

Abstract
The synthesis, photophysical properties, and structural characterization of a photodimerizable ditopic ligand are described. Upon irradiation at 366 nm, ligand 1 dimerizes to the head-to-tail tetra-bpy ligand 2. This thermally stable photodimer can be dissociated back to 1 using higher energy irradiation (254 nm).
Bridging ligands, capable of coordinating multiple metal ions, have been attracting considerable interest due to their potential use as building blocks for the fabrication of functional multimetallic assemblies.1 Of particular interest are systems that can respond to external stimuli, such as photons,2,3 protons,4,5 electrons6 or other reactive species. Such responsive ligands and their complexes can, in principle, be employed for the on/off switching or fine-tuning of the interactions between the coordinating centers. In contrast to the abundance of unreactive polypyridine multitopic ligands, the repertoire of responsive ditopic bridging ligands that can be modulated by external stimuli is limited. Here we report the synthesis and reactivity of a new photodimerizable ligand 1, where a diimine core is incorporated into an anthracene unit. Near ultraviolet irradiation (λ ∼ 360 nm) results in exclusive formation of a head-to-tail tetra-bpy ligand 2.
Anthracene photodimerization is one of the oldest photochemical reactions known.7 Since it was first observed in 1867, it has been employed in numerous responsive systems, including metal coordinating ones.8 Most anthracene-containing ligands, however, consist of isolated coordinating sites that are covalently linked to an anthracene unit, where the latter serves as the dimerizing module only. Examples include anthracene-containing crown ethers or cryptands,9,10 as well as systems with pendant polypyridyl ligands.11,12 In contrast to the majority of these systems, ligand 1 encompasses a conjugating diazaanthracene unit that serves as the common core of the ditopic bis-bpy ligand. As such, the electronic properties of the ligand and its metal complexes are expected to change upon photodimerization.
To facilitate the synthesis of the bridging ligand 1, a concise synthesis of the hitherto unknown dibromo-diazaanthracene building block 7 was needed (Scheme 1). It was prepared from the diester 3 which, in turn, can be made from resorcinol using established procedures.13,14 Following activation of the phenolic groups as triflates to give 4, a Sonogashira cross coupling reaction with trimethylsilylacetylene yielded 5. A sealed tube reaction with saturated ethanolic ammonia afforded the bislactam 6.15 As 6 is rather insoluble, a simple filtration provided the pure product. Bromination of the lactam using phosphorous oxybromide and potassium carbonate in acetonitrile16 produced the desired 1,8-dibromo-2,7-diazaanthracene 7. These milder conditions were necessary since refluxing the bislactam in pure phosphorous oxybromide led primarily to the tribrominated derivative. Final assembly of the bridging ligand 1 was achieved by a Negishi cross coupling reaction17 between 2-bromopyridine and the dibromo derivative 7 to give the desired product in 88% yield (Scheme 1).14,18,19
Scheme 1.

Synthesis of ligand 1
Single crystals of 1, suitable for X-ray crystallography, were obtained by slowly diffusing diethylether into a solution of ligand in chloroform in the dark. As seen in the solved structure (Figure 1), the bipyridine units adopt the expected trans geometry, thus minimizing the global dipole moment of the molecule and the electrostatic interaction between the nitrogen atoms' lone pairs. In addition, the diazaanthracene core and the individual pyridine rings are also out of plane (torsion angles: 134.1° and 151.5°). Inspection of the crystal lattice reveals intermolecular contacts between the pyridinic moieties of the anthracene unit. The average distance between planes is 3.44 Å, allowing for an efficient π-π stacking along the b axis. Note, however, the absence of stacked diazaanthracene units, an arrangement that is likely to be necessary for photodimerization in the solid state.20
Figure 1.
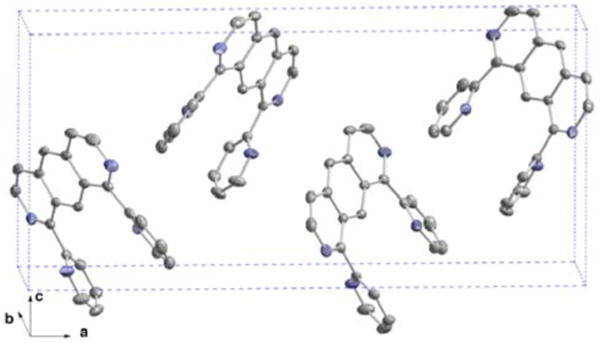
Crystal structure of 1.
The absorption spectrum of ligand 1 is shown in Figure 2a. It is characterized by an intense absorption at 250 nm with a shoulder centered around 290 nm. There is also a broad, featureless band tailing into the visible region with a maximum at 392 nm. The molecule's emission spectrum (Figure 2a, insert) lacks the fine features associated with typical anthracene derivatives, exhibiting a broad band with a λmax at 470 nm.21 The excitation spectrum agrees well with the absorption spectrum, with a peak maximum at 397 nm (Figure 2a, insert).
Figure 2.
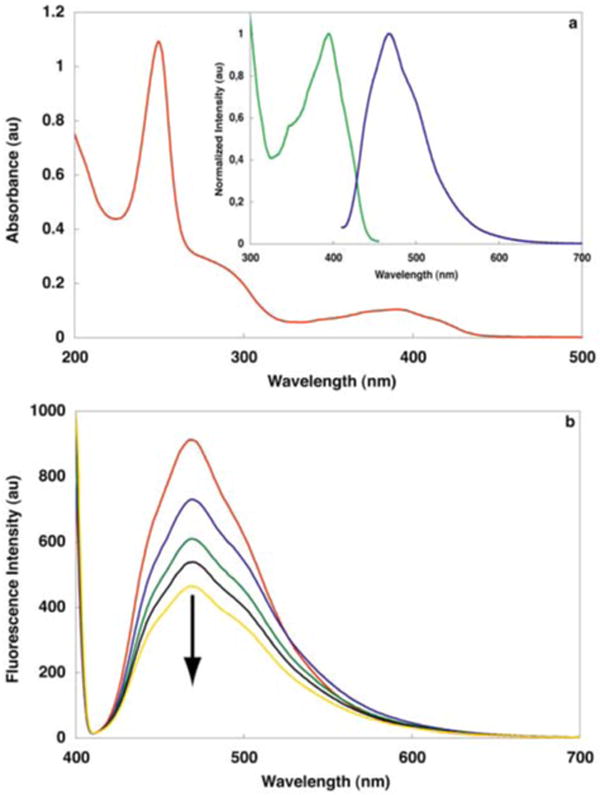
(a) Absorption spectrum of ligand 1 in CH3CN. Insert: emission (
 ) (excitation at 395 nm) and excitation (
) (excitation at 395 nm) and excitation (
 ) (monitoring at 470 nm) spectra of 1 in CH3CN; (b) Fluorescence intensity continuously decreases upon irradiation at 366 nm: t = 0
) (monitoring at 470 nm) spectra of 1 in CH3CN; (b) Fluorescence intensity continuously decreases upon irradiation at 366 nm: t = 0
 ; t = 45 min
; t = 90 min
; t = 45 min
; t = 90 min
 ; t = 180 min —; t = 360 min
; t = 180 min —; t = 360 min
 . Sample temperature is initially stabilized at 25 °C. A photostationary state can be estimated around 50%. Irradiation was performed in degassed acetonitrile solution in a quartz cell with a portable UV lamp 4 W at 366 nm.
. Sample temperature is initially stabilized at 25 °C. A photostationary state can be estimated around 50%. Irradiation was performed in degassed acetonitrile solution in a quartz cell with a portable UV lamp 4 W at 366 nm.
When crystallization attempts were carried out in the presence of light the photodimer 2, less soluble than 1, crystallized out exclusively. The head-to-tail dimeric structure of the photoproduct could be unambiguously determined by X-ray crystallography (Figure 3). As with the parent ligand, the bipyridine units adopt an expected trans geometry with significant deviation from coplanarity (torsion angles: 143.0° and 165.4°). In the crystal structure, the dimer displays an inversion center located in the center of the molecule. The crystal packing is ensured mainly by two π-π interactions (Figure 4). The principal π-π stacking is observed between two adjacent bipyridine fragments with a distance of 3.53 Å between the planes. Additional π-π interaction involves the other pyridyl substituent stacking with its analogue of the neighboring molecule. This effect is weaker as the interplanar separation is 3.62 Å.
Figure 3.
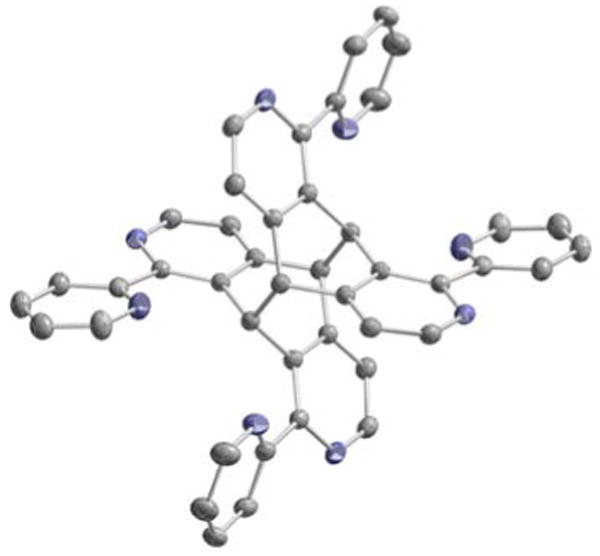
Crystal structure of ligand 2.
Figure 4.
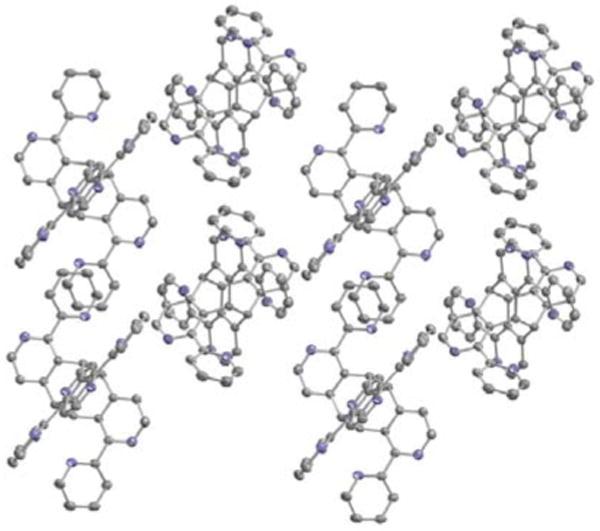
Packing of ligand 2 in the crystal.
Upon irradiation at 366 nm, ligand 1 undergoes dimerization at the 9 and 10 carbons. This photochemical transformation can be easily monitored by fluorescence spectroscopy as the anthracene core responsible for the molecule's emission at 470 nm is disrupted upon dimerization, thereby providing a probe for following the progress of the reaction (Figure 2b).22 A considerable amount of dimer 2 was prepared by irradiation (366 nm) of ligand 1 in a degassed acetonitrile solution followed by purification using preparative TLC.14 Most signals in the 1H NMR of the dimer 2 feature an up-field shift when compared to the corresponding protons in ligand 1 (Figure 5). As expected, protons 9 and 10 display the most dramatic shift as their hybridization changes from sp2 to sp3 (Figure 5, blue line). Their multiplicity also changes from singlets to doublets, as expected for a head-to-tail dimer. A group of three protons (4′, 5′ and 6′) are only slightly shifted (red line), consistent with a minimal change in their environment upon dimerization. Protons 3 and 4, along with proton 3′, experience a significant shift (0.4–0.6 ppm) (green line). For protons 3 and 4, the significant shielding can be attributed to ring current effects of the aromatic rings located directly below. On the other hand, the shift of proton 3′ could be attributed to the absence of the deshielding cone produced by the central ring as it loses aromaticity upon dimerization.
Figure 5.
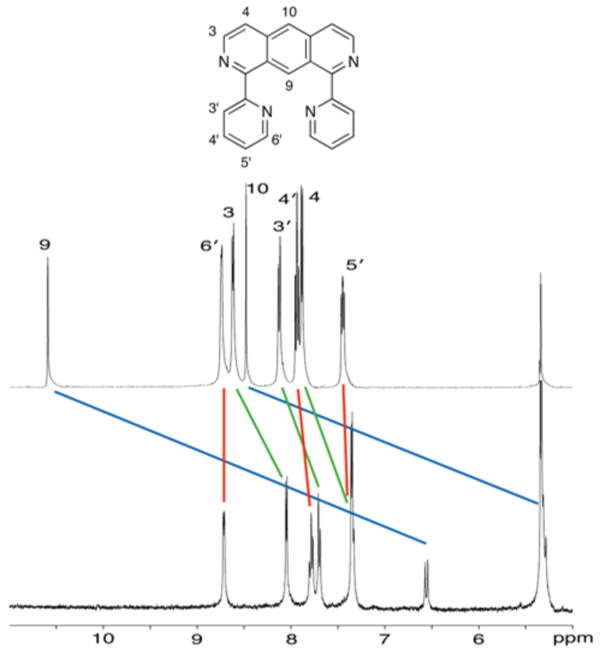
1H NMR spectra in CD2Cl2 and assignment of ligand 1 (top) and the photodimer 2 (bottom).
The photodimer 2 was found to be thermally stable, with no indication for dissociation to monomer 1 over 12 hours in solution at room temperature. Preliminary results suggest, however, that the dimerization may be reversed by irradiation at 254 nm (Scheme 2 and Figure S.12).14 This finding is in agreement with previous literature reports of photoreversible dimerization.11,23 Note, however, that further optimization is required since under the current unoptimized conditions the reaction is only partially reversible and repetitive cycling appears to result in sample degradation.14
Scheme 2.

Reversible photoinduced dimerization and dissociation reactions
To the best of our knowledge, ligand 1 is the first example of a photodimerizable ligand, in which the diimine core is embedded within the bridging anthracene unit. Akin to such systems that undergo photoinduced structural modifications (e.g., cys-trans isomerization,24 photocyclization25) this paradigm system could be considered as a photoswitch to which added properties may eventually be incorporated upon binding to metal ions.
Supplementary Material
Supporting Information Available: Full synthetic details and analytical data for all new compounds; photoinduced dimerization and dissociation; crystal data and refinment information for 1 and 2; spectra of mono- and dinuclear Ru2+ complexes of 1; and reactions of 1 and 2 with Fe2+. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank the National Institutes of Health (GM 069773) and NSF (CHE 0213323) for support.
References
- 1.(a) Ruben M, Rojo J, Romero-Salguero FJ, Uppadine LH, Lehn JM. Angew Chem, Int Ed. 2004;43:3644–3662. doi: 10.1002/anie.200300636. [DOI] [PubMed] [Google Scholar]; (b) Sauvage JP, Collin JP, Chambron JC, Guillerez S, Coudret C, Balzani V, Barigelletti F, De Cola L, Flamigni L. Chem Rev. 1994;94:993–1019. [Google Scholar]
- 2.Collin JP, Jouvenot D, Koizumi M, Sauvage JP. Eur J Inorg Chem. 2005:1850–1855. doi: 10.1021/ic050246a. [DOI] [PubMed] [Google Scholar]
- 3.Brouwer A, Frochot C, Gatti FG, Leigh DA, Mottier L, Paolucci F, Roffia S, Wurpel GWH. Science. 2001;291:2124–2128. doi: 10.1126/science.1057886. [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto A, Hirooka A, Namiki K, Kurihara M, Murata M, Sugimoto M, Nishihara H. Inorg Chem. 2005;44:7547–7558. doi: 10.1021/ic051184r. [DOI] [PubMed] [Google Scholar]
- 5.Haga MA, Takasugi T, Tomie A, Ishizuya M, Yamada T, Hossain MD, Inoue M. J Chem Soc Dalton Trans. 2003;10:2069–2079. [Google Scholar]
- 6.Zelikovich L, Libman J, Shanzer A. Nature. 1995;374:790–792. [Google Scholar]
- 7.Becker HD. Chem Rev. 1993;93:145–172. [Google Scholar]
- 8.Bouas-Laurent H, Castellan A, Desvergne JP, Lapouyade R. Chem Soc Rev. 2000;29:43–55. [Google Scholar]
- 9.(a) Hiraga H, Morozumi T, Nakamura H. Eur J Org Chem. 2004:4680–4687. [Google Scholar]; (b) Desvergne JP, Lahrahar N, Gotta M, Zimmermann Y, Bouas-Laurent H. J Mater Chem. 2005;15:2873–2880. [Google Scholar]
- 10.Another system based on a [2.2](9,10)anthracenophane is used to coordinate [Ru(Cp)]+ in an η6 fashion. See: Glatzhofer DT, Liang Y, Khan MA. J Chem Soc, Chem Commun. 1993:742–743.
- 11.Beyerler A, Belser P, De Cola L. Angew Chem Int Ed. 1997;36:2779–2781. [Google Scholar]
- 12.Trouts TD, Tyson DS, Pohl R, Kozlov DV, Waldron AG, Castellano FN. Adv Funct Mater. 2003;13:398–402. [Google Scholar]
- 13.Zeng H, Miller RS, Flowers RA, Gong B. J Am Chem Soc. 2000;122:2635–2644. [Google Scholar]
- 14.See Supporting Information for additional experimental details and data.
- 15.Sakamoto T, An-naka M, Kondo Y, Yamanaka H. Chem Pharm Bull. 1986;34:2754–2759. [Google Scholar]
- 16.Janin YL, Roulland E, Beurdeley-Thomas A, Decaudin D, Monneret C, Poupon MF. J Chem Soc, Perkin Trans. 2002;1:529–532. [Google Scholar]
- 17.Loren JC, Siegel JS. Angew Chem, Int Ed. 2001;40:754–757. [PubMed] [Google Scholar]
- 18.A succesful double cross coupling reaction requires thoroughly dried zinc dichloride (24 h at 55 °C under reduce pressure).
- 19.Interestingly, the usual stain employed to visualize bipyridines (Fe(NH4)2(SO4)2 in water) does not yield the typical red color, but rather a bright green color. See Figure S.15.
- 20.Single-crystal-to-single-crystal reactions are very rare for anthracene derivatives; however, dimerization can be performed in the crystalline state followed by recrystallization. See Turowska-Tyrk I, Grześniak K. Acta Cryst. 2004;C60:o146–o148. doi: 10.1107/S0108270103026775.
- 21.The emission quantum yield was determined to be 0.02.
- 22.Note the photodimer 2 is non-emissive under these conditions.
- 23.European Patent Application 0 394 846 A2
- 24.Shinkai S, Nakaji T, Nishida Y, Ogawa T, Manabe O. J Am Chem Soc. 1980;102:5860–5865. [Google Scholar]
- 25.(a) Irie M, Fukaminato T, Sasaki T, Tamai N, Kawai T. Nature. 2002;420:759–760. doi: 10.1038/420759a. [DOI] [PubMed] [Google Scholar]; (b) Fraysse S, Coudret C, Launay JP. Eur J Inorg Chem. 2000:1581–1590. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Full synthetic details and analytical data for all new compounds; photoinduced dimerization and dissociation; crystal data and refinment information for 1 and 2; spectra of mono- and dinuclear Ru2+ complexes of 1; and reactions of 1 and 2 with Fe2+. This material is available free of charge via the Internet at http://pubs.acs.org.