

Abstract
The carboxy-terminal subdomains of the homodimeric EcoRV restriction endonuclease each bear a net +4 charge, and are positioned on the inner concave surface of the 50° DNA bend that is induced by the enzyme. A complete kinetic and structural analysis of a truncated EcoRV mutant lacking these domains was performed to assess the importance of this diffuse charge in facilitating DNA binding, bending, and cleavage. At the level of enzyme-DNA complex formation, the association rate for the dimeric mutant enzyme was sharply decreased by 103-fold, while the equilibrium dissociation constant was weakened by nearly 106-fold as compared with wild-type EcoRV. Thus, the C-terminal subdomains strongly stabilize the enzyme-DNA ground-state complex in which the DNA is known to be bent. Further, the extent of DNA bending as observed by fluorescence resonance energy transfer was also significantly decreased. The crystal structure of the truncated enzyme bound to DNA and calcium ions at 2.4 Å resolution reveals that the global fold is preserved, and suggests that a divalent metal ion crucial to catalysis is destabilized in the active site. This may explain the 100-fold decrease in the rate of metal-dependent phosphoryl transfer observed for the mutant. These results show that diffuse positive charge associated with the C-terminal subdomains of EcoRV plays a key role in DNA association, bending, and cleavage.
Keywords: DNA-protein interactions, electrostatics, restriction endonuclease, asymmetric phosphate neutralization
DNA bending is a common attribute of protein-nucleic acid complexes despite the expenditure of free energy required to achieve the bent state (1). Examples of DNA bending in biologically important contexts include the relatively smooth curvature in nucleosomes that facilitates packaging, and the often sharp and localized bending by transcription factors to juxtapose two segments of DNA that are far apart in sequence (2-6). DNA bending is also known to be important to DNA replication, repair, recombination and methylation (7-17).
Many DNA bending proteins are also site-specific enzymes, so that the bending event is coupled to selective enhancement of the reaction rate at cognate sites. For example, restriction enzymes enhance the rate of DNA strand scission at specific sites by an estimated 1015-fold (18). The specificity of these enzymes is exquisite - a single incorrect base pair in a 4 to 6 base-pair target site reduces kcat/KM by 106-fold or more (19-21). Furthermore, these enzymes must also bind to nonspecific sites, prior to the one- and three-dimensional search for the cognate sequence within a vast molar excess of nonspecific DNA (22-24). In this context, the extent of DNA bending at specific but not nonspecific sites likely provides a basis for sequence discrimination, because enzyme complexes at nonspecific sites provide insufficient interaction energy to drive the unfavorable bending transition. Thus, free energy liberated in the highly complementary specific complex can be used to drive the conformational change which assembles the active site, thereby lowering the energy of the transition state for cleavage. In contrast, the incomplete conformational change associated with binding to nonspecific sites may result in the improper juxtaposition of reactive moieties for the subsequent catalytic steps (25, 26).
EcoRV restriction endonuclease cleaves the phosphodiester backbone of DNA at the center TA step of its recognition sequence 5’-GATATC (27, 28). For some years the enzyme has served as an important model system to explore the importance of DNA bending in specificity and catalysis. EcoRV sharply bends its specific DNA site by approximately 50° directly at the center TA step (29), helping to facilitate proper juxtaposition among the scissile phosphate, the catalytic side chains Asp90, Asp74 and Lys92, and divalent metal ions. DNA bending also produces a severe narrowing of the major groove at the center TA step, preventing the enzyme functional groups from penetrating to make discriminating hydrogen bonds with the bases. Instead, the center step is recognized primarily by indirect readout (30). The operation of indirect readout as a mechanism that underlies specificity provides another distinctive feature driving the sustained interest in EcoRV as a model system.
Comparisons among a number of EcoRV structures bound to specific DNA reveal that, as the bend angle of the DNA increases, more surface area is buried at the protein-nucleic acid interface (31). In this analysis, it was suggested that direct contacts between the DNA binding domains of EcoRV and DNA provide a force to facilitate DNA bending, via direct contacts with symmetrically disposed ribose sugars in the minor groove. However, in EcoRV electrostatic interactions on the concave surface of the DNA may also induce DNA bending into the major groove via asymmetric phosphate neutralization (32-36). By this mechanism, appropriately positioned positive charge along one face of the duplex promotes bending of the DNA in that direction, by allowing compensation of the unfavorable closer juxtaposition of phosphates on the inside of the bend. In the proposed two-stage model for EcoRV bending, the DNA is first bent into the major groove by the operation of asymmetric phosphate neutralization, which could function even at nonspecific sites during the course of target localization. After the specific site is reached, formation of the complementary enzyme-DNA interface, including the discriminatory contacts with base-specific moieties, would drive formation of the complete sharp localized bend at the center TA step (31).
A significant proportion of the positively charged surface of EcoRV is found in the 29 amino acid C-terminal subdomains, which possess a total net +4 charge (Figure 1). Two basic amino acids from the subdomain, Arg221 and Arg226, make direct contacts with phosphates within and flanking the GATATC target site. The subdomains are located on the concave DNA major groove side, and are thus in an appropriate position to contribute to asymmetric phosphate neutralization both by direct phosphate contacts and by the influence of diffuse charge. To understand the role of the C-terminal subdomain in DNA binding, bending, and catalysis, we carried out a thorough analysis of the structural and kinetic properties of a mutant enzyme in which the subdomains were removed in their entirety (EcoRVΔC). Remarkably, the cleavage rate of the truncated enzyme is diminished by only 100-fold. However, both the rates of DNA association and dissociation, and the total extent of equilibrium DNA bending, were strongly affected. The X-ray crystal structure of EcoRVΔC bound to DNA and calcium ions showed no global perturbations in the quaternary or tertiary fold as compared with wild-type EcoRV, but suggested that divalent metal binding at the active site may be significantly destabilized. The experiments suggest a significant role for asymmetric phosphate neutralization in promoting the induced-fit transition of the EcoRV-DNA complex, at the level of both protein-DNA association and DNA bending.
Figure 1. Structure of EcoRV endonuclease.
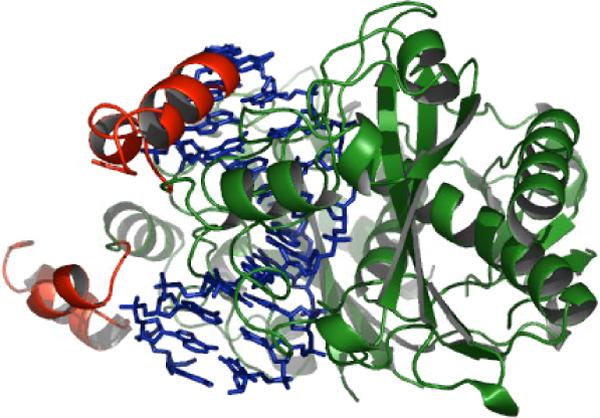
The DNA backbone trace is shown in blue. The carboxy-terminal subdomain of each monomer, comprising residues Arg221-Lys245, is shown in red. Within these subdomains, Arg221, Arg226, Lys229, Arg237, Arg242, Arg244 and Lys245 contribute positive charge, while Asp228 and Glu235, and the C-terminal carboxylate group likely contribute negative charge at physiological pH. Hence each subdomain carries a net +4 positive charge. Note the position of the C-terminal subdomains on the inner surface of the bent DNA.
EXPERIMENTAL PROCEDURES
Mutagenesis and preparation of enzyme and DNA
Mutagenesis was performed with the QuikChange approach (Stratagene) as previously described (37). Oligonucleotides for PCR were purchased from Integrated DNA Technologies (IDT; Coralville, IA); enzymes were purchased from New England Biolabs (Beverly, MA). The carboxy-terminal domain was deleted by including the stop codon UAA in place of Arg221, so that the C-terminal residue of EcoRVΔC is Glu220. This stop codon in E.coli is less susceptible to readthrough than is UGA (38). The entire coding region of the plasmid DNA was sequenced to verify that only the correct mutation was introduced.
Wild type and mutant EcoRV enzymes were purified by a two-column procedure (39). Enzyme preparations in 10% (v/v) glycerol, 0.4 M NaCl, 20 mM potassium phosphate (pH 7.5), 1 mM DTT were concentrated to approximately 4 mg/ml. Purified enzyme was analyzed by nanoelectrospray mass spectroscopy to ensure that the major product was the deletion mutant of the correct size, as also established by SDS-PAGE. Aliquots were flash frozen and stored at −80 °C. Enzyme was concentrated further for crystallization by precipitation with ammonium sulfate. Ammonium sulfate pellets were resuspended in 10 mM potassium phosphate (pH 7.5), 1.0 M NaCl, 1 mM EDTA, 1 mM DTT at approximately 10 mg/mL and dialyzed exhaustively in the same buffer before use.
DNA oligonucleotides for fluorescence were purchased from IDT. The DNA sequence used for fluorescence studies was AGAAGATATCTTGA. Carboxyfluorescein was attached to the 5’ end, and tetramethylrhodamine to the 5’ end of the complementary strand, through a C6 linker by IDT. Labeled oligonucleotides were gel purified by IDT, and HPLC purified in our laboratory on a Vydac C4 reversed-phase column (40). Single strands were annealed in an approximately 1:1 ratio, as determined by UV absorbance, by heating to 90 °C and cooling to room temperature over several hours. Annealed duplex was then purified from excess single strand on a nondenaturing 20% polyacrylamide gel. The band colored with both fluorescein and tetramethylrhodamine was cut out and crushed by squeezing through a 5 mL syringe. Several mLs of buffer containing 50 mM Tris (pH 7.5), 1 mM EDTA, and 100 mM NaCl (TEN buffer) were added and the DNA was allowed to diffuse out overnight with light shaking. The solution was then reduced to approximately 1 mL by butanol extraction before precipitating with ethanol. Pellets were resuspended at approximately 20 μM in TEN buffer.
DNA oligonucleotides for crystallization were purchased from the Midland Certified Reagant Company with the dimethoxyltrityl (DMT) group left on. The self-complementary DNA sequence AAAGATATCTT was used for crystallization. DNA was purified as described previously (31) and stored in 50 mM Tris (pH 7.5), 1 mM EDTA at −20°C.
Cleavage assay
Cleavage at the EcoRV site was assayed at 37°C under single-turnover conditions. Enzyme and 5’-end labeled DNA were combined at a molar ratio of 5:1, at concentrations of 2500 nM and 500 nM respectively. Reactions at higher concentrations of enzyme and DNA gave the same rates, indicating the DNA was fully bound. Enzyme and DNA were incubated separately with 10 mM MgCl2 in assay buffer (50 mM BTP (pH 7.5), 100 mM NaCl, 200 mg/mL BSA, 1 mM DTT) at 37 °C for 5 minutes. The reaction was initiated by adding the enzyme solution to the DNA. 4 μl aliquots were mixed with 12 μl of quench solution (8 M urea, 50 mM EDTA (pH 8.0)) at specific timepoints. Reaction products were separated on 8 M urea 20% polyacrylamide gels and quantitated by analysis on a Molecular Dynamics Storm 840 Phosphoimager. The fraction of DNA cleaved versus time was fit to a single exponential to give the observed rate for the cleavage step using the program Kaleidagraph.
Equilibrium FRET
Fluorescence emission scans were made on a Perkin-Elmer Luminescence Spectrometer (LS50B) at 22 °C. The buffer used contained 50 mM Tris (pH 7.5), 100 mM NaCl, and 10 mM CaCl2. Excitation and emission slit widths were 8 and 10 mm, respectively. Energy transfer was computed from the increase in acceptor fluorescence, as described previously (40). Fluorescein was excited at 485 nm and emission intensity collected from 500 to 650 nm. The fluorescence intensity of the acceptor tetramethylrhodamine due to energy transfer was determined by calculating the area under the curve from 575 to 585 nm and subtracting the contribution from fluorescein. The acceptor was then directly excited at 555 nm, and emission intensity collected from 570 to 650 nm. The area under the curve from 575 to 585 nm was again calculated. The value ratioA is equal to the intensity from energy transfer divided by the intensity when directly excited. FRET is then calculated via the following expression:
| (1) |
where εD(λ) and εA(λ) are the extinction coefficients at wavelength λ of the donor and acceptor, respectively (59).
For equilibrium titrations, increasing amounts of enzyme were added to 25 nM DNA. FRET efficiency versus substrate concentration was fit to a hyperbolic binding equation, since the approximation [S]free=[S]0 holds:
| (2) |
where Eo is the concentration of enzyme added, Kd is the equilibrium dissociation constant, Fo is the energy transfer for unbound DNA, and ΔF is the total change in energy transfer upon complete binding.
Stopped-flow FRET
Stopped flow fluorescence was carried out on an Applied Photophysics SX.18MV stopped flow reaction analyzer. Acceptor emission was monitored through a 570 nm cutoff filter. In all cases, at least 5 runs were averaged together and fit to a single exponential function to obtain the observed rate constant at each concentration. Plots of observed rate versus enzyme concentration were fit to the following equation:
| (3) |
This equation applies to second-order events with enzyme in significant excess of substrate (41). For each enzyme/substrate pair, the full range of kobs versus concentration was collected in triplicate, and each set was fit separately to eq 2. The mean and standard error of the three trials are reported.
The rate of DNA dissociation was also determined by a substrate trapping approach. 10 nM DNA was pre-incubated with excess and saturating enzyme in the presence of Ca2+ to allow for specific binding. This solution was mixed with an excess (>10-fold) of unlabeled cognate DNA in the stopped flow instrument and the dissociation monitored by the decrease in acceptor emission. Each dissociation rate was determined in triplicate; the mean and standard error of the three trials are reported.
Crystallization and X-ray structure determination
EcoRVΔC and DNA were mixed to give a final concentration of 8 mg/mL enzyme and a 1.5 molar excess of DNA. 1 μL of this solution was mixed with 1 μL of well solution on a siliconized coverslip placed over the well solution. Crystals were grown by vapor diffusion at ambient temperature. Well solutions contained 100 mM Hepes (pH 6.5), 250 mM NaCl, 8−12% PEG 4000.
Diffraction data was collected at the Stanford Synchrotron Radiation Laboratory. Data was processed using CCP4 (42) for integration and scaling and CNS (43) for molecular replacement and refinement. Phases were determined by molecular replacement using the wild-type structure (pdb 1EOP) as the model, omitting amino acids 221−245, all solvent, and DNA. Coordinates have been submitted to the Protein Data Bank as entry 2GE5.
The superposition of this structure with the structures of EcoRV determined previously (39) was performed with Insight II, using the backbone atoms of residues 4−9, 18−66, 71−77, 86−96, 104−137, 166−181 and 188−216 of each monomer for superposition of the DNA binding domains, and residues 21−26, 29−31 and 151−153 from both monomers for superposition of the dimerization interface. DNA helical parameters were determined using the program 3DNA (44). Electrostatic potentials were determined using the Adaptive Poisson-Bolztman Solver (45) run as a plugin for Pymol (Michael G. Lerner and Heather A. Carlson, personal communication) (46). All DNA, solvent, and metal ions were removed, and missing backbone and sidechain atoms were modeled into each structure. The charge of each atom at pH 7.5 was determined using the program pdb2pqr (47). All settings were left at their default values, including a protein dielectric of 20 and solvent dielectric of 80. Figures of the structure were made in Pymol.
RESULTS
DNA binding, bending, and cleavage by EcoRVΔC
Crystal structures of EcoRV endonuclease bound to DNA have revealed an extensive, complementary protein-DNA interface that includes interactions both within and adjacent to the 5’-GATATC-3’ target site (29, 48). A key feature of the specific enzyme-DNA complex is a sharp 50° DNA bend into the major groove, which is accomplished entirely by base-pair roll into the major groove at the center TA step of the recognition site. While the outer two base-pairs in each half-site are recognized by direct, specific hydrogen-bonding interactions with base-specific moieties in the major groove, no such hydrogen bonds are made at the center TA step. Together with base-analog studies, this observation points to the importance of indirect readout of the information at this position, and to the central role of DNA bending in this phenomenon (30, 48).
A set of extensive conformational changes in the EcoRV protein occurs concomitantly with the DNA bending, in a mutual induced-fit transition that is essential to assembly of the two active sites (29). Here, we address the importance of the C-terminal subdomains of the protein in facilitating this induced fit. A series of crystal structures of the EcoRV-DNA complexes in different lattice environments have suggested a two-stage mechanism to facilitate the DNA bending (31). First, flanking interactions made by the positively-charged C-terminal subdomains are proposed to facilitate an initial DNA bend in the correct direction, by virtue of their ability to selectively neutralize the negative charge on one face of the duplex. Such selective neutralization is possible because of the position of these domains adjacent to the concave inner side of the DNA bend (Figure 1). In the second proposed step, the partially bent DNA is driven into the precise conformation needed for phosphate cleavage by virtue of specific binding force exerted in the minor groove, on either side of the scissile phosphates.
The C-terminal subdomains of EcoRV feature seven positively charged residues together with two negatively charged residues, for a net charge of +5 (the charge is +4 when the C-terminal carboxylate is also considered; Figure 1). Two of the positively charged residues, Arg221 and Arg226, interact directly with phosphates. To investigate the role of this subdomain in facilitating the initial steps of DNA bending, we constructed a truncated enzyme in which 25 amino acids at the C-terminus of each subunit are deleted. The truncated enzyme was purified and studied by a variety of biophysical techniques previously developed for the study of the wild-type enzyme in its interaction with wild-type and modified DNA target sites (25, 30, 40). These approaches allow us to evaluate the effects of mutation on the microscopic rate constant for each step along the reaction pathway.
The magnesium-dependent rate of phosphodiester bond cleavage by EcoRVΔC was determined with a gel-based single turnover assay utilizing radiolabeled substrate DNA (Figure 2). This assay measures the rate of formation of product from free enzyme and substrate. At high concentrations of enzyme the early steps of the reaction (enzyme-DNA association and induced fit) do not limit the rate of product formation (see below). Therefore, this assay measures the rate of conversion of bound and bent DNA substrate to bound product on the surface of the enzyme. The rate of phosphodiester bond cleavage by EcoRVΔC is decreased by 100-fold compared with wild-type EcoRV (Table 1). While this decrease is significant, it is much smaller than the effects of a number of point mutations at residues in and around the active site, that play either direct or indirect roles in facilitating the bond-making and bond-breaking steps(49-52). Thus, the truncated enzyme, when assayed under conditions of binding saturation where the initial steps of association and induced-fit are bypassed, retains remarkably good activity as a catalyst.
Figure 2. Single-turnover kinetics.

The time course of the cleavage reaction for EcoRVΔC is depicted. Left: Denaturing polyacrylamide gel electrophoresis showing a reaction time course: the separation of 5’-end labeled 16-mer substrate strands (top bands; the two substrate strands run with slightly differing mobilities because of differing purine and pyrimidine content) from 9-mer and 7-mer cleavage products (bottom bands). Right: Plot of the extent of cleavage over time used to derive the first-order rate constant for cleavage (kchem; Table 1).
Table 1.
Kinetic and Thermodynamic Parameters of EcoRVΔC
| WT | ΔC | |
|---|---|---|
| KD | N/A | 381 ± 106 |
| (nM) | ||
| kon | 0.11 ± 0.02 | 7.5 ± 1.4 × 10−5 |
| (nM−1 s−1) | ||
| koff | 9.0 ± 0.4 × 10−5 | 5.6 ± 0.3 × 10−2a |
| (s−1) | 2.6 ± 0.3 × 10−2b | |
| koff/kon | 8.5 ± 1.6 × 10−4 | 750 ± 150a |
| (nM) | 350 ± 80b | |
| ΔFRETc | 0.157 ± 0.013 | 0.098 ± 0.016 |
| kchem | 0.60 ± 0.09 | 5.4 ± 0.2 × 10−3 |
| (s−1) |
Obtained from the y-intercept of concentration dependence
Obtained from substrate trapping
Change in FRET efficiency from unbound to saturated DNA
We next measured the equilibrium association constant of EcoRVΔC with specific DNA. This was accomplished by the use of a fluorescence resonance energy transfer (FRET) binding assay, in which the equilibrium was measured in the presence of calcium ions. Calcium promotes specific binding and bending of EcoRV to DNA, but inhibits cleavage, and has been shown to be an excellent analog of magnesium for the early steps of the reaction (40, 53). To determine the equilbrium dissociation constant of DNA with EcoRVΔC, steady-state emission spectra of a 14-mer duplex DNA substrate containing the specific EcoRV site were first measured in the presence and absence of enzyme (Figure 3A). These data revealed a much smaller decrease in donor emission for EcoRVΔC, indicating a smaller bend than is induced by wild-type EcoRV (see below). Next, fluorescently-labeled DNA was incubated with increasing amounts of EcoRVΔC enzyme, and the change in FRET was observed (Figure 3B). Strikingly, these data show that the equilibrium affinity for EcoRVΔC is reduced by nearly 106-fold relative to the wild-type enzyme (Table 1). The equilibrium dissociation constant for wild-type EcoRV cannot be directly determined by this technique, because of the very tight affinity. However, the picomolar binding constant derived from the ratio koff/kon (Table 1) is similar to equilibrium measurements made by gel mobility shift and competitive fluorescence anisotropy (53, 58). The substantially weakened affinity for DNA observed in EcoRVΔC demonstrates that the C-terminal domains play a strong role in stabilizing the ground state.
Figure 3. Equilibrium FRET titrations.
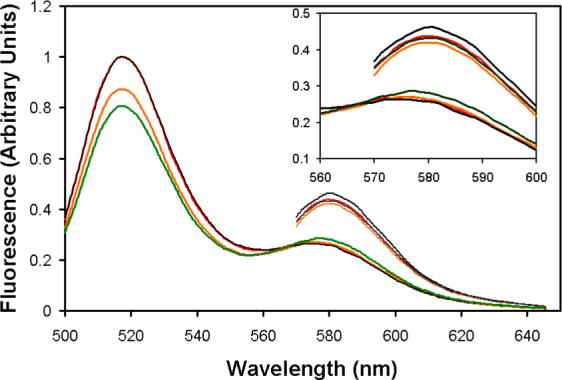
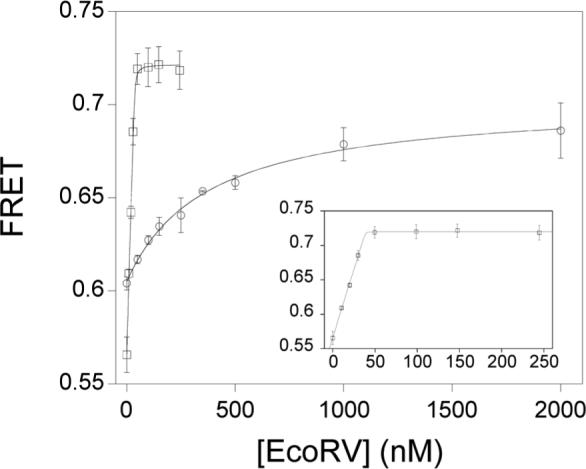
A) Steady-state emission scans. Excitations at 485 nm produce the 520 nm fluorescein emission peak, which is decreased by resonance energy transfer to tetramethylrhodamine. Excitations at 555 nm produce the 580 nm peak. Black trace: 25 nM doubly-labeled DNA; green trace: 25 nM doubly-labeled DNA in the presence of 50 nM wild-type EcoRV; red trace: 25 nM doubly-labeled DNA; orange trace: 25 nM doubly-labeled DNA in the presence of 1μM EcoRVΔC. The inset is an expansion of the scans around the 580 nm tetramethylrhodamine acceptor peak. B) A constant amount of labeled DNA was incubated with increasing concentrations of WT (squares) or EcoRVΔC (circles) and the change in FRET was monitored. Note that the asymptote for EcoRVΔC is substantially lower than for WT. Inset: Expansion of the ordinate for the wild-type enzyme showing stoichiometric binding. The concentration of the DNA used is 25 nM.
The fact that FRET could be used to monitor the binding equilibrium by EcoRVΔC shows that the DNA is bent in the ground state, as is the case in the wild-type EcoRV complex. In both cases, the experiment measures the total extent of bending averaged over all the DNA molecules. Since FRET is on an absolute scale, different equilibrium values determined in this experiment directly correspond to changes in the dye-to-dye distances in each complex. The identical substrate preparation was used for titrations of wild-type EcoRV and EcoRVΔC; thus, errors from incomplete labeling and other substrate-dependent factors, as well as variability in the orientation of the 5’-appended fluorescent dyes, can be neglected. In addition, our measurements of the variation in fluorescence intensity with emission wavelength show that the extent of quenching is identical in EcoRV and EcoRVΔC (data not shown). Therefore, any change that may occur in the local dielectric constant arising from removal of the subdomains does not influence the behavior of the fluorophores.
A significant decrease in the FRET change upon DNA binding to EcoRVΔC compared with wild-type EcoRV is observed (Figures 3A, 3B). This change is significantly greater than the experimental error in the measurements, despite the increased uncertainty in the EcoRVΔC endpoint due to the high concentrations of enzyme required to saturate DNA. The lower FRET value observed for EcoRVΔC could indicate that the bend angle has decreased in all complexes. Alternatively, there could be a distribution of complexes including some that are fully bent and others featuring little or no bending. Regardless, it appears clear that the C-terminal subdomains play an important role in facilitating the ability of the enzyme to induce the full 50° bend of the specific DNA site. Quantitative estimation of the extent to which the equilibrium DNA bending by EcoRVΔC is attenuated based on these data was not attempted, because of the high degree of uncertainty associated with establishing the precise positions of the fluorophores (40).
Next, the kinetics of DNA binding and bending were measured using the same fluorescence assay in a stopped-flow instrument. To measure the association of EcoRV and EcoRVΔC with cognate DNA, substrate was mixed with an excess (>5-fold) of enzyme, and the rate of change in FRET was monitored (Figure 4A). A concentration dependent phase was observed, the slope of which corresponds to the association rate (Figure 4B). For both wild-type EcoRV and EcoRVΔC, the concentration dependence of the observed rate of increase in FRET with enzyme in excess of DNA reveals a linear slope (Figures 4B, 4D). This indicates that the process being monitored is the second-order association step in both cases, and that the unimolecular bending step must be much faster than binding (40). Measurements of kobs for the wild-type reaction using singly-labeled DNA give identical association rates, further underlining the conclusion that the binding step is being monitored (40). Remarkably, in EcoRVΔC this association rate is reduced by over 103-fold compared with wild type (Table 1). Thus, the C-terminal domain plays an important role in the formation of the initial enzyme-DNA complex.
Figure 4. Association and dissociation kinetics of EcoRVΔC.

A) Upper left: Stopped flow time courses monitoring FRET. Varying concentrations of EcoRVΔC and labeled DNA were mixed and the change in acceptor fluorescence was monitored: red, blue, green, grey, and purple curves represent 200 nM, 400 nM, 600 nM, 800 nM and 1 μM EcoRVΔC, respectively. B) Upper right: Concentration dependence of the observed rates determined in (A). C) Lower left: Substrate trapping. Preincubated EcoRVΔC and labeled substrate were mixed with an excess of unlabeled DNA and the change in fluorescence was monitored. D) Lower right: Concentration dependence of the DNA bending rate for wild-type EcoRV. The intercept very near the origin reflects the very tight binding in this case.
The DNA dissociation rate was determined in a separate experiment by using a substrate trapping approach (Figure 4c). Enzyme was preincubated with labeled substrate, and an excess of unlabeled cognate DNA was subsequently added while monitoring FRET. Deletion of the C-terminal domain was found to increase the dissociation rate of DNA by 200-fold. For a single-step association process, the dissociation rate can also be determined from the y-intercept of the concentration dependence. By this measure, dissociation increases 500-fold. Systematic residuals are observed when each binding curve is fit to a single exponential function, suggesting that these processes may be biphasic. However, the amplitude of this second phase is extremely small, and its origin is not clear. A second phase may account for the discrepency between the dissociation rate determined from substrate trapping and that determined from the y-intercept of the concentration dependence, since the y-intercept is greater in a biphasic process.
Crystal structure of EcoRVΔC bound to cognate DNA
To gain further insight into the structural perturbations responsible for the decreased association and cleavage rates, the increased dissociation rate, and the decreased equilibrium bending, EcoRVΔC was crystallized bound to an 11-mer duplex oligonucleotide containing the cognate site 5’-GATATC-3’ in the presence of calcium ions (Table 2). Crystals formed in the space group P43212, in a unit cell which has not been previously characterized in any EcoRV X-ray structure. This represents the sixth distinct lattice environment in which the EcoRV-specific DNA complex has been crystallized, and only the second in which a ternary metal-bound complex is captured in the uncleaved state. Structure determination at 2.4 Å resolution by molecular replacement revealed that electron density is continuous across both scissile phosphates, indicating that the inclusion of calcium ions allowed an uncleaved complex to be trapped, as expected. Clear density was also observed for the backbone of the amino acids at the new C-termini of each chain, as well as for the Glu220 side-chain in one of the monomers.
Table 2.
Crystallographic data collection and refinement statistics
| EcoRVΔC/Cognate DNA/Ca2+ | |
|---|---|
| Space Group | P43212 |
| Cell Dimensions | a = b = 67.1 Å; c = 259.9Å α = β = γ = 90° |
| Resolution | 2.7 Å |
| # Observed Reflections | 28443 |
| Completeness (%) | 81.8 (45.1) |
| Average Intensity / σ | 8.6 (3.1) |
| Refinement range | 50.0 Å - 2.4 Å |
| Rcryst (%) | 22.6 |
| Rfree (%) | 27.4 |
| B-factor, Ca2+ ion (Å2) | 86 |
| # of waters | 25 |
| Rms bond lengths (Å) | 0.008 |
| Rms bond angles (°) | 1.23 |
Values in parentheses are for the 2.7 Å shell. However, data to 2.4 Å was included in refinement.
The following side chains did not have interpretable electron density and were modeled as alanine. Subunit A: N15, Q16, E57, K67, Q68, K85, K98, E99, R144, K145, S146, E155, K197, K203, E220. Subunit B: K17, S35, K38, E45, K54, E57, K98, E99, N100, T143, R144, K145, S146, S147, K149, N154, K197.
To analyze differences in conformation associated with the removal of the C-terminal subdomains, the structure of EcoRVΔC was superimposed on the previously determined wild-type EcoRV structure complexed with cognate DNA and calcium ions in the P1 lattice (pdb 1az0) (Figure 5a). No global rearrangements in conformation between the two complexes are observed. The root mean square deviations (r.m.s.d.) in position of the backbone atoms in the DNA-binding domains are 0.392 Å and 0.394 Å for the two monomeric subunits, within the coordinate error. Similarly, the dimerization interfaces of the wild-type and mutant complexes adopt identical conformations (r.m.s.d. of 0.251 Å). A small, roughly 1° reorientation of the DNA binding domains relative to each other is observed in EcoRVΔC, but this change is well within the variability observed among the many different structures of wild-type complexes (31), and is not attributable to the effects of removing the C-terminal subdomains. Similarly, the conformational parameters of the specific DNA bound in the wild-type and EcoRVΔC structures do not differ significantly. In particular, the base-pair roll into the major groove at the center TA step remains at approximately 50° in the truncation mutant, despite the observation that in solution, the equilibrium extent of bending is diminished (Figure 3; see Discussion).
Figure 5.

A) Superposition of wild type EcoRV (pdb 1az0) (enzyme in red, DNA in green) and EcoRVΔC (enzyme in blue, DNA in yellow). B) Active site superposition of wild type EcoRV (red) and EcoRVΔC (blue). Filled spheres show the positions of the calcium ion in the two structures. The side-chain of Glu45 is disordered in the wild-type structure used for comparison.
While no large conformational changes in the protein or DNA were observed, a small but significant rearrangement in the active site was detected (Figure 5b). In both wild-type and EcoRVΔC structures a single calcium ion is bound to one subunit, where it bridges between Asp74, Asp90, and the scissile phosphate. This position has been identified as site III in the wild-type complexes (54). However, in EcoRVΔC, this calcium ion is shifted in position by approximately 1.4 Å. The effect of the shift is to substantially lengthen the distances at which the nearby negatively-charged ligands are bound in the inner sphere. Inner-sphere distances of approximately 3 Å are found in the mutant active site, representing a significant weakening of the contacts. Further, only two waters are bound to the calcium ion instead of three (Figure 6). Finally, while the B-factor for the calcium ion at the equivalent position in the wild type structure is 29 Å2, in EcoRVΔC the B-factor increases to 86 Å2, suggesting significant destabilization in the new position. It is important to note that the pathway through which the C-terminal subdomain interactions stabilize the precise active site conformation required for optimal catalysis is not evident from the structural comparisons, and that the precise configuration of the metal-bound phosphate in the transition state is likely to require a further localized conformational change by which the phosphate is moved deeper into the binding cleft (54). The propagation of the effects of the C-terminal domain deletion may arise from small structural perturbations that are beyond the resolution of our structures, from changes in protein or DNA dynamics, from alterations in second-shell solvent structure, or for other reasons or a combination of these. Regardless of the mechanism, the destabilization of the calcium ion observed in EcoRVΔC points to the possibility that the 100-fold decrease in phosphoryl transfer rate has its origin in a destabilization of metal binding in the active site. Similar destabilization or repositioning of the active-site metal ions has previously been observed in other EcoRV mutants possessing decreased catalytic rates (39, 49). Because the global positioning of the DNA binding/catalytic domains is preserved in these variants as compared with wild-type EcoRV, it is unlikely that the altered positions of the metals arise from differences in crystal packing forces.
Figure 6.

Inner-sphere ligand distances for the active-site calcium ion bound in the wild-type (top) and EcoRVΔC (bottom) active sites. Crystallographic B-factors for the calcium ions are indicated.
DISCUSSION
Transient kinetic approaches have allowed a complete dissection of the role of the positively-charged carboxy-terminal subdomain of EcoRV endonuclease (amino acids 221−245) at each step along the catalytic pathway. This detailed analysis, using a combination of chemical-quench and fluorescence techniques, is essential to the critical evaluation of structure-based hypotheses regarding its possible function. The position of the subdomain in the context of the overall enzyme-DNA complex (Figure 1) suggested a role in the early stages of DNA bending (31). Another possibility was that distal contacts with DNA flanking regions might be crucial to transition-state stabilization, as was previously suggested for manganese ion-mediated flanking interactions in the minor groove (37). Evaluating such hypotheses clearly requires comparative evaluation of the relevant microscopic rate constants. By contrast, a mutational study in which variants are evaluated by steady-state kinetics provides only very general insight into the overall effect of a mutation. A C-terminal truncation of EcoRV has been constructed before, but in that study the C-terminal 30 residues (amino acids 216−245) were removed, and the enzyme was found to be inactive (52). The lack of reported activity may have been due to the relatively insensitive assays utilized. However, both Trp216 and Trp219, which were deleted in the earlier study, make apparently stabilizing hydrophobic interactions internal to the DNA-binding domains, suggesting that the larger truncation may have produced an enzyme that was unstable or unfolded in solution.
Electrostatic effects on enzyme-DNA association
An unusual aspect of the behavior of EcoRVΔC is the 1500-fold reduction in the second-order DNA association rate constant as compared with wild-type EcoRV (Table 1). This finding was unexpected because the association rates of macromolecular partners are unaffected by the stabilities of the final formed complexes. Thus, effects on association must arise from changes in the initial transient binary association that forms en route to assembly of the final ternary ground-state complex that also includes properly positioned divalent metal ions (55). In the transitory initial association of the enzyme with DNA, specific van der Waals contacts and hydrogen bonds prominent in the ground-state Michaelis complex have likely not yet been made. By contrast, electrostatic effects are probably significant, because the strength of the electrostatic force falls off only linearly with distance, and so is not as greatly affected by the absence of well-defined molecular complementarity.
Association rate constants for macromolecular complexes have been measured to be as high as 5 × 109 M−1s−1 in the case of the barnase-barstar interaction, and similar values have been reported in other cases (Schreiber & Fersht, 1996). However, such high values include an estimated 103 to 104-fold contribution from favorable electrostatic interactions, which help to overcome the orientational constraints associated with complex formation. That is, in the absence of favorable electrostatic forces, a basal association rate approximately on the order of 105 M−1s−1 would be obtained. In this context, it is of considerable interest to note that the 103-fold reduction in kon measured for EcoRVΔC is roughly equivalent to the estimated contribution of electrostatic guidance in providing for increased association constants generally. We suggest then that a primary role for the C-terminal subdomains of EcoRV is to facilitate DNA association by providing a complementary electrostatic environment for the docking of the DNA. Electrostatic surface potential calculations for EcoRV and EcoRVΔC support this notion, since it is clear that the removal of the C-terminal subdomain eliminates a substantial proportion of the positively charged enzyme surface (Figure 7). Because the truncation removes only some 10% of each subunit, it is highly unlikely that the reduced size of the enzyme alone plays a major role in contributing to the decreased association rate constant.
Figure 7.

Electrostatic potential at the van der Waals surface of (a) wild-type EcoRV and (b) EcoRVΔC. Scale is in units of kT/e. Note the negatively charged surface exposed at the new C-terminus of EcoRVΔC.
The importance of the complementary electrostatic surfaces to facilitating the association of EcoRV with DNA is consistent with our previous proposal, made on the basis of a series of crystal structures, that asymmetric phosphate neutralization is important to DNA bending (31). Of course, we do not know the nature of the transitory association complex in any structural detail, so it is not possible to state with confidence that bending of the DNA has already occurred at this stage. However, because asymmetric phosphate neutralization is based on the electrostatic force, it is reasonable to suggest that it operates in facilitating the very rapid approach to the initial association complex by EcoRV, where enzyme and DNA are proximal but remain loosely bound. A subdomain possessing significant diffuse positive charge may be especially well-suited to play this role. We emphasize that these observations are purely correlative, and that a definitive exploration of the importance of asymmetric phosphate neutralization requires further experimentation (see below).
Role of the C-terminal subdomain in DNA bending and cleavage
In addition to the sharp decrease in association, the removal of the C-terminal subdomains in EcoRV also both substantially increases the dissociation rate constant, and decreases the extent of bending present at equilibrium (Figures 3, 4; Table 1). Together, these measurements demonstrate that the EcoRVΔC-DNA complex, once formed, is significantly destabilized as compared with the wild-type complex. These observations are similar in nature to those made in our previous study of the divalent metal-ion dependence of DNA bending by the enzyme, where we demonstrated that the absence of divalent metal ions promoted dissociation of the enzyme-DNA complex as compared with the forward progress to the transition state (40). In both contexts, the lack of a full complement of interactions forming in the early stages of the association and induced-fit process makes it more difficult for the complex to reach the high-energy transition state, in which the scissile phosphates are stably and precisely juxtaposed with catalytic groups on the enzyme.
The crystal structure of EcoRVΔC bound to DNA shows the full 50° DNA bend into the major groove, suggesting that crystal lattice interactions drive the equilibrium of the ternary enzyme-DNA-metal ion complex into the fully bent state.
The discrepancy between the Xray structure and equilibrium FRET measurements may be explained by postulating that the fully bent DNA conformation observed in the EcoRVΔC crystal is sampled by substantially less than the full molecular population in solution — although, of course, time-resolved experiments would be necessary to distinguish between a similar and decreased overall bend in a large proportion of the complex, versus the presence of a full bend in a small proportion of complexes. The decreased equilibrium FRET value as measured by decreased dye-to-dye distances is very likely reliable: differences in labeling efficiencies are excluded by the use of the same labeled DNA preparation for both mutant and wild type titrations. Also, it is unlikely that the change in dye-to-dye distance is an artifact arising from altered orientation of the dyes, because the bound structure is compared in both cases. Therefore, the only required assumption is that any effects on dye orientation upon enzyme binding are the same for both the wild type and mutant. This seems reasonable, because the dyes are appended to opposite ends of a 14-mer duplex which is larger than the footprint of EcoRV. Therefore, it is likely that different FRET values correspond to a smaller extent of bending in the case of EcoRVΔC.
The EcoRVΔC mutant is also compromised in its ability to cleave DNA as measured in single-turnover experiments. In these experiments, the molar ratio of enzyme is maintained in excess of DNA, and saturation of the complex in terms of both DNA and metal ions is maintained. Carrying out the experiments in this manner isolates the cleavage event on the enzyme, bypassing the association and bending steps. Thus, while the conditions used are highly artificial as compared with the cellular milieu, they do allow for a clear evaluation of the role of the distal interactions in facilitating approach to the cleavage transition state. It is seen here that this effect, while significant, is nonetheless relatively modest as compared to the much greater lesions in binding and bending. Interestingly, contributions of roughly 102-fold on cleavage rate constants were also measured for the interactions made by the enzyme with flanking DNA in the minor groove (37).
Diffuse charge and localized electrostatic contacts
Variant EcoRV enzymes, in which Arg221, Ser223 and Arg226 within the C-terminal subdomains were individually mutated to alanine, have previously been studied by steady-state kinetics (56, 57). These experiments showed that mutations at positions Arg221 and Ser223 were without significant effect on either kcat or Km, while the R226A mutant was unaffected in kcat but possessed Km elevated by 600-fold. The rate-limiting step in catalysis for R226A was not determined, and it was not possible to directly evaluate the meaning of the increased Km in terms of an effect at a particular mechanistic step. Nonetheless, this finding suggests the likelihood that the initial binding and bending steps are affected in this mutant. Thus, the specific absence of the direct salt bridge formed between the Arg226 guanidinium group and the 5’-flanking cleavage-site phosphate P-5 at 5’-NpNpNpGATATC-3’ may contribute significantly to the diminished capacity of EcoRVΔC to bind and bend DNA.
Given the large 103-fold effects found in both the DNA association and dissociation steps, as well as the 102-fold effect on kchem, it is quite unlikely that the absence of the R226 interaction alone accounts for the properties of the truncation mutant. Instead, other portions of the C-terminal subdomains must also be playing important roles in facilitating the overall cleavage reaction. Further detailed studies of ion-pair interactions at the EcoRV-DNA interface by a structure-perturbation approach that combines DNA methylphosphonate substitutions with mutants in the enzyme are in progress, and are expected to yield substantial insights into the importance of specific interactions in facilitating DNA binding and binding by EcoRV. In particular, the pairing of site-directed mutants with neutral methylphosphonates will provide a thorough appraisal of the role of asymmetric phosphate neutralization in facilitating DNA bending within the context of a functional enzyme-DNA complex.
ACKNOWLEDGEMENTS
The authors thank Tim Bullock, Stephanie Snyder, and Scott Hauenstein for assistance with experimental design and crystallography, Jonathan Fogg for helpful discussions, and Elise Kleeman for critical reading of the manuscript.
Supported by NIH grant GM53763 (to J.J.P.)
ABBREVIATIONS
- FRET
fluorescence resonance energy transfer
- EcoRVΔC
mutant of EcoRV endonuclease in which amino acids 221−245 are removed from each chain
- DTT
dithiothreitol
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- EDTA
ethylenediammine tetra acetate
- BTP
bis-tris propane
REFERENCES
- 1.Maher LJ., 3rd Mechanisms of DNA bending. Curr Opin Chem Biol. 1998;2:688–694. doi: 10.1016/s1367-5931(98)80104-x. [DOI] [PubMed] [Google Scholar]
- 2.Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN. The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci U S A. 1991;88:10148–10152. doi: 10.1073/pnas.88.22.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luger K, Richmond TJ. DNA binding within the nucleosome core. Curr Opin Struct Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Martin J, Espinosa M. Protein-induced bending as a transcriptional switch. Science. 1993;260:805–807. doi: 10.1126/science.8387228. [DOI] [PubMed] [Google Scholar]
- 5.Perez-Martin J, de Lorenzo V. Clues and consequences of DNA bending in transcription. Annu Rev Microbiol. 1997;51:593–628. doi: 10.1146/annurev.micro.51.1.593. [DOI] [PubMed] [Google Scholar]
- 6.van der Vliet PC, Verrijzer CP. Bending of DNA by transcription factors. Bioessays. 1993;15:25–32. doi: 10.1002/bies.950150105. [DOI] [PubMed] [Google Scholar]
- 7.Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. Embo J. 2003;22:3749–3760. doi: 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mysiak ME, Bleijenberg MH, Wyman C, Holthuizen PE, van der Vliet PC. Bending of adenovirus origin DNA by nuclear factor I as shown by scanning force microscopy is required for optimal DNA replication. J Virol. 2004;78:1928–1935. doi: 10.1128/JVI.78.4.1928-1935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koepsel RR, Khan SA. Static and initiator protein-enhanced bending of DNA at a replication origin. Science. 1986;233:1316–1318. doi: 10.1126/science.3749879. [DOI] [PubMed] [Google Scholar]
- 10.Shi Q, Thresher R, Sancar A, Griffith J. Electron microscopic study of (A)BC excinuclease. DNA is sharply bent in the UvrB-DNA complex. J Mol Biol. 1992;226:425–432. doi: 10.1016/0022-2836(92)90957-l. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Zhu L, Reid BR, Drobny GP, Hopkins PB. Solution structure of a cisplatin-induced DNA interstrand cross-link. Science. 1995;270:1842–1845. doi: 10.1126/science.270.5243.1842. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, Lee SD, Larson ED, Drummond JT, Alani E, Hsieh P, Erie DA. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc Natl Acad Sci U S A. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stenzel TT, Patel P, Bastia D. The integration host factor of Escherichia coli binds to bent DNA at the origin of replication of the plasmid pSC101, Cell. 1987;49:709–717. doi: 10.1016/0092-8674(87)90547-2. [DOI] [PubMed] [Google Scholar]
- 14.Snyder UK, Thompson JF, Landy A. Phasing of protein-induced DNA bends in a recombination complex. Nature. 1989;341:255–257. doi: 10.1038/341255a0. [DOI] [PubMed] [Google Scholar]
- 15.Moitoso de Vargas L, Kim S, Landy A. DNA looping generated by DNA bending protein IHF and the two domains of lambda integrase. Science. 1989;244:1457–1461. doi: 10.1126/science.2544029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson CA, Nash HA. Bending of the bacteriophage lambda attachment site by Escherichia coli integration host factor. J Biol Chem. 1988;263:3554–3557. [PubMed] [Google Scholar]
- 17.Garcia RA, Bustamante CJ, Reich NO. Sequence-specific recognition of cytosine C5 and adenine N6 DNA methyltransferases requires different deformations of DNA. Proc Natl Acad Sci U S A. 1996;93:7618–7622. doi: 10.1073/pnas.93.15.7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 19.Taylor JD, Halford SE. Discrimination between DNA sequences by the EcoRV restriction endonuclease. Biochemistry. 1989;28:6198–6207. doi: 10.1021/bi00441a011. [DOI] [PubMed] [Google Scholar]
- 20.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the Eco RI endonuclease--DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 21.Thielking V, Alves J, Fliess A, Maass G, Pingoud A. Accuracy of the EcoRI restriction endonuclease: binding and cleavage studies with oligodeoxynucleotide substrates containing degenerate recognition sequences. Biochemistry. 1990;29:4682–4691. doi: 10.1021/bi00471a024. [DOI] [PubMed] [Google Scholar]
- 22.Mossing MC, Record MT., Jr. Thermodynamic origins of specificity in the lac repressor-operator interaction. Adaptability in the recognition of mutant operator sites. J Mol Biol. 1985;186:295–305. doi: 10.1016/0022-2836(85)90106-8. [DOI] [PubMed] [Google Scholar]
- 23.von Hippel PH, Berg OG. On the specificity of DNA-protein interactions. Proc Natl Acad Sci U S A. 1986;83:1608–1612. doi: 10.1073/pnas.83.6.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Hippel PH. Protein-DNA recognition: new perspectives and underlying themes. Science. 1994;263:769–770. doi: 10.1126/science.8303292. [DOI] [PubMed] [Google Scholar]
- 25.Hiller DA, Rodriguez AM, Perona JJ. Non-cognate Enzyme-DNA Complex: Structural and Kinetic Analysis of EcoRV Endonuclease Bound to the EcoRI Recognition Site GAATTC. Journal of Molecular Biology. 2005;354:121–136. doi: 10.1016/j.jmb.2005.09.046. [DOI] [PubMed] [Google Scholar]
- 26.Viadiu H, Aggarwal AK. Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol Cell. 2000;5:889–895. doi: 10.1016/s1097-2765(00)80329-9. [DOI] [PubMed] [Google Scholar]
- 27.Schildkraut I, Banner CD, Rhodes CS, Parekh S. The cleavage site for the restriction endonuclease EcoRV is 5'-GAT/ATC-3'. Gene. 1984;27:327–329. doi: 10.1016/0378-1119(84)90078-7. [DOI] [PubMed] [Google Scholar]
- 28.D'Arcy A, Brown RS, Zabeau M, van Resandt RW, Winkler FK. Purification and crystallization of the EcoRV restriction endonuclease. J Biol Chem. 1985;260:1987–1990. [PubMed] [Google Scholar]
- 29.Winkler FK, Banner DW, Oefner C, Tsernoglou D, Brown RS, Heathman SP, Bryan RK, Martin PD, Petratos K, Wilson KS. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. Embo J. 1993;12:1781–1795. doi: 10.2210/pdb4rve/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin AM, Sam MD, Reich NO, Perona JJ. Structural and energetic origins of indirect readout in site-specific DNA cleavage by a restriction endonuclease. Nat Struct Biol. 1999;6:269–277. doi: 10.1038/6707. [DOI] [PubMed] [Google Scholar]
- 31.Horton NC, Perona JJ. Crystallographic snapshots along a protein-induced DNA-bending pathway. Proc Natl Acad Sci U S A. 2000;97:5729–5734. doi: 10.1073/pnas.090370797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mirzabekov AD, Rich A. Asymmetric lateral distribution of unshielded phosphate groups in nucleosomal DNA and its role in DNA bending. Proc Natl Acad Sci U S A. 1979;76:1118–1121. doi: 10.1073/pnas.76.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauss JK, Maher LJ., 3rd DNA bending by asymmetric phosphate neutralization. Science. 1994;266:1829–1834. doi: 10.1126/science.7997878. [DOI] [PubMed] [Google Scholar]
- 34.Strauss-Soukup JK, Maher LJ., 3rd Role of asymmetric phosphate neutralization in DNA bending by PU.1. J Biol Chem. 1997;272:31570–31575. doi: 10.1074/jbc.272.50.31570. [DOI] [PubMed] [Google Scholar]
- 35.Tomky LA, Strauss-Soukup JK, Maher LJ., 3rd Effects of phosphate neutralization on the shape of the AP-1 transcription factor binding site in duplex DNA. Nucleic Acids Res. 1998;26:2298–2305. doi: 10.1093/nar/26.10.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardwidge PR, Zimmerman JM, Maher LJ., 3rd Charge neutralization and DNA bending by the Escherichia coli catabolite activator protein. Nucleic Acids Res. 2002;30:1879–1885. doi: 10.1093/nar/30.9.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sam MD, Horton NC, Nissan TA, Perona JJ. Catalytic efficiency and sequence selectivity of a restriction endonuclease modulated by a distal manganese ion binding site. J Mol Biol. 2001;306:851–861. doi: 10.1006/jmbi.2000.4434. [DOI] [PubMed] [Google Scholar]
- 38.Bjornsson A, Mottagui-Tabar S, Isaksson LA. Structure of the C-terminal end of the nascent peptide influences translation termination. Embo J. 1996;15:1696–1704. [PMC free article] [PubMed] [Google Scholar]
- 39.Perona JJ, Martin AM. Conformational transitions and structural deformability of EcoRV endonuclease revealed by crystallographic analysis. J Mol Biol. 1997;273:207–225. doi: 10.1006/jmbi.1997.1315. [DOI] [PubMed] [Google Scholar]
- 40.Hiller DA, Fogg JM, Martin AM, Beechem JM, Reich NO, Perona JJ. Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry. 2003;42:14375–14385. doi: 10.1021/bi035520w. [DOI] [PubMed] [Google Scholar]
- 41.Kozlov AG, Lohman TM. Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry. 2002;41:6032–6044. doi: 10.1021/bi020122z. [DOI] [PubMed] [Google Scholar]
- 42.Bailey S. The Ccp4 Suite - Programs for Protein Crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. PNAS. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 47.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kostrewa D, Winkler FK. Mg2+ binding to the active site of EcoRV endonuclease: a crystallographic study of complexes with substrate and product DNA at 2 A resolution. Biochemistry. 1995;34:683–696. doi: 10.1021/bi00002a036. [DOI] [PubMed] [Google Scholar]
- 49.Horton NC, Otey C, Lusetti S, Sam MD, Kohn J, Martin AM, Ananthnarayan V, Perona JJ. Electrostatic contributions to site specific DNA cleavage by EcoRV endonuclease. Biochemistry. 2002;41:10754–10763. doi: 10.1021/bi020305l. [DOI] [PubMed] [Google Scholar]
- 50.Hancox EL, Halford SE. Kinetic analysis of a mutational hot spot in the EcoRV restriction endonuclease. Biochemistry. 1997;36:7577–7585. doi: 10.1021/bi970156k. [DOI] [PubMed] [Google Scholar]
- 51.Selent U, Ruter T, Kohler E, Liedtke M, Thielking V, Alves J, Oelgeschlager T, Wolfes H, Peters F, Pingoud A. A site-directed mutagenesis study to identify amino acid residues involved in the catalytic function of the restriction endonuclease EcoRV. Biochemistry. 1992;31:4808–4815. doi: 10.1021/bi00135a010. [DOI] [PubMed] [Google Scholar]
- 52.Thielking V, Selent U, Kohler E, Wolfes H, Pieper U, Geiger R, Urbanke C, Winkler FK, Pingoud A. Site-directed mutagenesis studies with EcoRV restriction endonuclease to identify regions involved in recognition and catalysis. Biochemistry. 1991;30:6416–6422. doi: 10.1021/bi00240a011. [DOI] [PubMed] [Google Scholar]
- 53.Martin AM, Horton NC, Lusetti S, Reich NO, Perona JJ. Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry. 1999;38:8430–8439. doi: 10.1021/bi9905359. [DOI] [PubMed] [Google Scholar]
- 54.Horton NC, Perona JJ. DNA cleavage by EcoRV endonuclease: two metal ions in three metal ion binding sites. Biochemistry. 2004;43:6841–6857. doi: 10.1021/bi0499056. [DOI] [PubMed] [Google Scholar]
- 55.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 56.Wenz C, Jeltsch A, Pingoud A. Probing the indirect readout of the restriction enzyme EcoRV. Mutational analysis of contacts to the DNA backbone. J Biol Chem. 1996;271:5565–5573. doi: 10.1074/jbc.271.10.5565. [DOI] [PubMed] [Google Scholar]
- 57.Wenz C, Hahn M, Pingoud A. Engineering of variants of the restriction endonuclease EcoRV that depend in their cleavage activity on the flexibility of sequences flanking the recognition site. Biochemistry. 1998;37:2234–2242. doi: 10.1021/bi9719197. [DOI] [PubMed] [Google Scholar]
- 58.Parry D, Moon SA, Liu HH, Heslop P, Connolly BA. DNA recognition by the EcoRV restriction endonuclease probed using base analogues. J Mol Biol. 2003;331:1005–1016. doi: 10.1016/s0022-2836(03)00861-1. [DOI] [PubMed] [Google Scholar]
- 59.Clegg RM, Murchie AIH, Zechel A, Carlberg C, Diekmann S, Lilley DMJ. Fluorescence resonance energy transfer analysis of the structure of the four-way DNA junction. Biochemistry. 1992;31:4846–4856. doi: 10.1021/bi00135a016. [DOI] [PubMed] [Google Scholar]