

Abstract
In skeletal muscle, coupling between the 1,4-dihydropyridine receptor (DHPR) and the type 1 ryanodine receptor (RyR1) underlies excitation-contraction (EC) coupling. The III-IV loop of the DHPR α1S subunit binds to a segment of RyR1 in vitro, and mutations in the III-IV loop alter the voltage dependence of EC coupling, raising the possibility that this loop is directly involved in signal transmission from the DHPR to RyR1. To clarify the role of the α1S III-IV loop in EC coupling, we examined the functional properties of a chimera (GFP-α1S[III-IVa]) in which the III-IV loop of the divergent α1A isoform replaced that of α1S. Dysgenic myotubes expressing GFP-α1S[III-IVa] yielded myoplasmic Ca2+ transients that activated at ∼10 mV more hyperpolarized potentials and that were ∼65% smaller than those of GFP-α1S. A similar reduction was observed in voltage-dependent charge movements for GFP-α1S[III-IVa], indicating that the chimeric channels trafficked less well to the membrane but that those that were in the membrane functioned as efficiently in EC coupling as GFP-α1S. Relative to GFP-α1S, L-type currents mediated by GFP-α1S[III-IVa] were ∼40% smaller and activated at ∼5 mV more hyperpolarized potentials. The altered gating of GFP-α1S[III-IVa] was accentuated by exposure to ±Bay K 8644, which caused a much larger hyperpolarizing shift in activation compared with its effect on GFP-α1S. Taken together, our observations indicate that the α1S III-IV loop is not directly involved in EC coupling but does influence DHPR gating transitions important both for EC coupling and activation of L-type conductance.
The skeletal muscle L-type Ca2+ channel (1,4-dihydropyridine receptor; DHPR)3 serves as the voltage sensor for excitation-contraction (EC) coupling, activating Ca2+ release from the sarcoplasmic reticulum via the type 1 ryanodine receptor (RyR1) in response to depolarization of the plasma membrane (1). Skeletal muscle DHPRs are heteromultimeric channels composed of a primary α1S subunit and auxiliary β1a, α2δ-1, and γ1 subunits (2). Neither genetic ablation of DHPR γ1 subunits (3, 4) nor small interfering RNA knockdown of DHPR α2δ-1 subunits (5–7) seem to have pronounced effects on coupling of the DHPR with RyR1. On the other hand, the DHPR α1S and β1 subunits are essential for EC coupling (8, 9). Mice null for either the α1S or β1 subunit die perinatally because of respiratory paralysis. EC coupling can be restored in myotubes cultured from the respective null mouse pups, as well as from β1 null zebrafish embryos, by heterologous expression of the missing subunit (1, 9–17).
The inability of heterologously expressed cardiac (α1C) DHPRs to restore skeletal-type (i.e. Ca2+ entry-independent) EC coupling in dysgenic (α1S null) myotubes (18, 19) enabled Tanabe et al. (10) to employ chimeric DHPRs as a means to probe regions of α1S that are essential for skeletal-type EC coupling. In that study, the chimera containing the α1S II-III loop was the only chimera with a single loop substitution that was capable of restoring EC coupling similar to that observed for wild-type α1S when expressed in dysgenic myotubes. Subsequently, the experimental approach of expressing progressively more complex α1S/α1C chimeras in dysgenic myotubes identified a subdomain within the α1S II-III loop (residues 720–765, the “critical domain”) that was necessary to support skeletal-type EC coupling (“orthograde coupling”) (20–22). Similarly, the critical domain also has been found to be a major determinant for RyR1-dependent enhancement of the L-type current (“retrograde coupling”) (23).
Although an essential role for the critical domain in skeletal EC coupling has been established (Refs. 20–22, 24, and 25; but see Ref. 26), roles for other intracellular loops of the α1S subunit have also been proposed (27). For example, the α1S carboxyl terminus facilitates expression and targeting of the DHPR (28, 29), most likely by interacting with other junctional proteins (e.g. RyR1) (30–34). The α1S I-II loop is essential for EC coupling because it is the site of interaction with β1a (35–37). The α1S amino terminus does not play a major role in EC coupling, because deletion of the bulk of the amino terminus has little effect on EC coupling (38).
In contrast to the relatively well defined roles of the other intracellular regions of α1S in EC coupling, little is known about the contribution of the α1S III-IV loop. A potentially important role for the α1S III-IV loop in EC coupling was first raised by its identification as the locus for the only DHPR mutations (R1086H and R1086C) currently linked to malignant hyperthermia (39). Functional analysis of the R1086H mutant channel expressed in a dysgenic cell line showed that the α1S R1086H mutation causes a hyperpolarizing shift in the activation of myoplasmic Ca2+ transients (40). In addition, the α1S III-IV loop has also been identified as a potential site of DHPR-RyR1 protein-protein interaction by virtue of the ability of an α1S III-IV loop-glutathione S-transferase fusion protein to bind a purified fragment of RyR1 (residues 922–1112) in vitro (41).
Previous studies seeking to identify α1S cytoplasmic domains important for EC coupling have made use of chimeras of α1S and α1C (10, 42). Although these studies have been invaluable, they have not been revealing about the III-IV loop because it is highly conserved between α1S and α1C (46/53 residues conserved). To test the contribution of the α1S III-IV loop to EC coupling more stringently, we examined the functional characteristics of an α1S-based chimera that incorporated the relatively nonconserved (29 of 53 residues) III-IV loop of the neuronal P/Q-type channel α1A in place of the α1S III-IV loop. Our results indicate that the α1S III-IV loop influences conformational transitions of the DHPR, including transitions important for EC coupling. However, it is unlikely to represent a DHPR-RyR1 site of contact that is necessary for EC coupling.
EXPERIMENTAL PROCEDURES
Myotube Culture and Expression of cDNA—All of the procedures involving mice were approved by the University of Colorado-Denver Institutional Animal Care and Use Committee. Primary cultures of dysgenic myotubes were prepared from newborn mice as described previously (43). For electrophysiological experiments, myoblasts were plated on 35-mm ECL-coated, plastic culture dishes (number 353801; Falcon, San Jose, CA). Cultures were grown for 6–7 days in a humidified 37 °C incubator with 5% CO2 in Dulbecco's modified Eagle's medium (number 15-017-CM; Mediatech, Herndon, VA), supplemented with 10% fetal bovine serum/10% horse serum (Hyclone Laboratories, Logan, UT). This medium was then replaced with differentiation medium (Dulbecco's modified Eagle's medium supplemented with 2% horse serum). Two to four days after the switch to differentiation medium, single nuclei were microinjected with cDNA (400 ng/μl) encoding either GFP-α1S or GFP-α1S[III-IVa]. GFP-α1S (GenBank™ accession number X05921) and GFP-α1S[III-IVa] were constructed as previously described (44, 29).
Measurement of Ionic Currents—Myotubes were used in electrophysiological experiments 2 days following injection. Pipettes were fabricated from borosilicate glass and had resistances of ∼1.5 MΩ when filled with internal solution, which consisted of 140 mm cesium aspartate, 10 mm Cs2-EGTA, 5 mm MgCl2, and 10 mm HEPES, pH 7.4, with CsOH. The external solution contained 145 mm tetraethylammonium chloride, 10 mm CaCl2, 0.003 mm tetrodotoxin, and 10 mm HEPES, pH 7.4, with tetraethylammonium-OH. In some experiments, L-type currents were recorded in the continuous presence of racemic Bay K 8644 (10 μm; kindly supplied by Dr. A. Scriabine, Miles Laboratories Inc., New Haven, CT) in the bath solution. Linear capacitative and leakage currents were determined by averaging the currents elicited by eleven 30-mV hyperpolarizing pulses from a holding potential of -80 mV. Test currents were corrected for linear components of leak and capacitive current by digital scaling and subtraction of this average control current. Electronic compensation was used to reduce the effective series resistance (usually to <1 mΩ) and the time constant for charging the linear cell capacitance (usually to <0.5 ms). Ionic currents were filtered at 2 kHz (eight pole Bessel filter; Frequency Devices, Inc.) and digitized at 10 kHz. To measure macroscopic L-type current in isolation, a 1-s prepulse to -20 mV followed by a 50-ms repolarization to -50 mV was administered before the test pulse (prepulse protocol) (11) to inactivate T-type Ca2+ channels. Cell capacitance was determined by integration of a transient from -80 mV to -70 mV using Clampex 8.0 (Axon Instrument, Foster City, CA) and was used to normalize current amplitudes (pA/pF). Current-voltage (I-V) curves were fitted using the following Boltzmann expression,
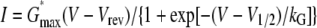 |
(Eq. 1) |
where I is the current for the test potential V, Vrev is the reversal potential, Gmax is the maximum Ca2+ channel conductance, V½ is the half-maximal activation potential, and kG is the slope factor. Tail-current amplitude (Itail) was measured 1 ms after the onset of the repolarization from the test pulse to -50 mV (45).
Measurement of Intracellular Ca2+ Transients—Changes in intracellular Ca2+ were recorded with Fluo-3 (F-3715; Molecular Probes, Eugene, OR) in the whole cell configuration. The salt form of the dye was added to the standard internal solution for a final concentration of 200 nm. After entry into the whole cell configuration, a waiting period of >5 min was used to allow the dye to diffuse into the cell interior. A 100-W mercury illuminator and a set of fluorescein filters were used to excite the dye present in a small rectangular region of the voltage-clamped myotube. A computer-controlled shutter was used to block illumination in the intervals between test pulses. Fluorescence emission was measured by means of a fluorometer apparatus (Biomedical Instrumentation Group, University of Pennsylvania, Philadelphia, PA). The average background fluorescence was quantified before bath immersion of the patch pipette. Fluorescence data are expressed as ΔF/F, where ΔF represents the change in peak fluorescence from base line during the test pulse, and F is the fluorescence immediately prior to the test pulse minus the average background (non-Fluo-3) fluorescence. Unless otherwise noted, the peak value of the fluorescence change (ΔF/F) for each test potential (V) was fitted according to the following equation,
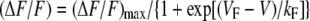 |
(Eq. 2) |
where (ΔF/F)max is the maximal fluorescence change, VF is the potential causing half the maximal change in fluorescence, and kF is a slope parameter.
Electrically Evoked Contractions—Contractions were elicited by 20-ms, 100-V stimuli applied via an extracellular pipette that contained 150 mm NaCl and was placed near intact myotubes expressing constructs of interest. The myotubes were bathed in rodent Ringer's solution (146 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 11 mm glucose, pH 7.4, with NaOH). Contractions were assayed by the movement of an identifiable portion of a myotube across the visual field.
Measurement of Charge Movements—For recordings of immobilization-resistant intramembrane charge movements as a measure of functional DHPR channel expression, ionic currents were blocked by the addition of 0.5 mm CdCl2 + 0.1 mm LaCl3 to the standard extracellular recording solution. All of the charge movements were corrected for linear cell capacitance and leakage currents using a -P/8 subtraction protocol (46). Filtering was at 2 kHz, and digitization was at 20 kHz. Voltage clamp command pulses were exponentially rounded with a time constant of 50–500 μs, and the prepulse protocol (see above) was used to reduce the contribution of gating currents from voltagegated Na+ channels and T-type Ca2+ channels. The integral of the depolarization transient (Qon) for each test potential (V) was fitted according to the following equation,
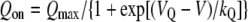 |
(Eq. 3) |
where Qmax is the maximal Qon, VQ is the potential causing movement of half the maximal charge, and kQ is a slope parameter.
Analysis—The figures were made using the software program SigmaPlot (version 7.0, Systat Software, Inc., San Jose, CA). All of the data are presented as the means ± S.E. Statistical comparisons were by unpaired, two-tailed t test (as appropriate), with p < 0.05 considered significant.
RESULTS
The primary experimental approach that has been used to determine the contributions of the α1S cytoplasmic domains to skeletal-type EC coupling is to characterize Ca2+ transients produced by chimeras of α1S and α1C (10, 20, 42). However, this approach is of limited value for the III-IV loop because it is highly conserved between α1S and α1C (Fig. 1A). In the present study, we examined the functional characteristics of a chimera in which the less conserved III-IV loop of the α1A subunit of the neuronal high voltage-activated P/Q type channel was incorporated into the corresponding region of α1S (chimera GFP-α1S[III-IVa]; Fig. 1B).
FIGURE 1.

Rationale for examination of the functional properties of the GFP-α1S[III-IVa] chimera. A, sequence comparison of the III-IV loops of rabbit α1S (GenBank™ accession number X05921), rabbit α1C (GenBank™ accession number X15539), and rabbitα1A (GenBank™ accession number X57477). Residues of α1C or α1A identical to those of α1S are shown boxed in black, and residues conserved with those of α1S are shown boxed in gray. Note that the residue corresponding to α1S R1086 (indicated with a red arrow) that, if exchanged to H or C is linked to the MH phenotype, is conserved in all three channels. B, schematic representation of GFP-α1S[III-IVa].
Ca2+ Release Is Reduced in Myotubes Expressing GFP-α1S[III-IVa]—Myoplasmic Ca2+ transients were measured in the whole cell patch clamp configuration to determine whether replacement of the α1S III-IV loop by that of α1A affects EC coupling. As shown in Fig. 2A, GFP-α1S expressing dysgenic myotubes produced robust Ca2+ transients with an average ΔF/Fmax of 0.75 ± 0.16 ΔF/F (n = 5). In contrast, dysgenic myotubes expressing GFP-α1S [III-IVa] yielded Ca2+ transients that were considerably smaller in magnitude (ΔF/Fmax = 0.27 ± 0.09; n = 5; p < 0.05; Fig. 2B). The reduction in ΔF/Fmax observed for GFP-α1S[III-IVa] was accompanied by a substantial (∼10 mV; p < 0.01) hyperpolarizing shift in voltage dependence of Ca2+ release (Fig. 2C and Table 1). This shift became more apparent when the respective ΔF/F-V curves were normalized to the average ΔF/F measured at +50 mV (Fig. 2D). Although the magnitude of Ca2+ release in response to depolarization from the sarcoplasmic reticulum was reduced in comparison with GFP-α1S, GFP-α1S[III-IVa] was still capable of producing myotube contractions evoked by extracellular electrical stimulation (10 of 16 myotubes tested; Table 1).
FIGURE 2.

EC coupling is reduced in myotubes expressing GFP-α1S[III-IVa]. Simultaneous recordings of myoplasmic Ca2+ transients (top) and L-type Ca2+ currents (bottom), elicited by 50-ms depolarizations from -50 mV to the indicated test potentials are shown for dysgenic myotubes expressing either GFP-α1S (A) or GFP-α1S[III-IVa] (B). C, average ΔF/F-V relationships. D, ΔF/F-V relationships normalized to average ΔF/F at +50 mV to allow direct comparison of voltage dependence. The smooth curves are plotted according to Equation 2, with fit parameters presented in Table 1. Throughout, the error bars represent ± S.E.
TABLE 1.
ΔF/F – V parameters and electrically-evoked contractions The parameters for ΔF/F – V were obtained by best fits of the raw data according to Equation 2. The values are given as the means ± S.E., with the number of myotubes tested indicated in parentheses. ND, not determined.
|
Construct
|
ΔF/F – V
|
Contracting cells/tested
|
||
|---|---|---|---|---|
| ΔF/Fmax | VF | kF | ||
| ΔF/F | mV | mV | ||
| GFP-α1S | 0.75 ± 0.16 (5) | 8.1 ± 2.1 | 7.1 ± 2.1 | 22/39c |
| GFP-α1S[III-IVa] | 0.27 ± 0.09 (5)a | –2.0 ± 1.9b | 8.1 ± 1.1 | 10/16 |
| Uninjected dysgenic myotubes | ND | 0/91 | ||
Significant difference from GFP-α1S (p < 0.05)
Significant difference from GFP-α1S (p < 0.01)
Data from Ref. 24
L-type Ca2+ Currents Are Reduced in Myotubes Expressing GFP-α1S[III-IVa]—As expected, GFP-α1S produced large, slowly activating L-type currents (-4.7 ± 0.6 pA/pF at +40 mV; n = 13; Fig. 3A, left panel) when expressed in dysgenic myotubes. By comparison, L-type currents were smaller in dysgenic myotubes expressing GFP-α1S[III-IVa] (-3.1 ± 0.3 pA/pF; n = 20; p < 0.05; Fig. 3A, right panel). Based on fitting with Equation 1, there was also a hyperpolarizing shift in the voltage dependence of activation for GFP-α1S[III-IVa] in comparison with GFP-α1S (V½ = 28.9 ± 1.0 mV versus 32.5 ± 1.5 mV, respectively; p < 0.05; Fig. 3B and Table 2). Based on this same analysis with Equation 1, the reduction in Gmax was ∼25%. It should be noted that the reversal potential for GFP-α1S[III-IVa] was routinely shifted by ∼10 mV to more hyperpolarized potentials (Fig. 3B). Such a shift in reversal potential is typical of low magnitude L-type currents recorded under similar conditions (47–50), which most likely occurs because of contamination by outward currents. Thus, we also used tail currents (Itail) to estimate relative conductances because these are unaffected by changes in apparent reversal potential (Fig. 3C). Tail currents at -50 mV upon repolarization from +40 mV (“Itail(40)”) were -11.2 ± 1.8 pA/pF in dysgenic myotubes expressing GFP-α1S (n = 13) and -6.3 ± 0.8 pA/pF in dysgenic myotubes expressing GFP-α1S[III-IVa] (n = 11; p < 0.05).
FIGURE 3.

l-type Ca2+ currents are reduced in myotubes expressing GFP-α1S[III-IVa]. A, recordings of L-type Ca2+ currents, elicited by 200-ms depolarizations to the indicated test potentials are shown for dysgenic myotubes expressing GFP-α1S or GFP-α1S[III-IVa]. B, comparison of average peak I-V relationships. The dashed curve represents the I-V relationship for GFP-α1S[III-IVa] scaled to match the peak current density for GFP-α1S. The currents were evoked at 0.1 Hz by test potentials ranging from -20 mV through +80 mV in 10-mV increments following a prepulse protocol (11). The current amplitudes were normalized by linear cell capacitance (pA/pF). The smooth I-V curves are plotted according to Equation 1. The best fit parameters for each plot are presented in Table 2. C, tail current amplitudes measured 1 ms after repolarization to -50 mV are plotted as a function of the preceding test potential.
TABLE 2.
Conductance and intramembrane charge movement The best fit parameters determined by fitting peak I-V data according to Equation 1 and Q-V data according to Equation 3. The values are given as the means ± S.E. with the number of myotubes tested given in parentheses. Itail(40) represents the average amplitude of the current measured 1 ms after repolarizing to –50 mV after a 200-ms depolarization to +40 mV. Q′ = Qmax – Qdys, using the average values for each given in the table. ND, not determined. For all of the data given, the calculated average voltage error was <5 mV.
|
G-V
|
Q-V
|
||||||
|---|---|---|---|---|---|---|---|
| Construct | Gmax | V½ | kG | Qmax | VQ | kQ | Itail(40)/Q |
| nS/nF | mV | mV | nC/microfarad | mV | mV | pA/fC | |
| GFP-α1S | 128 ± 10 (13) | 32.5 ± 1.5 | 8.1 ± 0.4 | 8.3 ± 1.3 (6) | –0.6 ± 3.0 | 13.5 ± 1.7 | 1.5 |
| GFP-α1S[III-IVa] | 96 ± 8 (20)a | 28.9 ± 1.0a | 8.4 ± 0.3 | 2.9 ± 0.5 (5)b | –5.2 ± 3.7 | 10.6 ± 2.6 | 3.2 |
| GFP-α1S + Bay K 8644 | 181 ± 24 (8)a | 29.2 ± 2.3 | 6.9 ± 0.3a | ND | ND | ||
| GFP-α1S[III-IVa] + Bay K 8644 | 148 ± 16 (7) | 19.9 ± 2.6b | 6.7 ± 0.9 | ND | ND | ||
| Uninjected dysgenic myotubes | No inward current (13) | 0.9 ± 0.2 (3)b | –22.8 ± 3.5b | 3.4 ± 2.9b | ND | ||
Significant difference from GFP-α1S (p < 0.05)
Significant difference from GFP-α1S (p < 0.01)
Charge Movements Are Reduced in Myotubes Expressing GFP-α1S[III-IVa]—Immobilization-resistant charge movements in GFP-α1S[III-IVa] expressing myotubes were reduced by 65% relative to GFP-α1S-expressing myotubes (Qmax = 2.9 ± 0.5 nC/μF; n = 5 versus 8.3 ± 1.3 nC/μF; n = 6, respectively; p = 0.0051; Fig. 4, A and B). A small hyperpolarizing shift was also observed for GFP-α1S[III-IVa], but this shift was not significant (Table 2). Importantly, the reduction in Qmax (Fig. 4B and Table 2) was nearly equal in magnitude to the reduction in maximal Ca2+ transients for GFP-α1S[III-IVa] (Fig. 2C and Table 1). Thus, GFP-α1S[III-IVa] appeared to have an ability to activate Ca2+ release via RyR1 that was similar to that of GFP-α1S (Fig. 4C, left panel). However, the reduction in peak Ca2+ current for GFP-α1S[III-IVa] (Fig. 3B) was smaller than the reduction in Qmax, suggesting that Po is higher for the chimeric channels. To assess the change in Po more directly, we calculated the ratio between Itail(40) and Q′, where Q′ is the measured, maximal charge (Qmax) corrected for the charge that is present in uninjected dysgenic myotubes (Qdys; 0.9 ± 0.2 nC/μF; n = 3). This ratio provides an indication of channel Po (11, 51). The ratio Itail(40)/Q′ was substantially larger for GFP-α1S[III-IVa] than for GFP-α1S (3.2 versus 1.5 pA/fC, respectively; Fig. 4C, right panel, and Table 2).
FIGURE 4.

GFP-α1S[III-IVa] releases Ca2+ as efficiently as GFP-α1S but has a higher l-type channel open probability (Po). A, recordings of immobilization-resistant charge movements elicited by 20-ms depolarizations from -50 mV to the indicated test potentials are shown for dysgenic myotubes expressing GFP-α1S or GFP-α1S[III-IVa]. B, comparison of Q-V relationships. Charge movements were evoked at 0.1 Hz by test potentials ranging from -40 mV through +60 mV in 10-mV increments, following a prepulse protocol (11). The smooth curves are plotted according to Equation 3, with best fit parameters presented in Table 2. C, (ΔF/F)max/Q′ and Itail(40)/Q′ (left and right, respectively) for GFP-α1S and GFP-α1S[III-IVa]. Q′ represents the charge attributable to heterologously expressed DHPRs in the membrane and was calculated as Qmax - Qdys (Table 2). The values for (ΔF/F)max are from Table 1. The values for Itail(40) are given in the text and Table 2.
GFP-α1S[III-IVa] Is Potentiated by ±Bay K 8644—We wondered whether the elevated Po for GFP-α1S[III-IVa] would affect the response of the chimeric channel to the L-type Ca2+ channel agonist Bay K 8644, because this agent is also known to increase the Po of L-type channels. In agreement with previous work on native skeletal muscle L-type channels (52), application of ±Bay K 8644 (10 μm) caused an increase in peak current for GFP-α1S (Fig. 5A) that was relatively modest compared with the effect on native α1C (53, 54). The most prominent effect was a slowing of tail current decay (Fig. 5A, inset), which was also seen for GFP-α1S[III-IVa] (Fig. 5B, inset). However, GFP-α1S[III-IVa] differed from GFP-α1S in that ±Bay K 8644 caused a pronounced leftward shift in activation (Fig. 5B and Table 2). This shift meant that the potentiation of peak current by ±Bay K 8644 was much greater for GFP-α1S[III-IVa] than for GFP-α1S at less depolarized potentials (Fig. 5C). The differential effect of ±Bay K 8644 is another indication that the III-IV loop influences gating transitions of the DHPR.
FIGURE 5.

GFP-α1S[III-IVa] is more sensitive to ±Bay K 8644 than GFP-α1S. A, average peak I-V relationships for GFP-α1S in the presence (▴, n = 8) and absence (•, n = 13) of 10 μm ± Bay K 8644. B, average peak I-V relationships for GFP-α1S[III-IVa] in the presence (▵, n = 7) and absence (○, n = 20) of 10 μm ±Bay K 8644. In both (A) and (B), peak I-V data obtained in the absence of ±Bay K 8644 are replotted from Fig. 3, and the smooth curves are plotted according to Equation 1 with best fit parameters given in Table 2. The insets illustrate representative currents obtained at a test potential of +30 mV in the presence of ±Bay K 8644. The vertical scale bar equals 5 pA/pF. The horizontal scale bar equals 50 ms. Note that these tail currents decay more slowly than currents in the absence of ±Bay K 8644 (Fig. 3A). C, potentiation ratios (IBay K 8644/Iuntreated) for GFP-α1S (▴) or GFP-α1S[III-IVa] (▵). The dashed line represents a potentiation ratio of 1.
DISCUSSION
In the current study, depolarization-triggered myoplasmic Ca2+ transients in dysgenic myotubes expressing GFP-α1S[III-IVa] were found to be activated ∼10 mV more negatively and to be reduced by ∼65% in amplitude, compared with those of GFP-α1S (Fig. 2 and Table 1). L-type Ca2+ currents were also reduced for GFP-α1S[III-IVa], but to a lesser extent (∼40%) than the Ca2+ transients, and were also shifted to more hyperpolarizing potentials (Fig. 3). Immobilization-resistant charge movements were reduced by ∼65% for GFP-α1S[III-IVa] (Fig. 4 and Table 2), similar to the reduction in the magnitude of Ca2+ transients but larger than the reduction in L-type Ca2+ currents. Calculation of the ratio between tail current amplitude (Itail(40)) and the charge attributable to DHPRs inserted into the membrane following construct cDNA injection (Q′) indicated that channel Po was increased about 2-fold for GFP-α1S[III-IVa] compared with GFP-α1S (Fig. 4). The hyperpolarizing shift in GFP-α1S[III-IVa] channel activation was accentuated by exposure to the L-type channel agonist ±Bay K 8644 (Fig. 5).
The observation of a hyperpolarizing shift in Ca2+ release for GFP-α1S[III-IVa] compared with GFP-α1S (Fig. 2 and Table 1) supports the idea that the α1S III-IV loop is in some way involved in EC coupling. Such an involvement was proposed by Weiss et al. (40) on the basis of a similar hyperpolarizing shift for an α1S construct containing the R1086H point mutation, which is located in the III-IV loop and has been linked to the malignant hyperthermia phenotype. One possibility for the involvement of the α1S III-IV loop in controlling sarcoplasmic reticulum Ca2+ release would be by direct interaction with RyR1, as suggested by the in vitro binding of this loop to a fragment (residues 922–1112) of RyR1 (41). If such a direct interaction were important, one might predict that the efficiency of EC coupling would be reduced for GFP-α1S[III-IVa] because the sequence of its III-IV loop differs significantly from that of wild-type α1S (Fig. 1). As an indicator of this efficiency, we calculated the ratio of ΔF/Fmax (the maximum change in Fluo-3 fluorescence elicited by a 50-ms depolarization) to Q′ (which equals Qmax - Qdys and is the charge attributable to recombinant DHPRs in the plasma membrane) (11). Because the value of (ΔF/Fmax)/Q′ was very similar for GFP-α1S[III-IVa] and GFP-α1S (Fig. 4C), it seems unlikely that an interaction between the α1S III-IV loop and RyR1 plays an important role in EC coupling. Furthermore, the fraction of cells contracting in response to focal extracellular stimulation was similar for cells expressing GFP-α1S[III-IVa] and GFP-α1S (Table 1). This result supports the idea that the reduced number of GFP-α1S[III-IVa] channels in the plasma membrane were effectively targeted to junctions with RyR1 (29) and elicited local Ca2+ release fluxes comparable with those elicited by GFP-α1S.
Although our data provide evidence against a direct interaction of the α1S III-IV loop with RyR1, they are consistent with the hypothesis that this loop is important for regulating gating transitions of other regions of the DHPR that are coupled directly to activation of RyR1. In particular, we observed that three features of L-type Ca2+ current were significantly altered for GFP-α1S[III-IVa]: 1) activation was shifted in the hyperpolarizing direction, 2) Po was increased, and 3) sensitivity to potentiation by ±Bay K 8644 was enhanced. Each of these effects indicate that substitution of the α1A III-IV loop for that of α1S promotes transitions of the channel from closed states into open states, which can be accounted for by the hypothesis that the wild-type α1S III-IV loop stabilizes resting states of the DHPR. Because some of the gating transitions of the DHPR that result in activation of L-type current may also be involved in activation of RyR1, it seems reasonable that the α1S III-IV loop might also inhibit depolarization-induced DHPR conformational changes that activate RyR1. This hypothesis would account for the hyperpolarizing shift in Ca2+ release observed for GFP-α1S[III-IVa] if substitution of the α1A III-IV loop removed the inhibitory effect of the wild-type α1S III-IV loop on depolarization-induced DHPR conformational changes.
The maximal Po is typically very low (<0.1) for L-type channels containing either α1S (55) or α1C (56) as the principle subunit, but quite high (∼0.5) for P/Q-type channels containing α1A (57) or N-type channels containing α1B (58) as the principle subunit. Previously, we had shown that a chimera containing α1C sequence for its amino-terminal half and α1A sequence for its carboxyl-terminal half had a maximal Po more like that of α1A than that of α1C (45). The increased Po for GFP-α1S[III-IVa] compared with GFP-α1S (Fig. 4C) now identifies the III-IV loop as an important determinant of Po. Indeed, our results with GFP-α1S[III-IVa] suggest that the III-IV loop has a quite wide-spread influence on channel gating. One important goal of future research will be to determine the mechanism whereby the III-IV loop influences the function of L-type channels. For example, do these effects depend only on the interaction of the III-IV loop with proximal portions of the primary sequence (IIIS6, IVS1), or do they depend on interaction of the loop with more distal portions of the primary sequence? Most likely, the interactions are complex because the α1S R1086H mutant displays a hyperpolarizing shift in activation of Ca2+ release and a depolarizing shift in activation of L-type Ca2+ current (40). In addition to determining the mechanism of III-IV loop effects on L-type channel gating, another important goal for future research will be to determine whether modifying the sequence of the III-IV loop affects gating of N- or P/Q-type channels. It seems possible that the III-IV loop represents a region of hitherto unrecognized importance for the regulation of all the high voltage-activated channels.
Acknowledgments
We thank Drs. N. M. Lorenzon, A. M. Payne, and D. C. Sheridan and J. D. Ohrtman for insightful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants NS24444 and AR44750 (to K. G. B.). This work was also supported by Fonds zur Förderung der wissenschaftlichen Forschung Grant P16098-B04 (to M. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: DHPR, 1,4-dihydropyridine receptor; EC, excitation-contraction; RyR1, type 1 ryanodine-sensitive intracellular Ca2+ release channel; GFP, green fluorescent protein.
References
- 1.Tanabe, T., Beam, K. G., Powell, J. A., and Numa, S. (1988) Nature 336 134-139 [DOI] [PubMed] [Google Scholar]
- 2.Beam, K. G., and Horowicz, P. (2004) in Myology (Engel, A. G., and Franzini-Armstrong, C., eds) pp. 257-280, McGraw Hill, New York
- 3.Freise, D., Held, B., Wissenbach, U., Pfeifer, A., Trost, C., Himmerkus, N., Schweig, U., Freichel, M., Biel, M., Hofmann, F., Hoth, M., and Flockerzi, V. (2000) J. Biol. Chem. 275 14476-14481 [DOI] [PubMed] [Google Scholar]
- 4.Ursu, D., Sebille, S., Dietze, B., Freise, D., Flockerzi, V., and Melzer, W. (2001) J. Physiol. 533 367-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obermair, G. J., Kugler, G., Baumgartner, S., Tuluc, P., Grabner, M., and Flucher, B. E. (2005) J. Biol. Chem. 280 2229-2237 [DOI] [PubMed] [Google Scholar]
- 6.García, K., Nabhani, T., and García, J. (2007) J. Physiol. 586 727-738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gach, M., Cherednichenko, G., Haarmann, C., Lopez, J. R., Beam, K. G., Pessah, I. N., Franzini-Armstrong, C., and Allen, P. D. (2008) Biophys. J. 94 3023-3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beam, K. G., Knudson, C. M., and Powell, J. A. (1986) Nature 320 168-170 [DOI] [PubMed] [Google Scholar]
- 9.Gregg, R. G., Messing, A., Strube, C., Beurg, M., Moss, R., Behan, M., Sukhareva, C., Haynes, S., Powell, J. A., and Coronado, R. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 13961-13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanabe, T., Beam, K. G., Adams, B. A., Niidome, T., and Numa, S. (1990) Nature 346 567-569 [DOI] [PubMed] [Google Scholar]
- 11.Adams, B. A., Tanabe, T., Mikami, A., Numa, S., and Beam, K. G. (1990) Nature 346 569-572 [DOI] [PubMed] [Google Scholar]
- 12.Strube, C., Beurg, M., Sukhareva, C., Ahern, C. A., Powell, J. A., Powers, P. A., Gregg, R. G., and Coronado, R. (1996) Biophys. J. 75 2531-2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beurg, M., Ahern, C. A., Vallejo, P., Conklin, M. W., Powers, P. A., Gregg, R. G., and Coronado, R. (1999) Biophys. J. 77 2953-2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheridan, D. C., Carbonneau, L., Ahern, C. A., Nataraj, P., and Coronado, R. (2003) Biophys. J. 85 3739-3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheridan, D. C., Cheng, W., Ahern, C. A., Mortensen, L., Alsammarae, D., Vallejo, P., and Coronado, R. (2003) Biophys. J. 84 220-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheridan, D. C., Cheng, W., Carbonneau, L., Ahern, C. A., and Coronado, R. (2004) Biophys. J. 87 929-942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schredelseker, J., Di Biase, V., Obermaier, G. J., Felder, E. T., Flucher, B. E., Franzini-Armstrong, C., and Grabner, M. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 17219-17224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanabe, T., Mikami, A., Numa, S., and Beam, K. G. (1990) Nature 344 451-453 [DOI] [PubMed] [Google Scholar]
- 19.García, J., Tanabe, T., and Beam, K. G. (1994) J. Gen. Physiol. 103 125-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakai, J., Tanabe, T., Konno, T., Adams, B., and Beam, K. G. (1998) J. Biol. Chem. 273 24983-24986 [DOI] [PubMed] [Google Scholar]
- 21.Kugler, G., Weiss, R. G., Flucher, B. E., and Grabner, M. (2004) J. Biol. Chem. 279 4721-4728 [DOI] [PubMed] [Google Scholar]
- 22.Takekura, H., Paolini, C., Franzini-Armstrong, C., Kugler, G., Grabner, M., and Flucher, B. E. (2004) Mol. Biol. Cell 15 5408-5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grabner, M., Dirksen, R. T., Suda, N., and Beam, K. G. (1999) J. Biol. Chem. 274 21913-21919 [DOI] [PubMed] [Google Scholar]
- 24.Wilkens, C. M., Kasielke, N., Flucher, B. E., Beam, K. G., and Grabner, M. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 5892-5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bannister, R. A., Papadopoulos, S., and Beam, K. G. (2005) Biophys. J. 88 640 (abstr.) [Google Scholar]
- 26.Ahern, C. A., Bhattacharya, D., Mortensen, L., and Coronado, R. (2001) Biophys. J. 81 3294-3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bannister, R. A. (2007) J. Musc. Res. Cell Motil. 28 275-283 [DOI] [PubMed] [Google Scholar]
- 28.Proenza, C., Wilkens, C. M., Lorenzon, N. M., and Beam, K. G. (2000) J. Biol. Chem. 275 23169-23174 [DOI] [PubMed] [Google Scholar]
- 29.Flucher, B. E., Kasielke, N., and Grabner, M. (2000) J. Cell Biol. 151 467-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slavik, K. J., Wang, J.-P., Aghdasi, B., Zhang, J.-Z., Mandel, F., Malouf, N., and Hamilton, S. L. (1997) Am. J. Physiol. 41 C1475-C1481 [DOI] [PubMed] [Google Scholar]
- 31.Sencer, S., Papineni, R. V., Halling, D. B., Pate, P., Krol, J., Zhang, J. Z., and Hamilton, S. L. (2001) J. Biol. Chem. 276 38237-38241 [DOI] [PubMed] [Google Scholar]
- 32.Papadopoulos, S., Leuranguer, V., Bannister, R. A., and Beam, K. G. (2004) J. Biol. Chem. 279 44046-44056 [DOI] [PubMed] [Google Scholar]
- 33.Lorenzon, N. M., Haarmann, C. S., Norris, E. E., Papadopoulos, S., and Beam, K. G. (2004) J. Biol. Chem. 279 44057-44064 [DOI] [PubMed] [Google Scholar]
- 34.Lorenzon, N. M., and Beam, K. G. (2007) J. Gen. Physiol. 130 379-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen, Y., Li, M., Zhang, Y., He, L., Yamada, Y., Fitzmaurice, A., Shen, Y., Zhang, H., Tong, L., and Yang, J. (2004) Nature 429 675-680 [DOI] [PubMed] [Google Scholar]
- 36.Opatowsky, Y., Chen, C.-C., Campbell, K. P., and Hirsch, J. A. (2004) Neuron. 42 387-399 [DOI] [PubMed] [Google Scholar]
- 37.Van Petegem, F., Clark, K. A., Chatelain, F. C., and Minor, D. L. (2004) Nature 429 670-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bannister, R. A., and Beam, K. G. (2005) Biophys. Biochem. Res. Commun. 336 134-141 [DOI] [PubMed] [Google Scholar]
- 39.Monnier, N., Procaccio, V., Stieglitz, P., and Lunardi, J. (1997) Am. J. Hum. Genet. 60 1316-1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss, R. G., O'Connell, K. M. S., Flucher, B. E., Allen, P. D., Grabner, M., and Dirksen, R. T. (2004) Am. J. Physiol. 287 C1094-C1102 [DOI] [PubMed] [Google Scholar]
- 41.Leong, P., and MacLennan, D. H. (1998) J. Biol. Chem. 273 29958-29964 [DOI] [PubMed] [Google Scholar]
- 42.Carbonneau, L., Bhattacharya, D., Sheridan, D. C., and Coronado, R. (2005) Biophys. J. 89 243-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beam, K. G., and Franzini-Armstrong, C. (1997) Methods Cell Biol. 52 283-306 [DOI] [PubMed] [Google Scholar]
- 44.Grabner, M., Dirksen, R. T., and Beam, K. G. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1903-1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkens, C. M., Grabner, M., and Beam, K. G. (2001) J. Gen. Physiol. 118 495-508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bannister, R. A., Colecraft, H. M., and Beam, K. G. (2008) Biophys. J. 94 2631-2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakai, J., Dirksen, R. T., Nguyen, H. T., Pessah, I. N., Beam, K. G., and Allen, P. D. (1996) Nature 380 72-75 [DOI] [PubMed] [Google Scholar]
- 48.Avila, G., and Dirksen, R. T. (2000) J. Gen. Physiol. 115 467-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Avila, G., O'Connell, K. M., Groom, L. A., and Dirksen, R. T. (2001) J. Biol. Chem. 276 17732-17738 [DOI] [PubMed] [Google Scholar]
- 50.Sheridan, D. C., Takekura, H., Franzini-Armstrong, C., Beam, K. G., Allen, P. D., and Perez, C. F. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 19760-19765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams, B. A., Mori, Y., Kim, M. S., Tanabe, T., and Beam, K. G. (1994) J. Gen. Physiol. 104 985-996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams, B. A., and Beam, K. G. (1989) J. Gen. Physiol. 94 429-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanguinetti, M. C., and Kass, R. S. (1984) J. Mol. Cell Cardiol. 16 667-670 [DOI] [PubMed] [Google Scholar]
- 54.Thomas, G., Chung, M., and Cohen, C. J. (1985) Circ. Res. 56 87-96 [DOI] [PubMed] [Google Scholar]
- 55.Dirksen, R. T., and Beam, K. G. (1995) J. Gen. Physiol. 105 227-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cachelin, A. B., de Peyer, J. E., Kokubun, S., and Reuter, H. (1983) Nature 304 462-464 [DOI] [PubMed] [Google Scholar]
- 57.Llinas, R. R., Sugimori, M., and Cherksey, B. (1989) Ann. N. Y. Acad. Sci. 560 103-111 [DOI] [PubMed] [Google Scholar]
- 58.Delcour, A. H., and Tsien, R. W. (1993) Science. 259 980-984 [DOI] [PubMed] [Google Scholar]