in the biosynthesis of mycaminose. J Am Chem Soc. 1998;120:9951–9952. [Google Scholar]
- 17.Cundliffe E, Bate N, Butler A, Fish S, Gandecha A, Merson-Davies L. The tylosin biosynthetic genes of Streptomyces fradiae. Antonie van Leeuwenhoek. 2001;79:229–234. doi: 10.1023/a:1012065300116. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Yeung SM, Que NLS, Muller T, Schmidt RR, Liu H-w. Expression, purification, and characterization of TylB, an aminotransferase involved in the biosynthesis of mycaminose. J Am Chem Soc. 1999;121:7166–7167. [Google Scholar]
- 19.Naudorf A, Klaffke W. Substrate specificity of native dTDP-D-glucose-4,6-dehydratase: chemo-enzymic syntheses of artificial and naturally occurring deoxy sugars. Carbohydr Res. 1996;285:141–150. doi: 10.1016/s0008-6215(96)90180-9. [DOI] [PubMed] [Google Scholar]
- 20.Stein A, Kula MR, Elling L, Versack S, Klaffke W. Synthesis of dTDP-6-deoxy-4-ketoglucose and analogs with native and recombinant dTDP-glucose-4,6-dehydratase. Angew Chem. 1995;34:1748–749. [Google Scholar]
- 21.Borisova SA, Zhao L, Sherman DH, Liu H-w. Biosynthesis of desoamine: construction of a new macrolide carrying a genetically designed sugar moiety. Org Lett. 1999;1:133–136. doi: 10.1021/ol9906007. [DOI] [PubMed] [Google Scholar]
- 22.Pfoestl A, Hofinger A, Kosma P, Messner P. Biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-D-galactose in Aneurinibacillus thermoaerophilus L420-91T. J Biol Chem. 2003;278:26410–26417. doi: 10.1074/jbc.M300858200. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Andrews P. Estimation of the molecular weights of proteins by Sephadex gel-filtration. Biochem J. 1964;91:222–233. doi: 10.1042/bj0910222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 27.Chen H, Agnihotri G, Guo Z, Que NLS, Chen XH, Liu H-w. Biosynthesis of mycarose: isolation and characterization of enzymes involved in the C-2 deoxygenation. J Am Chem Soc. 1999;121:8124–8125. [Google Scholar]
- 28.Waldron C, Matsushima P, Rosteck PR, Jr, Broughton MC, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH. Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa. Chem Biol. 2001;8:487–499. doi: 10.1016/s1074-5521(01)00029-1. [DOI] [PubMed] [Google Scholar]
- 29.Draeger G, Park SH, Floss HG. Mechanism of the 2-deoxygenation step in the biosynthesis of the deoxyhexose moieties of the antibiotics granatacin and oleandomycin. J Am Chem Soc. 1999;121:2611–2612. [Google Scholar]
- 30.Chen XMH, Ploux O, Liu H-w. Biosynthesis of 3,6-dideoxyhexoses: in vivo and in vitro evidence for protein-protein interaction between CDP-6-deoxy-L-threo-D-glycero-4-hexulose-3-dehydrase (E1) and its reductase (E3) Biochemistry. 1996;35:16412–16420. doi: 10.1021/bi961921i. [DOI] [PubMed] [Google Scholar]
- 31.Thorson JS, Lo SF, Ploux O, He X, Liu H-w. Studies of the biosynthesis of 3,6-dideoxyhexoses: molecular cloning and characterization of the asc (ascarylose) region from Yersinia pseudotuberculosis serogroup VA. J Bacteriol. 1994;176:5483–5493. doi: 10.1128/jb.176.17.5483-5493.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thorson JS, Kelly TM, Liu H-w. Cloning, sequencing, and overexpression in Escherichia coli of the α–D-glucose-1-phosphate cytidylyltransferase gene isolated from Yersinia pseudotuberculosis. J Bacteriol. 1994;176:1840–1849. doi: 10.1128/jb.176.7.1840-1849.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merson-Davies LA, Cundliffe E. Analysis of five tylosin biosynthetic genes from the tylIBA region of the Streptomyces fradiae genome. Mol Microbiol. 1994;13:349–355. doi: 10.1111/j.1365-2958.1994.tb00428.x. [DOI] [PubMed] [Google Scholar]
- 34.Vorholter FJ, Niehaus K, Puhler A. Lipopolysaccharide biosynthesis in Xanthomonas campestris pv. campestris: a cluster of 15 genes is involved in the biosynthesis of the LPS O-antigen and the LPS core. Mol Genet Genomics. 2000;266:79–95. doi: 10.1007/s004380100521. [DOI] [PubMed] [Google Scholar]
- 35.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 36.Jakimowicz P, Tello M, Freel Meyers CL, Walsh CT, Buttner MJ, Field RA, Lawson DM. The 1.6-A resolution crystal structure of NovW: a 4-keto-6-deoxy sugar epimerase from the novobiocin biosynthetic gene cluster of Streptomyces spheroides. Proteins: Struct, Funct, Bioinf. 2006;63:261–265. doi: 10.1002/prot.20818. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Thomas MG, Hubbard BK, Losey HC, Walsh CT, Burkart MG. Deoxysugars in glycopeptide antibiotics: enzymatic synthesis of TDP-L-epivancosmaine in chloroeremomycin biosynthesis. Proc Natl Acad Sci U S A. 2000;97:11942–11947. doi: 10.1073/pnas.210395097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang J, Biggins JB, Thorson JS. A general enzymatic method for the synthesis of natural and “unnatural” UDP- and TDP-nucleotide sugars. J Am Chem Soc. 2000;122:6803–6804. [Google Scholar]