

In this paper we develop a straightforward and general method to introduce the thiocyanate nitrile stretch as a site-specific electric field probe for proteins. Electrostatics affect nearly all aspects of protein function, but general, site-specific probes for these fields are not yet available. The nitrile stretch is a particularly attractive probe because the frequency is in a relatively uncluttered region of the IR spectrum ( – 2240 cm−1), is typically quite intense (ε∼ 50−1000 M−1cm−1), and is sensitive to electric fields, that is, it has a relatively large Stark tuning rate [∼ 0.4 − 1.1 cm−1 /(MV/cm)]1. In some cases it is possible to deliver the nitrile probe on a substrate or inhibitor2, and it may prove possible to introduce nitrile-containing amino acids such as 4-CN-Phe site-specifically into proteins by semi-synthesis or nonsense suppression3, but both methods often yield smaller quantities of modified protein than typically used for biophysical studies and are not readily compatible with diverse expression systems or multi-subunit protein assemblies. Since the introduction of cysteine residues is used widely as a site-specific labeling strategy (e.g. for spin4 or fluorescent labels), we exploit a well-known chemical modification of cysteine residues to form thiocyanates as a general method for introducing a very small IR-based probe of protein electric fields.
The strategy outlined in Scheme 1 for converting cysteine thiols into thiocyanates5,6 is routinely employed as the first step in the selective cleavage of peptide bonds at cysteine residues7. Briefly, the protein in buffer at pH 7 was reacted with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB or Ellman's reagent8) to form the mixed protein-thionitrobenzoic acid disulfide (PS-TNB), followed by displacement by cyanide (CN−), to form the protein-thiocyanate (PS-CN)9. The electronic absorption of 2-nitro-5-thiobenzoate (TNB) anion byproduct is conveniently monitored at 412 nm (ε412=13600 M−1cm−1)8 to follow the course of reaction. We observed that the PS-CN products in the examples that follow are stable when stored at 4°C over 4 days at pH 710, consistent with previous reports11.
SCHEME 1.
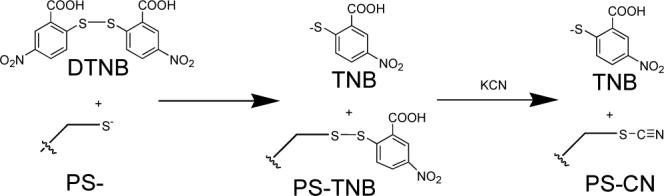
Cysteine Cyanylation.
We have chosen three very different systems to demonstrate the versatility and scope of this method: modification of S-peptide bound to the ribonuclease S-protein (RNase S)12, human aldose reductase (hALR2), which has multiple cysteine residues and for which a number of CN-containing inhibitors related to diabetes control are available2,13, and the bacterial photosynthetic reaction center (RC), which is a multi-subunit, integral membrane protein containing many prosthetic groups.
The sensitivity of a vibrational frequency to an electric field is calibrated by vibrational Stark effect (VSE) spectroscopy14-16. Thiocyanate was introduced into RNase S and the VSE spectrum recorded (Figure 1). RNase S is a non-covalent complex between residues 1−20 (S-peptide) and 21−124 (S-protein) of bovine ribonuclease A12 and is an extensively studied vehicle for the introduction of non-natural biophysical probes into proteins17,18. S-peptide was prepared by solid-phase peptide synthesis with homocysteine substituted at methionine 13, and the unique thiol of the peptide was labeled according to Scheme 1 (see supplemental for details) and combined with S-protein to form the labeled RNase S complex. Formation of the complex was observed by the shift of from 2161.2 cm−1 (FWHM = 11.4 cm−1, ε = 120 M−1cm−1) for free, labeled S-peptide in buffer solution to 2155.4 cm−1 (FWHM = 8.0 cm−1, ε = 130 M−1cm−1) for the complex. By analyzing the VSE spectrum the Stark tuning rate14,15,19 was determined to be 0.7 cm−1/(MV/cm), comparable to the value observed in simpler model compounds in organic glasses1. Both ε and the Stark tuning rate are quite large, demonstrating the utility of thiocyanate as a probe.
FIGURE 1.
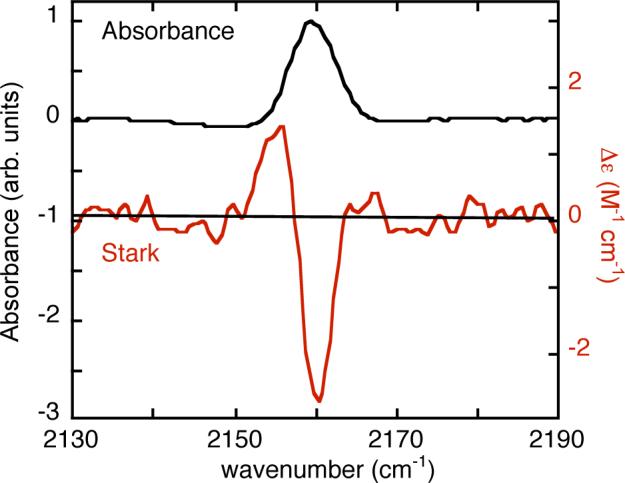
IR absorption (top, left axis) and VSE (red, bottom, right axis) spectra for 11 mM RNase S with homocysteine introduced at position 13 and labeled with CN. The VSE spectrum is the field-on minus field-off difference spectrum obtained at 77K in a 50/50 (v/v) glycerol/water glass. The spectrum is scaled to a pathlength of 1 cm and a field of 1 MV/cm (actual field was 0.67 MV/cm).
The thiocyanate electrostatic probe was introduced into reactive cysteine residues of the globular protein hALR2. hALR2 has seven cysteines, none of which participates in disulfide bonds20. Cysteine 298, which lies in the active site of the enzyme20,21, has been shown to be particularly reactive towards cysteine-thiol modifying reagents21,22. When reacted with 1.1 equivalents of DTNB for 10 minutes, 1 equivalent of TNB was released, and after displacement by cyanide, the LC-MS determined mass was 26 Da higher than that of the unmodified protein. The FTIR spectrum of the labeled protein (Figure 2, red), exhibited a narrow peak at 2159.4 cm−1 with approximately equal intensity, but very different peak position, to a nitrile on a bound inhibitor which makes a 1:1 complex with the protein (see supplemental and Figure 2). It is possible to modify additional labile cysteines by driving the reaction with the addition of 10 mM DTNB over 4 hrs, after which we measured between 2.2 and 2.8 total equivalents of TNB released23. The integrated area in the 2160 cm−1 region of multiply-labeled hALR2 is between 2 and 3 times that of the singly labeled protein (normalized to the inhibitor as an internal standard). Multiply-labeled hALR2 shows peaks at 2151.3 cm−1 and 2161.3 cm−1, the latter presumably the sum of contributions from the first labeled peak centered at 2159.4 cm−1 with another shifted to slightly higher energy. Importantly, this higher energy vibration contributing to the 2161.3 cm−1 peak and the peak at 2151.3 cm−1 both report environments in the enzyme that are distinct from that of the most reactive site. Detailed assignments based on removal of individual cysteines will be reported elsewhere.
FIGURE 2.
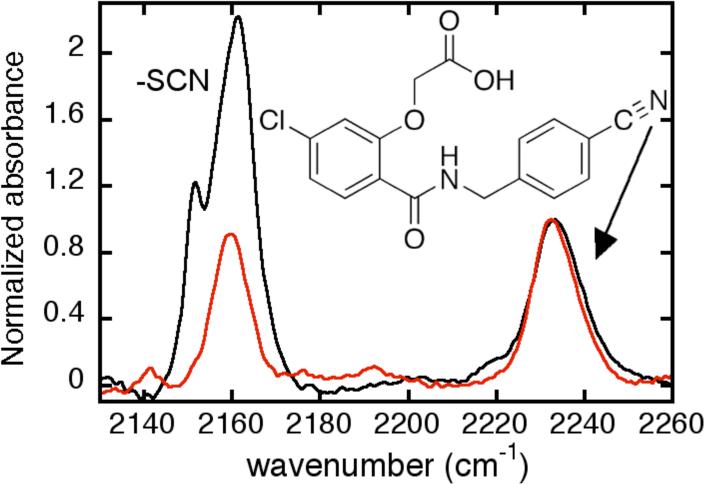
Room temperature IR absorption spectra for hALR2 in a 1:1 complex with the CN-containing inhibitor shown (2232 cm−1) and bearing one (red) or multiple (about 2−3 cysteines labeled, see text, black) thiocyanate labels (2160 cm−1 region). Both spectra are scaled to the absorption of the nitrile group in the inhibitor.
In a final demonstration of the utility of this method we employed Scheme 1 to label the RC of Rb. capsulatus. Native cysteines were removed, and a single cysteine replaced Ile(L150), a moderately buried residue located on a short helix on the RC periplasmic surface. Following successive reactions with DTNB and cyanide for 8−16 hrs each, the overall labeling efficiency was estimated at 60% (see supplemental). As shown in Figure 3, narrow peaks were observed at 2159 cm−1 for -S12CN and 2110 cm−1 for -S13CN with approximately equal intensities for identically treated RC samples, consistent with homogeneous, site-specific labeling. The isotope shift ( = 49 cm−1) is as expected; this simple isotope labeling strategy is especially useful for confirming that small peaks are real in the presence of large backgrounds or baseline offsets!as often occurs with aqueous protein samples.
FIGURE 3.
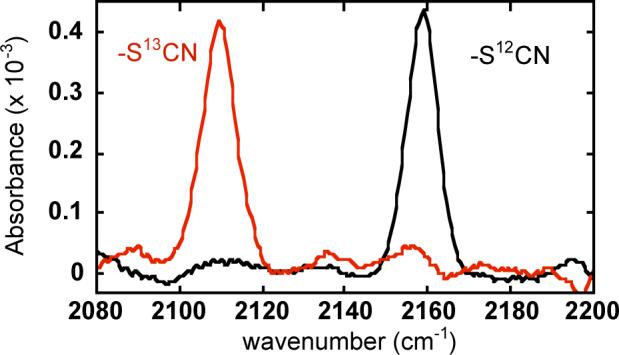
Room temperature IR spectra for the a Rb. capsulatus photosynthetic reaction center mutant with a unique cysteine [I(L150)C/C(L92/98/108/246/247)A] labeled with -S12CN (black) or -S13CN (red). Spectra are for samples of approximately equal concentration.
We have demonstrated the versatility of a simple reaction scheme to introduce very small, stable nitrile electrostatics probes into proteins with three very diverse systems. For each system work is underway to measure electric fields and changes that occur during function, and these will be reported in full publications.
Supplementary Material
Acknowledgment
We thank Ian Suydam for his material and intellectual input and Professor Alberto Podjarny (Strassbourg) for providing the gene for hALR2. LJW is supported by an NRSA postdoctoral fellowship 1F32GM076833-01 and JIC by a Hertz Graduate Fellowship. This work was supported in part by grants from the NIH and the NSF Chemistry Division.
Footnotes
Supporting Information Available: Experimental procedures and product characterization. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Suydam IT, Boxer SG. Biochemistry. 2003;42:12050. doi: 10.1021/bi0352926. [DOI] [PubMed] [Google Scholar]
- 2.Suydam IT, Snow CD, Pande VS, Boxer SG. Science. 2006;313:5784. doi: 10.1126/science.1127159. [DOI] [PubMed] [Google Scholar]
- 3.Schultz Peter. personal communication.
- 4.McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Biochemistry. 1996;35:7692. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 5.Vanaman TC, Stark GR. J Biol Chem. 1970;245:3565. [PubMed] [Google Scholar]
- 6.Degani Y, Neumann H, Patchornik A. J Am Chem Soc. 1970;92:6969. doi: 10.1021/ja00726a043. [DOI] [PubMed] [Google Scholar]
- 7.Wood JL, Catsimpoolas N. J Biol Chem. 1963;238:2887. [PubMed] [Google Scholar]
- 8.Ellman GL. Arch Biochem Biophys. 1959;82:70. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 9.A closely related reaction can be performed in one step using 2-nitro-5-thiocyanatobenzoic acid (NTCB) in the presence of excess cyanide. See: Degani Y, Patchorn A. J Org Chem. 1971;36:2727.
- 10.When subsequent peptide bond scission is the desired outcome, the pH is raised above 8, where scission competes with β-elimination of the thiocyanate. See: Stark GR. Methods Enzymol. 1977;47:129. doi: 10.1016/0076-6879(77)47015-0.
- 11.Doherty GM, Motherway R, Mayhew SG, Malthouse JP. Biochemistry. 1992;31:7922. doi: 10.1021/bi00149a025. [DOI] [PubMed] [Google Scholar]
- 12.Richards FM, Vithayathil PJ. J Biol Chem. 1959;234:1459. [PubMed] [Google Scholar]
- 13.Van Zandt MC, Sibley EO, McCann EE, Combs KJ, Flam B, Sawicki DR, Sabetta A, Carrington A, Sredy J, Howard E, Mitschler A, Podjarny AD. Bioorg Med Chem. 2004;12:5661. doi: 10.1016/j.bmc.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 14.Andrews SS, Boxer SG. J Phys Chem A. 2000;104:11853. [Google Scholar]
- 15.Bublitz GU, Boxer SG. Ann Rev of Phys Chem. 1997;48:213. doi: 10.1146/annurev.physchem.48.1.213. [DOI] [PubMed] [Google Scholar]
- 16.Park ES, Andrews SS, Hu RB, Boxer SG. J Phys Chem B. 1999;103:9813. [Google Scholar]
- 17.Goldberg JM, Baldwin RL. Proc Natl Acad Sci U S A. 1999;96:2019. doi: 10.1073/pnas.96.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huestis WH, Raftery MA. Biochemistry. 1971;10:1181. doi: 10.1021/bi00783a014. [DOI] [PubMed] [Google Scholar]
- 19.Andrews SS, Boxer SG. J Phys Chem A. 2002;106:469. [Google Scholar]
- 20.Wilson DK, Bohren KM, Gabbay KH, Quiocho FA. Science. 1992;257:81. doi: 10.1126/science.1621098. [DOI] [PubMed] [Google Scholar]
- 21.Liu SQ, Bhatnagar A, Ansari NH, Srivastava SK. Biochim Biophys Acta. 1993;1164:268. doi: 10.1016/0167-4838(93)90258-s. [DOI] [PubMed] [Google Scholar]
- 22.Cappiello M, Voltarelli M, Cecconi I, Vilardo PG, Dal Monte M, Marini I, Del Corso A, Wilson DK, Quiocho FA, Petrash JM, Mura U. J Biol Chem. 1996;271:33539. doi: 10.1074/jbc.271.52.33539. [DOI] [PubMed] [Google Scholar]
- 23.A similar maximum of 3 equivalents of TNB released was obtained with 10 mM DTNB: Liu SQ, Bhatnagar A, Das B, Srivastava SK. Arch Biochem Biophys. 1989;275:112. doi: 10.1016/0003-9861(89)90355-x.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.