

As part of a target-oriented synthetic study, we were interested in developing selective syntheses of linear- and cross-conjugated dienones via aldol cyclizations of diketones (eq 1). Methods for the regioselective aldol reaction of such diketones are scarce, especially in cases where the steric environments of the two carbonyl groups are very similar.1 1,5-Diketones such as 1 were of interest because they are readily prepared via the phosphine-catalyzed intramolecular vinylogous Morita-Baylis-Hillman (MBH) cyclization of bis-α,β-unsaturated carbonyl compounds,2 a reaction that was developed simultaneously in the Krische group3 and in our laboratory4,5. During the course of our studies of the vinylogous MBH reaction, competitive intramolecular aldol cyclizations were observed for vinylogous MBH products 1 bearing enolizable carbonyl units when these reactions were performed in protic solvents such as t-Amyl alcohol.4a This observation is consistent with the notion that the aldol reactions are catalyzed by alkoxide generated via deprotonation of alcoholic solvent by the zwitterionic phosphonium enolate intermediates.6 During efforts to optimize this tandem MBH/aldolization process, we discovered and report herein remarkable and unprecedented regioselectivity in the aldol step, resulting in extremely high selectivity for the less stable cross-conjugated dienones. We also provide evidence for the involvement of the phosphonium unit of the phosphine-enone Michael adduct (e.g., 6) in controlling the regiochemistry of these reactions.
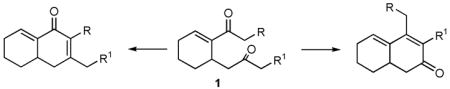 |
(1) |
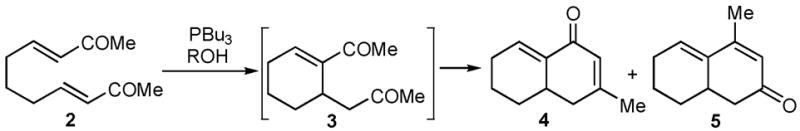
Symmetrical bisenone 2 was selected for initial reaction development studies (Table 1). The cyclization of 2 at room temperature in the presence of 1 equiv of PBu3 in MeOH as the solvent produced regioisomeric aldol condensation products 4 and 5 with excellent selectivity (4 : 5 = 94 : 6) for the cross-conjugated isomer 4 (entry 1). The Bu3P loading can be decreased to 0.25 equiv if the reaction is performed at 60 °C, and the selectivity is only slightly lower (4 : 5 = 93 : 7, entry 2). Unfortunately, these products were contaminated with inseparable adducts of MeOH-Michael addition to 4 and 5.7 Use of t-AmylOH as the reaction solvent resulted in inefficient aldol condensation (entry 3), while use of i-PrOH gave a very clean, high yield (80%) of products, but with poor selectivity (4 : 5 = 71 : 29, entry 4). Remarkably, however, use of CF3CH2OH as solvent for the tandem reaction gave clean aldol condensation product with exclusive selectivity for 4; isomer 5 was undetected by 1H NMR analysis (entry 5). PMe3 can also be used to promote this reaction with identical selectivity to that obtained using PBu3 (entry 6).
Table 1.
Survey of Solvents for the Tandem Cyclizationa
 | |||||
|---|---|---|---|---|---|
| entry | Solvent | T(°C) | time (h) | % 4+5 | Ratio (4 : 5) |
| 1b | MeOH | 25 | 24 | 83c | 94 : 6 |
| 2 | MeOH | 60 | 10 | 82c | 93 : 7 |
| 3 | t-AmylOH | 60 | 48 | 10d | 90 : 10 |
| 4 | i-PrOH | 60 | 3 | 80 | 71 : 29 |
| 5b | CF3CH2OH | 60 | 24 | 80 | >99 : 1 |
| 6e | CF3CH2OH | 60 | 22 | 76 | >99 : 1 |
Conditions: 0.25 equiv PBu3, 0.05 M 2.
1 equiv PBu3 was used.
Contaminated with 20% MeOH-adducts of 4 and 5.
Aldol product (19%) and 3 (42%) were also isolated.
1 equiv PMe3 was used.
The very high regioselectivity for 4 in these reactions was unexpected. A priori, we had anticipated that poor kinetic selectivity would occur in the aldol step due to the similarity in pKa and steric environment of the two ketone units in intermediate 3; moreover, the linearly-conjugated isomer 5 was expected to predominate if the reaction was subject to thermodynamic control.1 Therefore, it is noteworthy that we observe such high regioselectivity and that the reaction is highly selective for the less stable cross-conjugated isomer.8 We postulate that the high degree of selectivity derives from an interaction between the phosphonium unit and the adjacent carbonyl in intermediate 6. This would increase the acidity of the β-phosphonium-substituted methyl ketone such that it is deprotonated regioselectively by the alkoxide (6→7, eq 2).9
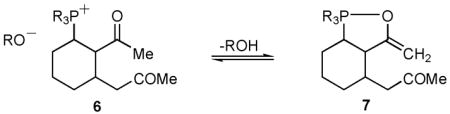 |
(2) |
Experimental evidence for this phosphine effect was obtained by subjecting MBH product 3 to a catalytic amount of CF3CH2ONa in CF3CH2OH (eq 3). No reaction was observed, indicating that CF3CH2ONa is not basic enough to deprotonate 3 in the absence of phosphine. While 3 undergoes efficient aldol cyclization when treated with i-PrONa (eq 4), the selectivity (4 : 5 = 18 : 82) is opposite to that observed using PBu3/i-PrOH (4 : 5 = 71 : 29). Furthermore, treatment of 3 with PBu3 in CF3CH2OH affords 4 exclusively (eq 5), demonstrating that 3 is a viable intermediate in the conversion of 2 to 4. These results support our proposal that 6 plays a key role in controlling the regioselectivity of the aldol step.
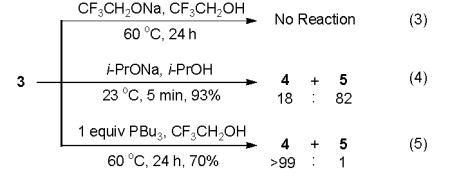 |
Other bisenone substrates cyclize to afford exclusively the cross-conjugated isomers (Table 2). Bisenone 8, bearing a shorter tether, cyclizes to dienone 9 in 76% yield (Table 2, entry 2). Substrate 10 can theoretically afford two vinylogous MBH adducts (c.f., 1), each of which in principle can cyclize to give two aldol regioisomers. Remarkably, 10 cyclized to afford only one out of four possible products (11) in 71% yield (entry 3). Evidently, the vinylogous MBH cyclization of 10 occurs with selectivity that is consistent with initial phosphine addition to the least hindered enone, while the selectivity of the aldol step is governed by the phosphine effect outlined above.
Table 2.
Substrate Scope of the Tandem Cyclization
| Entry | Substrate | product(s) | Yield(%)a |
|---|---|---|---|
| 1b |
2 |
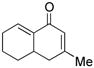
4 |
80 |
| 2b |
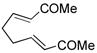
8 |
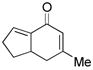
9 |
76 |
| 3b |
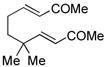
10 |
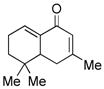
11 |
71 |
| 4c |
12 |
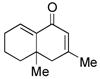
13 |
60d |
| 5c |
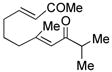
14 |
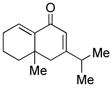
15 |
64d |
| 6c |
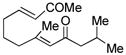
16 |
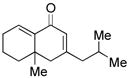
17 |
58d |
| 7c |
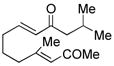
18 |
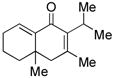
19 |
58d |
The only product isomers detected by 1H NMR analysis of the crude material are those indicated.
Method A: 1 equiv PMe3, 0.05 M substrate in CF3CH2OH, 60 °C.
Method B: 5 equiv PMe3, 0.05 M substrate in t- AmylOH, 80 °C.
The MBH intermediate (c.f., 1) was isolated in 7–8% yield.
Sterically differentiated bisenones 12, 14, 16, and 18 (Table 2, entries 7–10), which contain a hindered β,β-disubstituted enone, undergo efficient and selective MBH cyclization and subsequent aldol condensation to give the cross-conjugated products. Again, only one out of four possible products is formed. For these substrates, the optimal conditions involved use of 5 equiv of PMe3 in t-AmylOH at 80 °C; the MBH cyclization in these cases was unsuccessful using CF3CH2OH as solvent. In all cases, products bearing quaternary centers were generated in good yield (58–64%).
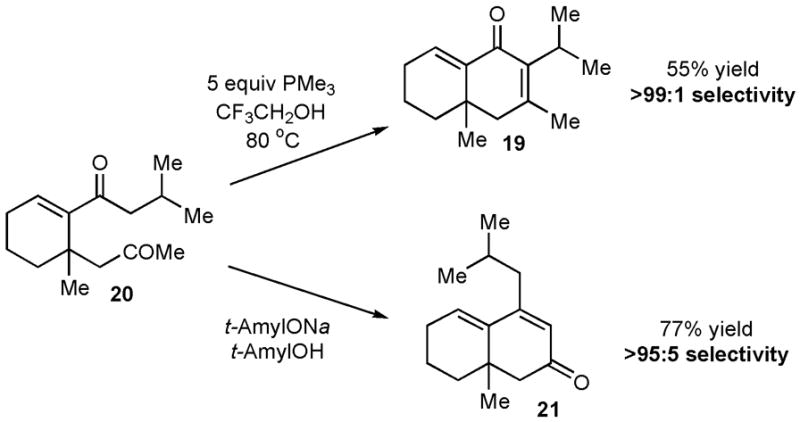
MBH product 20, which can be isolated from the cyclization of bisenone 18, undergoes phosphine-mediated aldol cyclization with the most hindered enolate serving as the nucleophile to generate isomer 19 (Scheme 1). Interestingly, a base-promoted aldol cyclization of 20 in the absence of phosphine results in a complete reversal of selectivity, and the linearly-conjugated isomer 21 is obtained in >95:5 selectivity. This example highlights the striking complementarity of the phosphine-mediated aldol condensation to a traditional aldol process.
Scheme 1.
Synthesis of Cross-Conjugated or Linear Dienones
In summary, we have discovered a phosphine-mediated intramolecular aldol cyclization of unsaturated diketones 1 that proceeds with extremely high levels of regioselectivity for the cross-conjugated bicyclic dienone products. The sense of regioselectivity observed in this reaction is unattainable using traditional aldol conditions, and is governed by the chemistry of the phosphine Michael adduct 6. Applications of this method to the synthesis of natural products will be reported in due course.
Supplementary Material
Supporting Information Available: Complete experimental details and spectroscopic data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health for financial support (GM26782) and a postdoctoral fellowship to R. K. T. (GM073325).
References
- 1.(a) For leading references on the regioselectivity of base-catalyzed aldol cyclizations of 1,4- and 1,5-diketones, see: Singh RK, McCurry PM. J Org Chem. 1974;39:2316. and Danishefsky S, Zimmer A. J Org Chem. 1976;41:4059. doi: 10.1021/jo00869a001. For a review: Heathcock CH. In: Comprehensive Organic Synthesis. Heathcock CH, editor. Vol. 2. Pergamon Press; New York: 1991. p. 181.
- 2.For selected reviews on the Morita-Baylis-Hillman reaction, see: Basavaiah D, Rao AJ, Satyanarayana T. Chem Rev. 2003;103:811. doi: 10.1021/cr010043d.Kim JN, Lee KY. Curr Org Chem. 2002;6:627.Langer P. Angew Chem Int Ed. 2000;39:3049. doi: 10.1002/1521-3773(20000901)39:17<3049::aid-anie3049>3.0.co;2-5.Ciganek E. Org React. 1997;51:201.Drewes SE, Roos GHP. Tetrahedron. 1988;44:4653. For a review of reactions involving phosphine organocatalysis: Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035. For selected examples of the related Rauhut-Currier reaction, see: Rauhut MM, Currier H. U.S. Patent, 3,074,999. :1963.McClure JDJ. U.S Patent 3,225,083. 1965Drewes SE, Emslie ND, Karodia N. Synthetic Commun. 1990;20:1915.Jenner G. Tetrahedron Lett. 2000;41:3091.
- 3.(a) Wang LC, Luis AL, Agapiou K, Jang HY, Krische MJ. J Am Chem Soc. 2002;124:2402. doi: 10.1021/ja0121686. [DOI] [PubMed] [Google Scholar]; (b) Agapiou K, Krische MJ. Org Lett. 2003;5:1737. doi: 10.1021/ol030035e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Frank SA, Mergott DJ, Roush WR. J Am Chem Soc. 2002;124:2404. doi: 10.1021/ja017123j. [DOI] [PubMed] [Google Scholar]; (b) Mergott DJ, Frank SA, Roush WR. Org Lett. 2002;4:3157. doi: 10.1021/ol026540d. [DOI] [PubMed] [Google Scholar]; (c) Methot JL, Roush WR. Org Lett. 2003;5:4223. doi: 10.1021/ol0357550. [DOI] [PubMed] [Google Scholar]
- 5.For a related study: Brown PM, Kappel N, Murphy PJ. Tetrahedron Lett. 2002;43:8707.
- 6.(a) Stewart IC, Bergman RG, Toste FD. J Am Chem Soc. 2003;125:8696. doi: 10.1021/ja035232n. [DOI] [PubMed] [Google Scholar]; (b) Inanaga J, Baba Y, Hanamoto T. Chem Lett. 1993:241. [Google Scholar]; (c) Trost BM, Li CJ. J Am Chem Soc. 1994;116:3167. [Google Scholar]; (d) Couturier M, Ménard F, Ragan JA, Riou M, Dauphin E, Anderson BM, Ghosh A, Dupont-Gaudet K, Girardin M. Org Lett. 2004;6:1857. doi: 10.1021/ol049392v. [DOI] [PubMed] [Google Scholar]
- 7.Solvent mixtures of THF, MeCN, or and MeOH significantly reduced CH2Cl2 the amount of MeOH Michael adducts (from 20% to 3%), but conversions and/or selectivities were lower in these solvent systems.
- 8.Isomers 4 and 5 do not equilibrate under the reaction conditions.
- 9.For known examples of structures related to 7, see: Bentrude WG, Johnson WD, Khan WA. J Am Chem Soc. 1972;94:923–932.Arbuzov BA, Zoroastrova VM, Tudrii GA, Fuzhenkova AV. Bull Acad Sci USSR Div Chem Sci. 1973;22:2513.Aksnes G, Frøyen P. Acta Chem Scand. 1968;22:2347.Ramirez F, Madan OP, Heller SR. J Am Chem Soc. 1965;87:731.Evans DA, Hurst KM, Takacs JM. J Am Chem Soc. 1978;100:3467.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Complete experimental details and spectroscopic data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.