

Abstract
Thymidylate synthase (TS) is a target in the chemotherapy of colorectal cancer and some other neoplasms. It catalyses the transfer of a methyl group from methylenetrahydrofolate to dUMP to form dTMP. Based on structural considerations, we have introduced 1,3-propanediphosphonic acid (PDPA) as an allosteric inhibitor of human TS (hTS); it is proposed that PDPA acts by stabilizing an inactive conformer of loop 181–197. Kinetic studies showed that PDPA is a mixed (noncompetitive) inhibitor vs dUMP. In contrast, vs methylenetrahydrofolate at concentrations lower than 0.25 μM PDPA is an uncompetitive inhibitor, while at PDPA concentrations higher than 1 μM the inhibiton is noncompetive, as expected. At the concentrations corresponding to uncompetitive inhibition, PDPA shows positive cooperativity with an antifolate inhibitor, ZD9331, which binds to the active conformer. PDPA binding leads to the formation of hTS tetramers, but not higher oligomers. These data are consistent with a model in which hTS exists preferably as an asymmetric dimer with one subunit in the active conformation of loop 181–197 and the other in the inactive conformation.
Thymidylate synthase (TS) catalyzes the reaction in which the nucleotide deoxyuridylate (dUMP) is reductively methylated by the folate co-substrate 5,10-methylenetetrahydrofolate (CH2H4folate) to form thymidylate (TMP) and dihydrofolate (1). Substrates are bound in an ordered manner, with dUMP binding at the active site prior to CH2H4PteGlu. A cysteine residue (Cys195 in hTS) at the active site attacks the 6-position of the pyrimidine base of the nucleotide, resulting in the formation of a covalent bond between TS and the nucleotide and activating the 5-position of the nucleotide for subsequent covalent-bond formation with the C-11 substituent of CH2H4folate (reviewed in 2–4). The enzyme is the sole source of de novo synthesized thymidylate and its inhibition leads to apoptosis of rapidly dividing cells such as cancer cells, an effect sometimes referred to as thymineless death (5). This phenomenon is exploited in therapeutic protocols utilizing TS inhibitors, such as raltitrexed, pemetrexed or pro-drugs such as 5-fluorouracil and 5-fluorodeoxyuridine that are metabolized to TS inhibitors. The inhibitors are either nucleotide analogs such as 5-fluorodeoxyuridylate (FdUMP) or folate analogues that are collectively referred to as antifolates. The effectiveness of TS-directed chemotherapy is often limited by emerging resistance, which usually arises from an increase in intracellular TS protein levels by a factor of 2–4 (reviewed in 6). Two major mechanisms leading to increased hTS levels have been proposed. In one mechanism, the intracellular turnover of hTS protein is decreased upon formation of inhibitory complexes with drugs (6, 7). The other mechanism is related to hTS protein binding to its own mRNA and inhibiting its translation. The formation of inhibitory complexes by hTS competes with mRNA binding and thus reduces the translational repression of hTS (reviewed in 8, 9). This effect is reversed in some other species (10).
Human TS differs from bacterial TS in three regions: the N-terminus of hTS is extended by 28–29 residues and two insertions of 12 and eight residues are present at positions 117 and 146 of hTS, respectively (2). The crystal structure of hTS has been initially determined using crystals obtained at high ammonium sulfate concentrations (11, 12). At these conditions the active-site loop 181–197 is in a conformation different from that observed in bacterial TS. Since this conformation places Cys195, a residue crucial for catalytic activity, outside the active site, the conformer must be inactive. Another characteristic feature is that loop 108–129, which contains one of the eukaryotic inserts, was disordered. There were four sulfate ions bound per subunit, which appeared to stabilize the inactive conformer. Studies of a truncated version of hTS (13) and an inhibitory complex of hTS with dUMP and raltitrexed (14) yielded high-resolution structures of hTS with loop 181–197 in the active conformation. In these structures, determined at low salt concentration, loop 108–129 was ordered. Later studies showed that also at low salt, 30 mM ammonium sulfate, hTS adopts the inactive conformation with loop 108–129 disordered (15). Intrinsic fluorescence studies of hTS showed that in solution there is equilibrium between the active and inactive conformers and that the presence of phosphate or sulfate ions drives the equilibrium towards the inactive conformation, while dUMP, a substrate, drives it towards the active conformation (12). It was proposed that the stabilization of the inactive conformation may be used to achieve hTS inhibition (11) and was argued that it may yield therapeutic outcomes superior to those of classical active-site-directed inhibitors as it may not lead to increased levels of TS (12; 16).
The enzyme is a dimer of two identical subunits, which generate an asymmetry upon substrate/ligand binding (17). The negative cooperativity between subunits strongly depends on inhibitors (18) and the source of the enzyme. Among many structural studies of inhibitory complexes of TS only one reports a structure of the enzyme with an inhibitor bound in only one subunit; Pneumocystis carini TS (PcTS) binds an antifolate, CB3717, with Kd values which differ by a factor of 8000 between the subunits and crystallized with one subunit forming a ternary inhibitory complex PcTS•dUMP• CB3717 while the other active site contained only loosely bound dUMP (19).
Here we report studies which indicate that cooperativity between the hTS subunits is not limited to substrate or substrate analogues binding but also is reflected in the binding of molecules that affect equilibrium between the active and inactive conformations of loop 181–197.
MATERIALS AND METHODS
Chemicals
Salts, isopropyl-β-D-thiogalactopyranoside (IPTG), 2-mercaptoethanol (2-ME), phenylmethylsufonyl fluoride (PMSF), ethanediol (EDO), polyethylene glycols (PEGs), D-fructose-1,6-bisphosphate (FBP) and folic acid were obtained from Sigma (St. Louis, MO). Ultra-pure ammonium sulfate was from ICN Biomedicals, Inc. (Aurora, OH). (6S)-5,6,7,8-tetrahydrofolic acid (H4folate) was prepared from folic acid and converted to (6R)-CH2H4folate as described previously (20). Diphosphonates were from Epsilon-Chimie, (Brest, France). ZD9331 was kindly provided by Astra Zeneca.
Expression and Purification of Human Thymidylate Synthase
Recombinant hTS was expressed and purified as described previously (12) with minor modifications. Briefly, the cell-free extract was first chromatographed on a Q-Sepharose anion exchange column with a 300 mL linear salt gradient (10–100% 1 M KCl) followed by buffer exchange of the active fractions. The resulting supernatant was then passed through a Blue Sepharose CL-6B column with a similar salt gradient. hTS usually elutes around 230 mM KCl. The active fractions were pooled and analyzed by SDS–PAGE for purity.
Expression and Purification of Murine Thymidylate Synthase
The mTS E. coli expression system was a gift from Dr. Lee F. Johnson, Ohio State University. Recombinant mTS was expressed and purified as described previously (21) with modifications. Similar to hTS, mTS cell-free extract was first chromatographed on a Q-Sepharose anion exchange column with a 300 mL linear salt gradient (10–100% 1 M KCl) followed by buffer exchange of the active fractions. The resulting supernatant was then passed through a Blue Sepharose CL-6B column with a similar salt gradient. The active fractions were pooled and analyzed by SDS–PAGE for purity.
Activity Assays
Enzyme activity was determined spectrophotometrically as previously described (22, 23). Measurements at 340 nM were made at 22 ºC using a UV210 Spectrophotometer with a temperature controlled 6 cell sample chamber. A typical assay uses 40 nM [TS], 100 μM [dUMP], and 150 μM [mTHF] in Morrison buffer (120 mM Tris, 60 mM 2-(N-morpholino)ethanesulfonic acid, 60 mM acetic acid, pH 7.2). One unit of enzyme activity is defined as the amount of enzyme required to synthesize 1 μmol of dTMP per minute. Enzyme concentration was determined by measurement of absorbance at 280 nm, as described previously (24).
Inhibition Assays
Enzyme inhibition was determined as previously described (25) with some modifications. The reaction mixture consisted of 40 nM [TS], 100 μM [dUMP], and 150 μM [mTHF] in Morrison buffer and varying concentrations of inhibitors. The enzyme was incubated with various concentrations of inhibitors along with dUMP and buffer for a minimum of 5 minutes before adding mTHF and measuring the change in absorbance at 340 nm over 3 minutes. For determination of mode of inhibition by PDPA, activity was measured varying the concentration of mTHF with constant dUMP and vice versa. The calculated ratio between measured velocities and those predicted by equation 1 (26) were used as a measure of cooperativity of the inhibitors.
| Equation 1 |
Where V1+2 is the theoretical value for reaction rate in the presence of two inhibitors, V0 is the rate with no inhibitors, V1 is the rate for one inhibitor, and V2 is the rate for the second inhibitor.
Dynamic Light Scattering
Changes in the approximate molecular weights of hTS were measured using dynamic light scattering (DLS). A sample of hTS (40 nM) was filtered and analyzed in a PDDLS/BatchPlus System from Precision Detectors. Measurements were taken on 20 μL samples at room temperature. A sample of hTS (40 nM) incubated with PDPA (1μM) for about 3 hours was measured in the same instrument.
Isothermal titration calorimetry
Binding enthalpy was recorded using an ITC instrument (Microcal Corporation, Northhampton, MA) with a 250 μL syringe. Measurements were carried out at 25 ºC in Morrison buffer; after degassing, the pH of the solutions was adjusted to 7.3 with a small amount of concentrated NaOH solution. The studies of ZD9331 binding were carried out with the protein concentration approximately 18 μM and the titrant, ZD9331, was 400 μM. Titrations with dUMP were carried out using 70 μM protein and 1500 μM titrant concentrations while PDPA binding was measured using 71 μM protein and 1500 μM titrant concentrations. The data were processed using the manufacturer provided ORIGIN software.
Crystallization
Crystals of hTS were grown as previously (15) under low-salt conditions (30 mM ammonium sulfate, 30–40% PEG 4K, 0.1 M Tris pH 8.5, 20 mM βME) by hanging-drop vapour diffusion. Crystals were soaked in artificial mother liquor containing 10 mM PDPA and 5% ethylene glycol for 5 to 10 minutes and then flash frozen in a cryo stream.
X-ray Diffraction Data Measurement and Processing
Crystallographic diffraction experiments were carried out at the SER-CAT BM beamline at APS, Argonne National Laboratory. Data were collected on a single crystal of wild type hTS with approximate dimensions of 0.30 × 0.45 × 0.45 mm. The data were indexed and processed with HKL2000 (27). Data collection and processing statistics are shown in Table 1.
Table 1.
Crystallographic data collection and refinement statistics for hTS•PDPA complex.
| X-Ray Source | APS SER-CAT 22BM | |
|---|---|---|
| Wavelength (Å) | 0.97251 | |
| Space group | P3121 | |
| Unit cell dimensions: | a, b (Å) | 95.689 |
| c (Å) | 82.503 | |
| Resolution (Å) | 2.5 | |
| Mosaicity (degree) | 0.26 | |
| Completeness (%) | 99.3 | |
| Total linear R-mergea (higest shell) | 0.115 (0.506) | |
| Rworkingb (highest shell, 2.60–2.63 Å ) | 22.1 (29.9) | |
| Rfree (highest shell, 2.60–2.63 Å) | 25.8 (35.4) | |
Structure determination and refinement
The structure of hTS (PDB code 1YPV) obtained at low salt conditions was used as the search model in the molecular replacement method carried out with the CNS software (28). Crystallographic refinements were carried out using the TURBO-FRODO graphics software (29) and CNS. The correctness of the structure was evaluated using PROCHECK (30).
Fluorescence studies
The effect of PDPA binding on hTS was measured using a similar approach to that used in studies of phosphate binding (12). The excitation wavelength was set at 295 nm and the emission was scanned from 305–450 nm at a rate of 5 nm/sec. An Ultra-Vu polystyrene cuvette (Baxter Diagnostics Inc., Deerfield, IL) was used to hold a 3-mL sample containing 4 μM protein in 50 mM Tris-base (pH 7.4). The purity of the protein (recombinant wild type TS) was better than 95%. The titration was carried out by addition of potassium phosphate (pH 7.4), PDPA (pH 7.4) or dUMP in increments up to 10% of the sample volume. No change in fluorescence was observed upon dilution of the sample up to 20% of the sample volume. No fluorescence signal was observed from the buffer (Tris-HCl), phosphate, or dUMP in the experiments. All measurements were done in triplicate to eliminate errors introduced by non-homogeneity of the sample and instrumental drift; standard deviations were below 2%.
RESULTS
Stability of the inactive conformation
The inactive conformation of loop 181–197 is stabilized by a pair of hydrogen bonds from Arg163 to the carbonyls of Ala191 and Leu192 (Fig. 1). Arginine in the position corresponding to human residue 163 is not present in rodents (and, in general, in known eukaryotic sequences except for primates) and in particular in mouse TS (mTS) in which a lysine is present. Despite similarity of these two amino-acids, modeling has indicated that a lysine residue in this position cannot mimic the hydrogen bonding system provided by Arg163 (not shown). Therefore it appears likely that mTS does not populate the inactive conformer. This hypothesis is corroborated by studies of mTS inhibition reported below and studies of TS variants with altered Arg163 (31).
Figure 1.
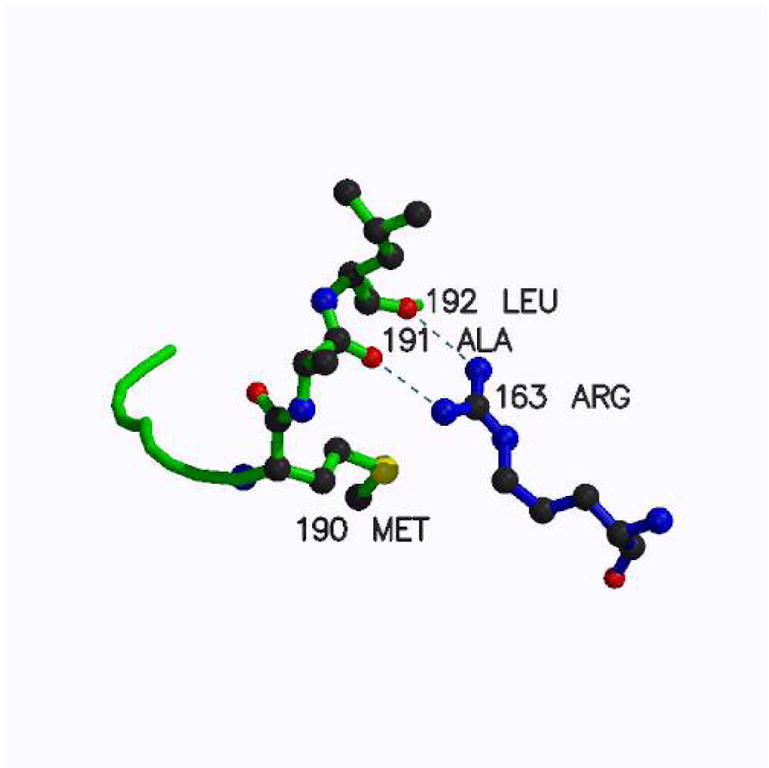
The inactive conformation of loop 181–197 is stabilized by a pair of hydrogen bonds from Arg163 to the carbonyls of Ala191 and Leu192
TS inhibition by phosphates and phosphonates
Inspection of the crystal structures of hTS showed that the inactive conformation of loop 181–197 is stabilized by three or four sulfate or phosphate ions (12, 15). The distances between these ions, 6.5 Å, 9.5 Å, and 9.9 Å, suggested that bifunctional ligands may have stronger propensity to stabilize the inactive conformer through the chelating effect. Diphosphonates with 3–6 carbon linkers, which have distances between phosphonate moieties in the desired range, and fructose bisphosphate were tested for inhibitory properties against hTS. The results are shown in Table 2. It is apparent that one of the inhibitors, propane-1,3-diphosphonic acid (PDPA), has significantly stonger inhibitory properties than the remaining compounds. Subsequently, we tested inhibition of mTS, which is unlikely to be driven into the inactive conformation (vide supra), by PDPA and Pi. Only weak inhibition was observed; this inhibition is most likely a result of the inhibitors binding in the site that accommodates the phosphate moiety of the substrate, dUMP. Bacterial TSs bind phosphate or sulfate in this site (12) and are weakly inhibited by phosphate, as are rodent TSs.
Table 2. Comparison of hTS and mTS Activity in the presence of various Inhibitors At 1 mM concentration.
Comparison of hTS and mTS activity in the presence of 1 mM inhibitors. The strongest inhibitor, PDPA, was tested against mTS. PDPA, 1,3-propanediphosphonic acid; BDPA 1,4-Butanediphosphonic acid; PeDPA 1,5-pentanediphoshonic acid; HDPA 1,6- hexanediphosphonic acid; FBP, fructose-1,6-bisphosphoric acid.
| Enzyme | No Inhibitor % act | Pi % act | PDPA % act | BDPA % act | PeDPA % act | HDPA % act | FBP % act |
|---|---|---|---|---|---|---|---|
| hTS | 100 | 13 | 3 | 11 | 32 | 29 | 23 |
| * mTS | 195 (100) | 73 (37) | 75 (39) | na | na | na | na |
These values are relative to hTS, values in parentheses are relative to mTS.
Fluorescence
The strong inhibition of hTS by Pi and PDPA and only weak inhibition of mTS suggested that the ligands stabilize the inactive conformation of loop 181–197 in hTS. The measurements of fluorescence (Fig. 2) support this hypothesis. PDPA induces an increase in hTS fluorescence in a similar manner as Pi and the arguments linking this phenomenon to the loop 181–197 conformational switching (12) should apply here as well. The lack of similar effects for the mouse enzyme (Fig. 2) and for the R163K mutant of hTS (L. Gibson, unpublished data) suggests that the increase in fluorescence is due to the conformational switching. dUMP lowers the fluorescence intensity of hTS and mTS as well as R163K and bacterial TSs (12). Thus this phenomenon is related to changes other than loop 181–197 conformational switching. Titration of the dUMP•hTS complex with either PDPA or Pi only partially reverses fluorescence decrease (data not shown) as would be expected for the formation of a mixed complex with PDPA in one subunit and dUMP in the other.
Figure 2.
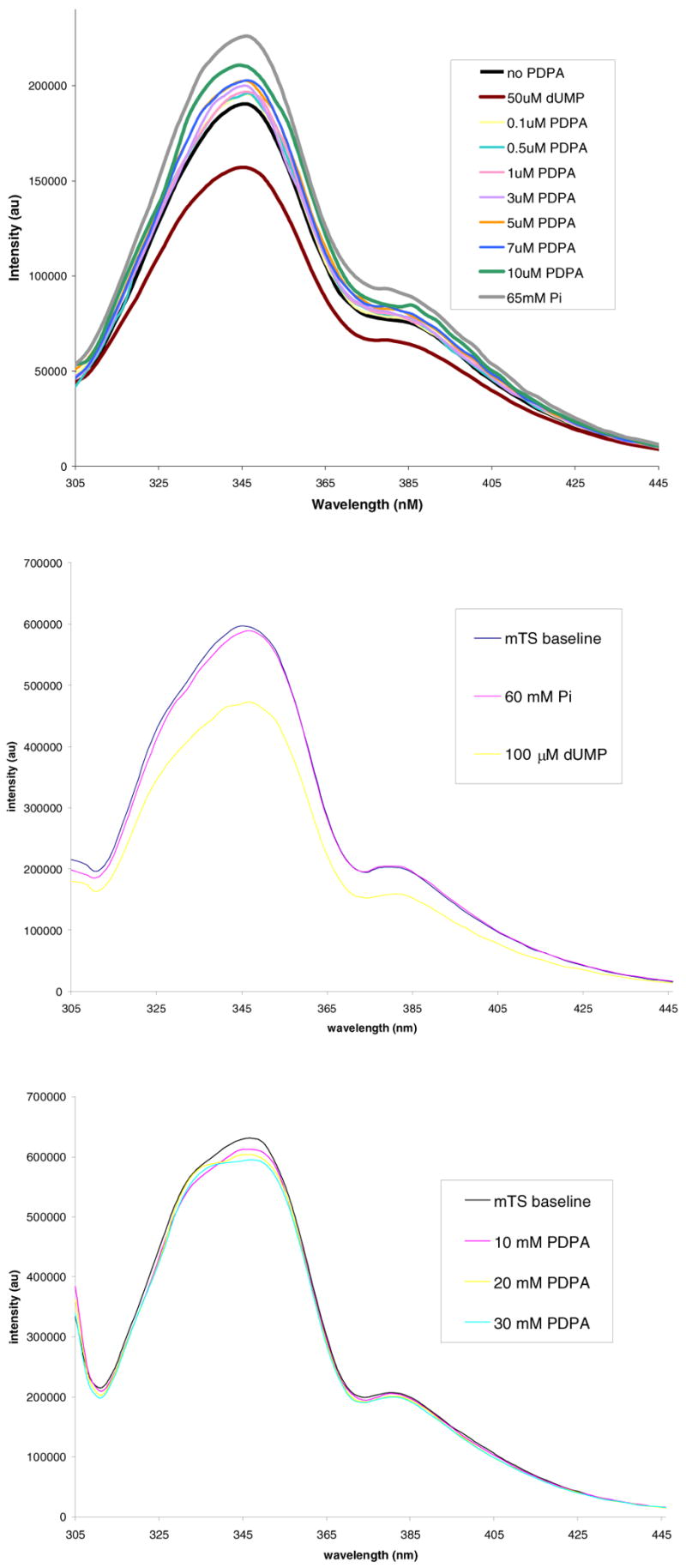
Fluorescence of hTS and mTS. Top panel: Enhancement effect of PDPA and phosphate ion on hTS fluorescence. Middle panel: No effect of phosphate ion on mTS. Bottom panel: No effect of PDPA on mTS.
Activity Assays and Kinetic Study of Inhibition by PDPA
Purified hTS exhibited a specific activity of 1.26 at 22ºC. This value is similar to the previously reported values of 1.31 (23). The proposed mechanism of hTS inhibition by PDPA via stabilization of the inactive conformation was expected to lead to non-competitive inhibition since both substrates binding sites in the inactive conformer are affected. For the studies at which CH2H4folate concentration was kept constant and dUMP concentration varied, at PDPA concentrations of 0.123 and 0.98 μM preincubation times of 10 min were needed to eliminate time dependence. At 0.66 μM PDPA 10 min incubation time was insufficient to achieve stability while prolonged exposures to PDPA led to hTS inactivation. Data shown in Fig. 3A indicate that the inhibition is mixed, and thus both dUMP binding and catalysis are affected.
Figure 3.
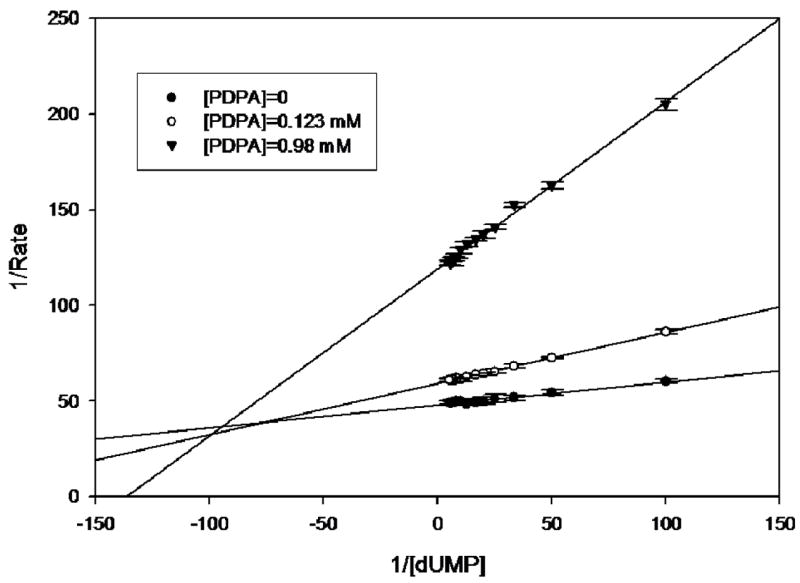
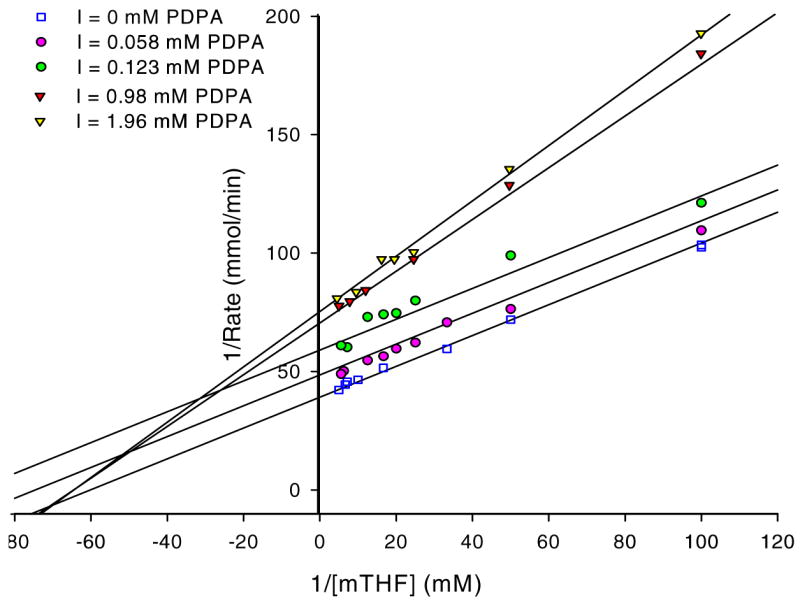
hTS inhibition by PDPA. The results are presented as a Lineweaver-Burke plot. (SigmaPlot v9.0.1) A) Versus dUMP. Error bars represent standard deviations based on five measurements for each point. Fig 3B) Versus the cosubstrate, CH2H4folate. The ratios of standard deviations to the rates are 0.013, 0.006, 0.032, 0.032, 0.018 for increasing concentrations of PDPA as shown in the inset. Numerical data are included as supplementary material.
Studies in which the cosubstrate, CH2H4folate, concentration was varied yielded more complicated results. It is apparent that at higher PDPA concentrations the mode of inhibition is indeed noncompetitive, as expected for allosteric inhibition, with a Ki of 2.7 μM. Somewhat surprisingly, at low concentrations of PDPA the inhibition mode is uncompetitive, indicating cooperativity between CH2H4folate and PDPA binding; the Ki is 0.26 μM. In between these concentrations the fit to either model is poor. Moreover, the reproducibility of measurements in this range of PDPA concentrations is much worse than for the measurements that fit to either uncompetitive or noncompetitive models indicating sensitivity of the system to some additional parameters.
The results presented in Fig. 3B represent measurements carried out with 5 min incubation of hTS with PDPA. At concentrations in the 0.2–0.6 μM range there is a dependence of inhibition on incubation time. Prolonged incubations of hTS with PDPA, in the range 1–5 hours, show complex behavior and activity fluctuations dependent on PDPA concentration. At higher concentrations of PDPA the inhibitory effect takes over and the observed activity is stable.
Heteroinhibition by mixtures of PDPA and ZD9331
The cooperativity of PDPA and CH2H4folate binding to hTS raised the possibility that other folates may actually produce even stronger effects. ZD9331 is a non-glutamylated antifolate, which passed second stage clinical trials (32); its structure is shown in Fig. 4. The mechanism of inhibition, with respect to CH2H4folate, is noncompetitive for hTS with a Ki of 0.38 nM (33). The effects of mixtures of PDPA and ZD9331 on hTS activity are presented in Table 3. As a reference we included activity calculated with the equation that predicts reaction velocities in the case of non-interacting inhibitors. The deviations from expected values may be considered a measure of cooperativity (26). In general, the observed velocities are lower than those predicted for non-interacting inhibitors indicating synergistic inhibition.
Figure 4.
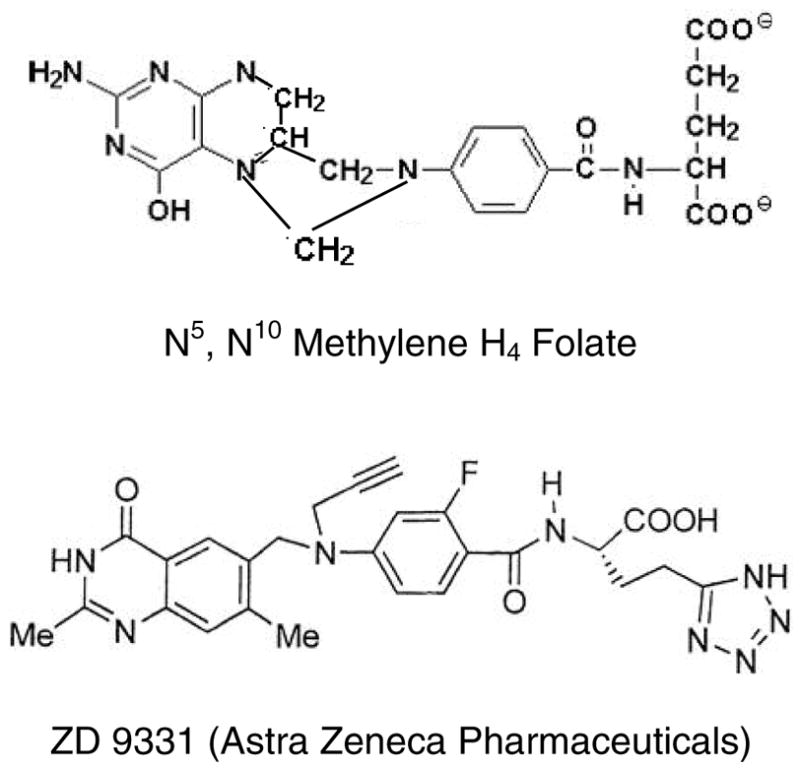
The structure of ZD9331, a non-glutamylated antifolate, compared to N5,N10 methylene-H4-folate (mTHF).
Table 3. The effects of mixtures of PDPA and ZD9331 on hTS activity.
The effects of mixtures of PDPA and ZD9331 on hTS activity. Measured values, relative to the maximum velocity, are given in Obs columns. Theoretical activities, V1,2, calculated with equation 1 that predicts reaction velocities for a mixture of non-interacting inhibitors are in Calc columns. The bottom row contains ratios of Vcalc/Vobs. The deviations from expected values may be considered a measure of inhibitors cooperativity (24).
| Constant [ZD9331] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 nM PDPA
10 nM ZD9331 |
100 nM PDPA
10 nM ZD9331 |
250 nM PDPA
10 nM ZD9331 |
1 μM PDPA
10 nM ZD9331 |
2 μM PDPA
10 nM ZD9331 |
10 μM PDPA
10 nM ZD9331 |
||||||
| Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | Calc. | Obs. |
| 54.7 | 42.6 | 53.3 | 29.5 | 49.75 | 23.8 | 44.09 | 18.1 | 43.67 | 17.2 | 37.77 | 16.1 |
| 1.28 | 1.81 | 2.09 | 2.44 | 2.54 | 2.35 | ||||||
| Constant [PDPA] | |||||||||||
| 1 nM ZD9331
1 μM PDPA |
10 nM ZD9331
1 μM PDPA |
100 nM ZD9331
1 μM PDPA |
1 mM ZD9331
1 μM PDPA |
10 mM ZD9331
1 μM PDPA |
|||||||
| Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | Calc. | Obs. | ||
| 71.6 | 27.3 | 54.9 | 17.9 | 31.7 | 15.1 | 12.1 | 12.1 | 7.3 | 6.9 | ||
| 2.09 | 1.00 | 2.62 | 3.06 | 1.06 | |||||||
Thermodynamics of Ligand Binding
Isothermal titration calorimetry showed binding of approximately one molecule of dUMP per hTS dimer (Fig. 5, top panel). The binding constant, 1.8 × 105 is in good agreement with the equilibrium dialysis data 1.3 × 105 reported previously (17). The binding of ZD9331 (Fig. 5, bottom panel) showed two non-equivalent binding sites with binding constants different by a factor of 51. Unfortunately, the enthalpic effects of PDPA binding (supplementary material) were too small to be interpreted.
Figure 5.
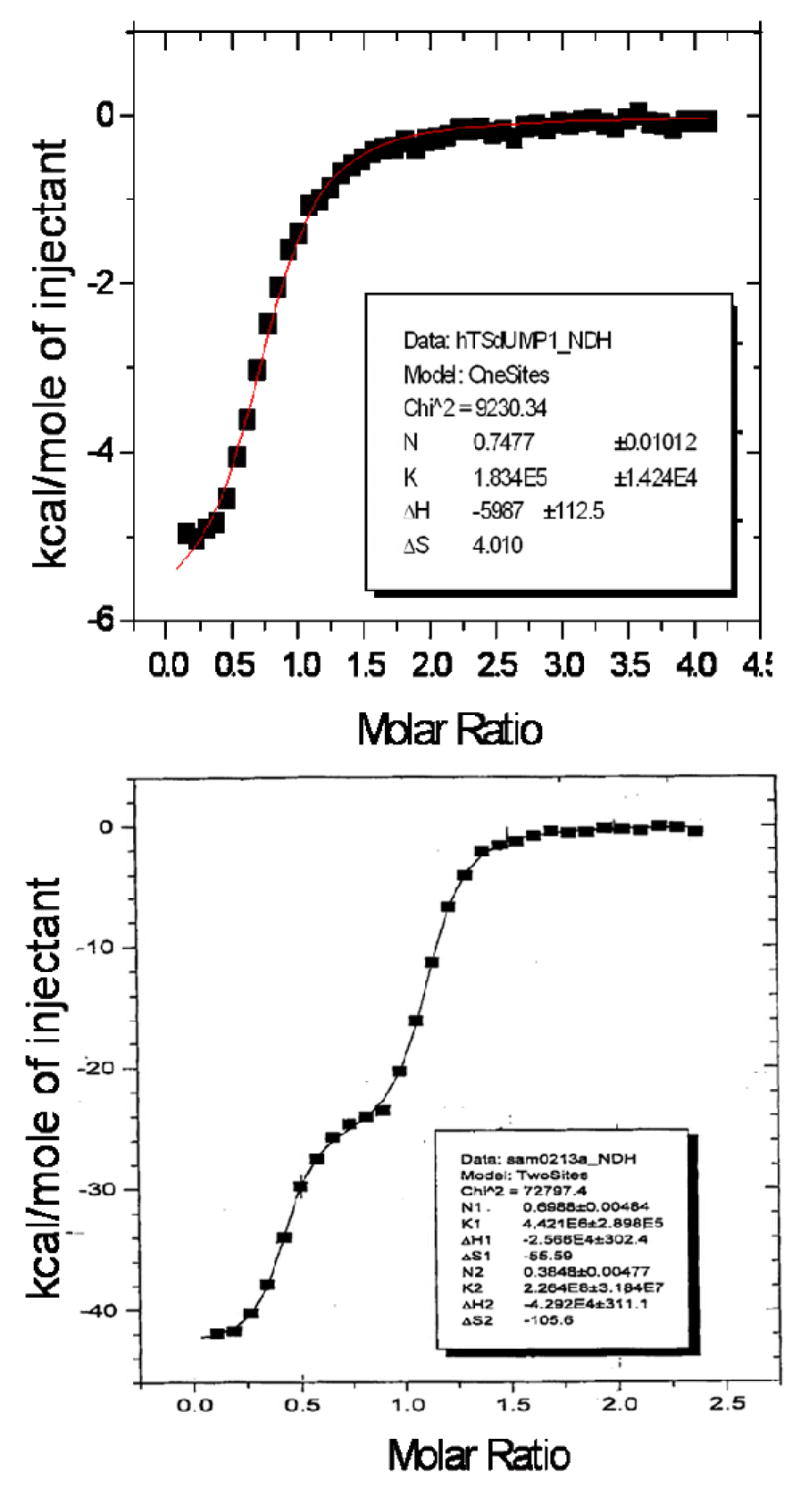
Isothermal titration calorimetry. Top panel: Binding of dUMP indicates one molecule per hTS dimer. Bottom Panel: The binding of ZD9331 shows two non-equivalent binding sites
Dynamic Light Scattering
The effects of PDPA on hTS were also investigated by monitoring the protein hydrodynamic radius. hTS is known to form a dimer and in the absence of PDPA data were consitent with the predicted dimer size. 97% of the mass in solution had a radius of approximately 3.8 nM, which converts to a molecular weight of approximately 75 kD, in good agreement with the 71 kD molecular weight of dimeric hTS. In contrast, in the presence of 1 μM PDPA 99.3% of the total mass gave an average molecular weight of approximately 148 kD. This value corresponds to the molecular weight of an hTS tetramer (142 kD). Results are summarized in Table 4.
Table 4.
Dynamic light scattering statistics. The molecular weight of dimeric hTS is 71 kD. Peak 1 values correspond to the samples; Peak 2 represents the highest noise observed.
| Sample | Item | R (nm) | MW-R (kDa) | %Mass |
|---|---|---|---|---|
| hTS and PDPA | Peak 1 | 5.0 | 148 | 99.3 |
| Peak 2 | 142.8 | 370219 | 0.7 | |
| hTS only | Peak 1 | 3.8 | 75 | 96.5 |
| Peak 2 | 19.8 | 3627 | 3.5 |
Structural studies of PDPA Binding
Our efforts to cocrystallize PDPA and hTS yielded crystals which did not diffract. Reported previously low salt hTS crystals contain hTS in the inactive conformation (15). These crystals were soaked in artifical mother liquors in which 30 mM sulfate ion was exchanged with PDPA or dUMP or their mixtures and their structures determined to 2.0 Å resolution. We did not observed dUMP binding in the crystals and loop 181–197 remained in the inactive conformation observed in the native crystals. However, PDPA apparently bound and its binding site is shown in Fig. 6. One of the phosphate moieties of PDPA binds in the sulfate binding site observed previously (15); the other moiety is disordered between two postions. Apparently the contacts between the dimers that lead to tetramer formation upon PDPA binding in solution are not present in the crystal and only one-pronged interaction is observed. The binding site corresponding to the other end of PDPA that induces the tetrameric species in solution could not be identified.
Figure 6.
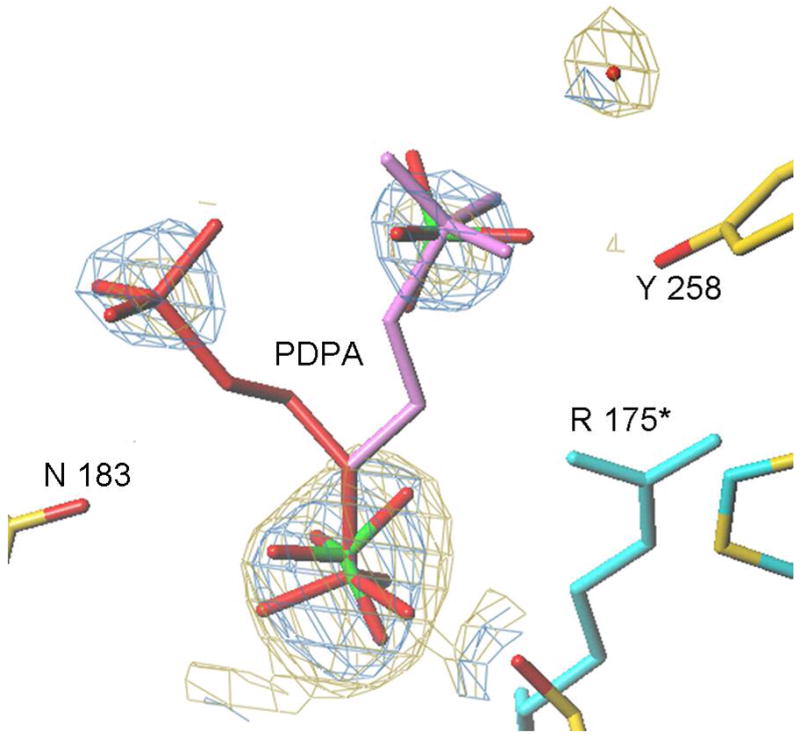
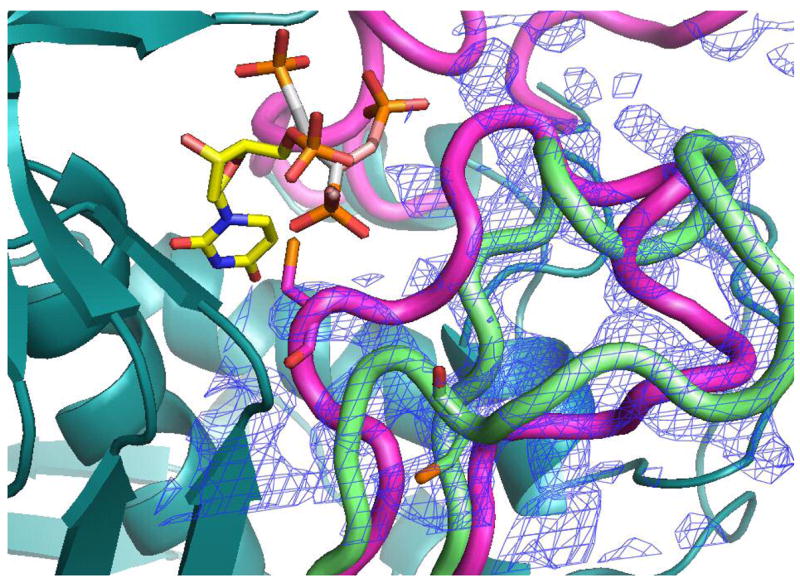
The PDPA binding site. A) One of the phosphonates occupies the same position as one the phosphate ions observed previously bound to the inactive conformer (15). The flexible 3-carbon linker allows the other phosphonate to occupy two positions as indicated. Residues from the other subunit are indicated by an asterisk (*) at the residue number. B) Superposition of the structures of hTS with bound PDPA (in brown) and with bound dUMP (in yellow). Constant parts of the molecule are in malachite. Loop 181–197 in the inactive conformation, observed in the PDPA complex, is shown in light green; corresponding electron density is represented by basket conturing in blue. In purple are loop 181–197 and loop 108–129 as observed in the active conformation, in complex with dUMP and raltitrexed (14).
Discussion
Mixed inhibition vs dUMP indicates that both substrate binding and catalysis are affected. The substrate binding may be affected by either binding in the phosphate binding site in the active conformation or by stabilization of the 181–197 loop in the inactive conformation. Much stronger inhibition of hTS by PDPA than by other phosphonate compounds indicates inhibitor specificity. Together with weak inhibition of mTS and the R163K this strongly suggests that the inhibitory effect is dominated by the stabilization of the inactive conformation. The changes in turnover rate are more difficult to explain; they may be perhaps linked to subunits cooperativity.
At low concentrations PDPA acts as an uncompetitive inhibitor versus CH2H4folate, which indicates its binding to the hTS•substrate complex. Since substrates binding is sequential (at the assay conditions CH2H4folate binds only after dUMP) (34), apparently at low concentrations PDPA binds to the hTS•dUMP•CH2H4folate complex. As PDPA concentration increases, the mode of inhibition switches from uncompetitive to non-competitive (mixed). It probably reflects PDPA binding in the second, lower affinity binding site. A model, which could be proposed, that is consistent with the uncompetitive inhibition observed at low concentrations of PDPA, is based on the half-of-the sites activity of TS (17). In this model PDPA binds in the other subunit than the substrates and apparently affects the hTS dimer enough to inhibit catalysis (Fig. 7). Several antifolates, including ZD9331, also inhibit the hTS dimer by binding in the other subunit than the substrates (33).
Fig. 7.
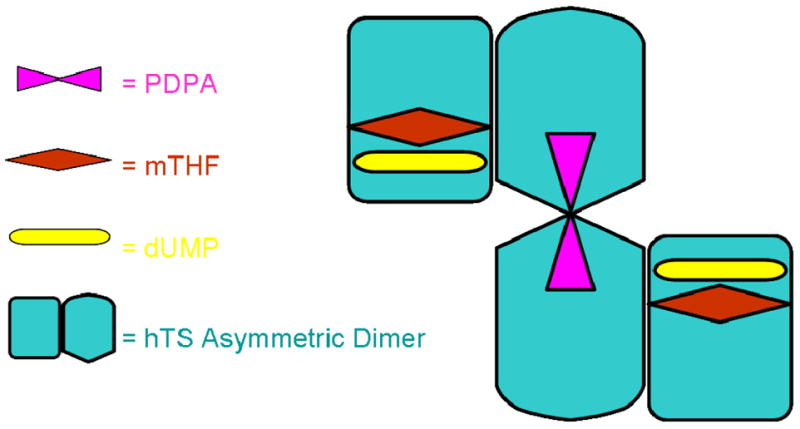
Model for hTS tetramer with one PDPA inhibitor bound to two subunits in the inactive conformation while dUMP molecules and folates are bound to subunits in the active conformation.
At higher PDPA concentrations, inhibition becomes mixed (noncompetitive). This is consistent with PDPA binding in both subunits, and preventing dUMP binding but not CH2H4folate attachment to one subunit.
Dynamic light scattering data show that when PDPA binds to hTS it induces tetramer formation, likely by bridging two dimers. No aggregates higher than tetramers are observed and this may suggest that a symmetric arrangement is formed with two PDPA ions bridging the tetramer.
Inhibition of hTS by mixtures of ZD9331 and PDPA at low antifolate concentrations, but not at higher concentrations, displays positive cooperativity. Again, such behavior is consistent with the model in which PDPA and a folate bind in alternative subunits of the dimer. The subunit that binds PDPA is in the inactive conformation while the one that binds folates is in the active conformation with already bound dUMP. At higher ZD9331 concentrations inhibition by the antifolate dominates. Similarly, a recent study has shown variable cooperativity between antifolates and F-dUMP (35).
It may be speculated that the cooperativity observed for PDPA and folates binding reflects the situation in which the minimum energy state of the hTS dimer is asymmetric: one subunit in the active conformation the other in the inactive conformation. Although such hTS has never been crystallized, pseudosymmetric molecules present more challenge to form well ordered crystal lattice than other systems.
The use of two inhibitors that bind with high affinity to non-equivalent sites of hTS yields synergy in which the binding of both is tighter than that of each individually and may provide therapeutic advantages. Also, targeting two inhibitors at non-equivalent sites may reduce the likelihood for the emergence of drug resistance through defects in folate transporters. This study also points to a relatively rare large difference in response to some inhibitors betweene murine TS and the human enzyme.
Supplementary Material
Isothermal titration of hTS with PDPA, Lineweaver plots with separated low and high concentrations of PDPA corresponding to uncompetitive and noncompetitive models and numerical data for the inhibition of hTS by PDPA are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. Catherine J. Murphy for making her fluorimeter available, Dr. Ramin Radfar for access to his dynamic light scattering instrument, Dr. Lee F. Johnson for the expression system of mouse TS, and Drs. Sondra H. Berger and R. Bruce Dunlap for many helpful discussions.
Footnotes
This work was supported by NIH Grant CA 76560 and the South Carolina Cancer Center.
The PDB file with atomic coordinates of hTS complex with propane-1,3-bisphosphonate have been deposited in the Protein Data Bank as entry xxxx. Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Abbreviations: TS, thymidylate synthase; hTS, human thymidylate synthase; EcTS, Escherichia coli thymidylate synthase; dUMP, 2’-deoxyuridine 5’-monophosphate; dTMP, 2’-deoxythymidine 5’-monophosphate; CH2H4folate or mTHF, 5,10-methylenetetrahydrofolate; ZD9331, [2S]-2-{o-fluoro-p-[N-(2,7-dimethyl-4-oxo-3, 4-dihydroquinazolin-6-ylmethyl)-N-prop-2-ynyl)amino]benzamido}-4-(tetrazol-5-yl) butyric acid; FdUMP, 5-fluoro-2’-deoxyuridine 5’-monophosphate; EDTA, ethylenediaminetetraacetic acid; β-ME, 2-mercaptoethanol; IPTG, isopropyl β-D-thiogalactopyranoside; PMSF, phenylmethylsulfonyl fluoride; DTT, dithiothreitol; Tris, tris(hydroxymethyl)aminomethane; H4folate, (+)-tetrahydrofolate; PEG, polyethylene glycol; FBP, D-fructose-1,6-bisphosphate; PDPA, 1,3-propanediphosphonic acid; BDPA, 1,4-butanediphosphonic acid; PDB, Protein Data Bank.
References
- 1.Humphries GK, Greenberg DM. Studies on the conversion of deoxyuridylic acid to thymidylic acid by a soluble extract from rat thymus. Arch Biochem Biophys. 1958;78:275–287. doi: 10.1016/0003-9861(58)90351-5. [DOI] [PubMed] [Google Scholar]
- 2.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 3.Stroud RM, Finer-Moore JS. Conformational dynamics along an enzymatic reaction pathway: thymidylate synthase, “the movie”. Biochemistry. 2003;42:239–247. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- 4.Finer-Moore JS, Santi DV, Stroud RM. Lessons and conclusions from dissecting the mechanism of a bisubstrate enzyme: thymidylate symthase mutagenesis, function and structure. Biochemistry. 2003;42:248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 5.Houghton PJ. Thymineless death. In: Jackman AL, editor. Antifolate drugs in cancer chemotherapy. Totowa (NJ): Humana Press, Inc; 1999. pp. 423–436. [Google Scholar]
- 6.Forsthoefel AM, Pena MMO, Xing YY, Rafique Z, Berger FG. Structural determinants for the intracellular degradation of human thymidylate synthase. Biochemistry. 2004;43:1972–1979. doi: 10.1021/bi035894p. [DOI] [PubMed] [Google Scholar]
- 7.Kitchens ME, Forsthoefel AM, Rafique Z, Spencer HT, Berger FG. Ligand-mediated induction of thymidylate synthase occurs by enzyme stabilization. J Biol Chem. 1999;274:12544–12547. doi: 10.1074/jbc.274.18.12544. [DOI] [PubMed] [Google Scholar]
- 8.Chu E, Allegra CJ. The role of thymidylate synthase in cellular regulation. Adv Enzyme Regul. 1996;36:143–163. doi: 10.1016/0065-2571(95)00004-6. [DOI] [PubMed] [Google Scholar]
- 9.Ciesla J. Metabolic enzymes that bind RNA: yet another level of cellular regulatory network? Acta Biochem Pol. 2006;53:11–32. [PubMed] [Google Scholar]
- 10.Ciesla J, Jagielska E, Skopinski T, Dabrowska M, Maley F, Rode W. Binding and repression of translation of the cognate mRNA by Trichinella spiralis thymidylate synthase differ from the corresponding interactions of the human enzyme. Biochem J. 2005;390:681–688. doi: 10.1042/bj4020601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiffer CA, Clifton IJ, Davisson VJ, Santi DV, Stroud RM. Crystal structure of human thymidylate synthase: a structural mechanism for guiding substrates into the active site. Biochemistry. 1995;34:16279–16287. doi: 10.1021/bi00050a007. [DOI] [PubMed] [Google Scholar]
- 12.Phan J, Steadman D, Koli S, Ding W, Minor W, Dunlap RB, Berger SH, Lebioda L. Structure of human thymidylate synthase suggests advantages of chemotherapy with noncompetitive inhibitors. J Biol Chem. 2001;276:14170–14177. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 13.Almog R, Waddling CA, Maley F, Maley GF, Van Roey P. Crystal structure of a deletion mutant of human thymidylate synthase Δ(7–29) and its ternary complex with Tomudex and dUMP. Protein Science. 2001;10:988–996. doi: 10.1110/ps.47601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phan J, Mahdavian E, Minor W, Berger S, Spencer HT, Dunlap RB, Lebioda L. Human thymidylate synthase complexed with dUMP and Tomudex, an antifolate drug, is in the closed conformation. Biochemistry. 2000;39:6969–6978. [Google Scholar]
- 15.Lovelace LL, Minor W, Lebioda L. Structure of human thymidylate synthase under low-salt conditions. Acta Crystallogr D. 2005;61:622–627. doi: 10.1107/S0907444905005895. [DOI] [PubMed] [Google Scholar]
- 16.Berger SH, Berger FG, Lebioda L. Effects of Ligand Binding and Conformational Switching on Intracellular Stability of Human Thymidylate Synthase. BBA Proteins and Proteomics. 2004;1696:15–22. doi: 10.1016/j.bbapap.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Danenberg KD, Danenberg PV. Evidence for a Sequential Interaction of the Subunits of Thymidylate Synthase. J Biol Chem. 1979;254:4345–4348. [PubMed] [Google Scholar]
- 18.Dev IK, Dallas WS, Ferone R, Hanlon M, McKee DD, Yates BB. Mode of binding of folate analogs to thymidylate synthase. Evidence for two asymmetric but interactive substrate binding sites. J Biol Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 19.Anderson AC, O’Neil RH, Delano WL, Stroud RM. The structural mechanism for half-the-sites reactivity in an enzyme, thymidylate synthase, involves a relay of changes between subunits. Biochemistry. 1999;38:13829–13836. doi: 10.1021/bi991610i. [DOI] [PubMed] [Google Scholar]
- 20.Steadman DJ, Zhao PS, Spencer HT, Dunlap RB, Berger SH. A structural role for glutamine 214 in human thymidylate synthase. Biochemistry. 1998;37:7089–7095. doi: 10.1021/bi9725428. [DOI] [PubMed] [Google Scholar]
- 21.Zhang HC, Cisneros RJ, Deng WL, Zapf JW, Johnson LF, Dunlap RB. Purification and characterization of recombinant mouse thymidylate synthase. Biochim Biophys Acta. 1991;1077:35–46. doi: 10.1016/0167-4838(91)90523-3. [DOI] [PubMed] [Google Scholar]
- 22.Steadman DJ, Spencer HT, Dunlap RB, Berger SH. Substitution at residue 214 of human thymidylate synthase alters nucleotide binding and isomerization of ligand-protein complexes. Biochemistry. 1999;38:5582–5587. doi: 10.1021/bi982910n. [DOI] [PubMed] [Google Scholar]
- 23.Felder T, Dunlap RB, Dix D, Spencer TH. Differences in natural ligand and fluoropyrimidine binding to human thymidylate synthase identified by transient-state spectroscopic and continuous variation methods. Biochim Biophys Acta. 2002;1597:149–156. doi: 10.1016/s0167-4838(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 24.Zapf JW, Weir MS, Emerick V, Villafranca JE, Dunlap RB. Substitution of glutamine for glutamic acid-58 in Escherichia coli thymidylate synthase results in pronounced decreases in catalytic activity and ligand binding. Biochemistry. 1993;32:9274–9281. doi: 10.1021/bi00087a003. [DOI] [PubMed] [Google Scholar]
- 25.Ciesla J, Golos B, Walajtys-Rode E, Jagielska E, Plucienniczak A, Rode W. The effect of Arg209 to Lys mutation in mouse thymidylate synthase. Acta Biochim Pol. 2002;49:651–658. [PubMed] [Google Scholar]
- 26.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–6442. [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Roussel A, Cambillau C. “Turbo Frodo” Silicon Graphics Geometry Partners Directory. Mountain View Ca Silicon Graphics. 1991:86. [Google Scholar]
- 30.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 31.Johnson SR. Ph.D. Thesis. University of South Carolina; Columbia, South Carolina: 2006. Stabilization of an Active and Inactive Conformation and Investigation of a Potential Proton Relay System of Human Thymidylate Synthase. [Google Scholar]
- 32.Benepal TS, Judson I. ZD9331: discovery to clinical development. Anticancer Drugs. 2005;16(1):1–9. doi: 10.1097/00001813-200501000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Boyle FT, Stephens TC, Averbuch SD, Jackman AL. ZD9331, preclinical and clinical studies. In: Antifolate drugs in cancer chemotherapy. Jackman AL, editor. Totowa (NJ): Humana Press, Inc.; 1999. pp. 243–60. [Google Scholar]
- 34.Montfort WR, Perry KM, Fauman EB, Finer-Moore JS, Maley GF, Hardy L, Maley F, Stroud RM. Structure, multiple site binding, and segmental accommodation in thymidylate synthase on binding dUMP and an anti-folate. Biochemistry. 1990;29:6964–6977. doi: 10.1021/bi00482a004. [DOI] [PubMed] [Google Scholar]
- 35.Van der Wilt CL, Smid K, Peters GJ. Effects of antifolates on the binding of 5-fluoro-2’-deoxyuridine monophosphate to thymidylate synthase. Biochem Pharmacol. 2002;64:669–675. doi: 10.1016/s0006-2952(02)01219-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Isothermal titration of hTS with PDPA, Lineweaver plots with separated low and high concentrations of PDPA corresponding to uncompetitive and noncompetitive models and numerical data for the inhibition of hTS by PDPA are included. This material is available free of charge via the Internet at http://pubs.acs.org.