

Abstract
Recent reports have suggested the involvement of interleukin-4 (IL-4) in glomerular pathophysiology. Using immunohistochemistry and reverse transcriptase polymerase chain reaction we investigated the renal lesions in transgenic (tg) mice with widely distributed IL-4 expression including the kidney, and measured the serum levels of the cytokines transforming growth factor-β (TGF-β) and IL-4 by ELISA.
Transgenic animals exhibited glomerular hypertrophy with progressive mesangial sclerosis leading to renal failure. Renal IL-4 transcript expression, mesangial accumulation of collagen types I, III, IV and V, and immune deposition accompanied by increased expression of TGF-β1 protein and mRNA were observed. Seven day-old transgenic animals showed early renal fibrotic changes in the absence of immune deposits or TGF-β1 upregulation. The sera of transgenic mice not only showed elevated levels of circulating IL-4 (tg: 76.6 pg/ml ± 7.1 vs wildtype (wt): < 3 pg/ml), but significantly decreased TGF-β1 levels (tg: 18.9 ng/ml ± 4.1 vs wt: 38.7 ng/ml ± 2.9; P < 0.005). The disease severity correlated with the serum IL-4/TGF-β1 ratio rather than with the IL-4 concentration.
These data suggest that renal IL-4 production results in matrix accumulation prior to any immunological insult, that increased circulating IL-4/TGF-β1 ratios are associated with renal immunopathological manifestations and that upregulation of renal TGF-β1 expression following glomerular Ig deposition accelerates the sclerosis and exacerbates disease development.
Keywords: interleukin-4, transforming growth factor-β, glomerulosclerosis, transgenic mice
Progressive glomerular sclerosis is a common feature of a variety of glomerular lesions. Several factors, both immunological and nonimmunological, contribute to the progression of disease (Wardle 1996). The exact pathogenic mechanisms involved in the process from the initial glomerular insult to the increased deposition of mesangial extracellular matrix are still largely unknown (Müller et al. 1996). However, there is increasing evidence that cytokines play a critical role as mediators of inflammation and as factors for the progression of various renal disorders (Takemura et al. 1994; Neale et al. 1995; Isbel & Atkins 1996).
Recently interleukin-4 (IL-4) has been implicated in the pathogenesis of experimental and human glomerulonephritis (Furusu et al. 1997; Nakajima et al. 1997). It has been shown that intrinsic human glomerular cells express mRNA for both IL-4 and IL-4 receptor, and that IL-4 expression is upregulated in various types of glomerulonephritis (Furusu et al. 1997). Furthermore, an in vitro study has suggested that IL-4 contributes to the pathological changes in glomerular diseases by stimulating collagen synthesis in human mesangial cells (Nakazato et al. 1996).
IL-4 is an approximately 20kD, secreted, pleiotropic cytokine, predominantly produced by CD4+ helper T lymphocytes, as well as by non-T, non-B cells of the mast cell lineage (Paul 1991). IL-4 mediates its many functions by binding to receptors expressed on target cells. Such receptors have been reported to exist on B and T lymphocytes, macrophages and various other cell types including lymphoid, mast and haematopoietic cells, fibroblast and stromal cell lines (Lowenthal et al. 1988), and also on intrinsic human glomerular cells (Furusu et al. 1997).
We have recently found that constitutive expression of IL-4 under the control of a MHC class I promoter leads to autoimmune-type disorders characterized by increased B cell-surface MHC class II expression, elevated responsiveness of B cells to polyclonal ex vivo stimulation, and increased IgG1 and IgE serum levels in mice (Erb et al. 1997). These mice showed increased production of autoantibodies, developed anaemia and manifested glomerulonephritis with immune deposition. In other IL-4 transgenic (tg) mouse models, where T cells and/or B cells express tg IL-4, no renal pathological changes have been reported (Tepper et al. 1990; Lewis et al. 1991; Müller et al. 1991). Interestingly, the glomerular lesions in MHC-I/IL-4 tg mice resembled the progressive renal disease observed in transforming growth factor-β1 (TGF-β1) tg mice which show elevated plasma TGF-β1 levels (Kopp et al. 1996). However, the extrarenal autoimmune-type manifestations in MHC-I/IL-4 tg mice were similar to the disorders seen in TGF-β1 deficient mice which did not show any renal changes histologically despite immunohistochemical detection of glomerular immune deposits (Yaswen et al. 1996). It has been well established that TGF-β exerts various effects, including immunosuppression and fibrogenesis, and acts in a context-dependent fashion (McCartney-Francis & Wahl 1994). The role of TGF-β in tissue fibrosis, particularly in connection with progressive renal disease, has been studied extensively in recent years (Border et al. 1991; Sharma & Ziyadeh 1994).
We have investigated the mechanism(s) that are responsible for the development of the renal lesions observed in MHC-I/IL-4 tg mice including the role of TGF-β. Kidneys of tg and littermate wildtype (wt) mice were analysed by light microscopy, immunohistochemistry and reverse transcriptase-polymerase chain reaction (RT-PCR). In addition, the concentrations of IL-4 and TGF-β1 in the sera from these animals were determined by ELISA.
Materials and methods
Mice
The IL-4 tg mice were originally established from B6C3F1/CrlBR mice and then bred with B6C3F1 mice (Erb et al. 1994). The use of an MHC class I promoter leads to a low level of IL-4 production in almost all cell types. The mice were tested for the presence of the transgene using genomic PCR (see below and Table 1). Littermates were used as wild-type controls. One to 12 week-old mice (5–6 litters each) were analysed in this study. All animals used in this project were bred, housed and maintained under conventional conditions in an isolation facility at the Wellington School of Medicine Animal Facility. Access to food and drinking water was available at all times. The Wellington School of Medicine Animal Ethics Committee approved the experiments conducted in this project.
Table 1.
Sense (+) and antisense (−) primers used for genomic PCR and RT-PCR

Histology
Sections (2–3 μm) of formalin-fixed paraffin-embedded kidney tissues from tg and wt mice were cut and stained with haematoxylin & eosin and Congo Red.
Immunohistochemistry
Immunoperoxidase labelling
Frozen sections from tg and wt mice (one to 12 week-old) were fixed in acetone for 5 min at room temperature (rt) and incubated with primary antibodies (diluted in 1% TBS/BSA) overnight at 4 °C. Paraffin sections were digested with bacterial protease XXIV (Sigma, St Louis, MO) prior to incubation with peroxidase-conjugated rabbit antimouse immunoglobulins (anti-total Igs, DAKO Corp., Carpinteria, CA) for 60 min at rt. Controls included both the omission of and the substitution of primary antibodies with either an irrelevant mAb of the same isotype, or with normal rabbit or goat serum. Endogenous peroxidase was blocked with 0.3% H2O2 in TBS for 20 min at rt, or by the addition of 1% d-glucose (BDH, Poole, UK) and 0.85 units/ml glucose-oxidase type VII (Sigma) (during incubation with secondary/bridging antibodies in 5% casein/TBS). Cross-reactivity of mouse Igs with Igs of other species that were present in the detection reagents was prevented by adding 5% normal mouse serum to each antibody mix.
Monoclonal antibodies to murine T cell subsets, CD4 and CD8 (PharMingen, San Diego, CA) were used at a 1 : 800 dilution each. Binding of mAb was detected with a sensitive three-step immunoperoxidase technique (Rüger et al. 1994, 1996), using peroxidase-conjugated rabbit antirat F(ab′)2 (Serotec, Oxford, England; incubation at room temperature for 45 min) and peroxidase- conjugated swine antirabbit Ig (DAKO; incubation at rt for 45 min), both antibodies diluted 1 : 100. For staining of macrophages, a biotinylated antibody (Mac-1, PharMingen, San Diego, CA, diluted 1 : 500) was used, followed by incubation with peroxidase-conjugated streptavidin (BioGenex, San Ramon, CA) (1 : 100 in 0.5% Casein/TBS) for 30 min at rt. A peptide-specific rabbit polyclonal antibody to TGF-β1 (Santa Cruz Biotech, Santa Cruz, CA), and goat polyclonal antibodies to types I, III, IV and V collagen (Southern Biotech, Birmingham, AL) were all used at 1 : 100 dilutions. The reactivity of these polyclonal antibodies was revealed using peroxidase-conjugated swine antirabbit Ig (1 : 100 dilution) and rabbit antigoat Ig (1 : 100) (both DAKO) as bridging reagents followed by peroxidase-conjugated rabbit antiswine Ig (1 : 100) (DAKO) and goat peroxidase antiperoxidase (DAKO) (1 : 50), respectively. The sections were exposed to 0.05% diaminobenzidine in 50 mm Tris/HCl, pH 7.5, for 10 min or until the desired colour reaction occurred, counterstained with Mayer's Hemalum for 1 min at rt, rinsed for 10 min with tap-water and mounted with Glycergel Mounting Medium (DAKO).
Immunofluorescence microscopy
Frozen sections were postfixed as for immunoperoxidase labelling, and goat serum (diluted 1 : 10 in TBS/BSA) applied for 30 min as a blocking reagent. Slides were drained and incubated for 60 min at rt with FITC-conjugated goat anti-complement 3 (C3) (Nordic, Tilburg, Netherlands) (diluted 1 : 200 in TBS/BSA). The slides were washed three times in TBS buffer, mounted with 10% glycerol/TBS and viewed under a Leitz fluorescence microscope.
Cell counting
To estimate the number of Mac-1-positive cells in kidneys of MHC-I/IL-4 tg and control wt mice, a minimum of 20 equatorially sectioned glomeruli were assessed per animal, and the results were expressed as macrophages per glomerular cross section (macs/gcs) (mean ± SEM). Statistical evaluation was carried out using Student's t-test with P-values > 0.05 being considered not significant.
PCR
DNA obtained from the tail was used for typing of MHC-I/IL-4 tg mice and wt littermates. After a ‘hot start’ (95 °C for two min), the protocol involved denaturation at 94 °C for 15 s, annealing of primers at 60 °C for 15 s, and extension at 72 °C for 40 s with a final extension step at 72 °C for 10 min. A total of 30 cycles were performed. PCR products were electrophoresed on 2% agarose gels in tris acetate ethylene diamine tetraacetic acid buffer. Ethidium bromide stained gels were documented using the Eagle Eye II Digital Imaging System (Stratagene, CA).
RT-PCR
Total RNA from snap-frozen kidneys obtained from the same mice used for immunohistochemistry was extracted using TRIzol™ Reagent (GIBCO, Life Technologies, Inc., Gaithersburg, MD) according to the manufacturer's instructions.
Prior to assessing the presence of TGF-β1 and IL-4 mRNA in renal tissue (primer sequences see Table 1), the amount of transcribable RNA in each sample was equalized by competitive RT-PCR as described previously (Rüger et al. 1996; Hasan et al., 1998) using gamma-glutamylcysteine synthetase (γGCS) as the control gene. Intron-spanning γGCS primers (for sequence see Table 1) based on the published sequence (Yan & Meister 1990) were designed to generate a predicted product of 391 bp. RNA was amplified using the Perkin Elmer (Norwalk, CT) rTth RT-PCR kit, with a 10-min RT step followed by 45 cycles of three stage PCR with annealing at 60 °C and 63 °C for IL-4 and TGF-β1, respectively, in a Perkin Elmer 9600 machine. RT-PCR products were analysed by agarose gel electrophoresis as described above.
Renal function
Serum urea levels were measured in a Hitachi 717 Automatic Analyser (Boehringer Mannheim, Mannheim, Germany). 20-h urine samples were collected from mice kept in metabolic cages. Proteinuria was determined by the Bradford assay (Bradford 1976).
IL-4 and TGF-β1 ELISAs
Quantification of IL-4 and TGF-β1 concentrations in the sera from three to 12 week-old MHC-I/IL-4 tg and wt mice was determined using commercially available kits (IL-4: R & D Systems, Minneapolis, MN; TGF-β1: Genzyme, Cambridge, MA). The assays were performed following the manufacturers' instructions. Absorbance was measured at 405 nm (for IL-4) and 450 nm (for TGF-β1) in a Bio-tek Microplate Autoreader.
Statistics
Statistical analyses of ELISA and biochemical results were performed using the Mann–Whitney nonparametric test. All data are expressed as mean ± SEM. Correlation analyses were undertaken using the StatView™ programme.
Results
Renal histology
On light microscopy the kidneys of MHC-I/IL-4 tg mice showed a disproportionate increase in glomerular size and an increase in mesangial matrix which resulted in severe progressive glomerulosclerosis (Figure 1a). The histological renal abnormalities were present to varying degrees in all tg mice from three weeks of age onwards. Structural changes included a thickened glomerular basement membrane, and an accumulation of eosinophilic material in the mesangium of affected glomeruli. As Congo Red staining of kidney sections was negative (data not shown) the diagnosis of amyloidosis was excluded. Most but not all glomeruli within each kidney showed a similar degree of scarring. The kidneys of seven day-old tg animals showed no obvious histological changes (Figure 2a). Disease progression accelerated generally after weaning (three weeks after birth) with the mean survival time being seven weeks. Renal interstitial fibrosis was not a major feature in this model and occurred only in mice which showed a slow disease progression and survived beyond 12 weeks (data not shown).
Figure 1.
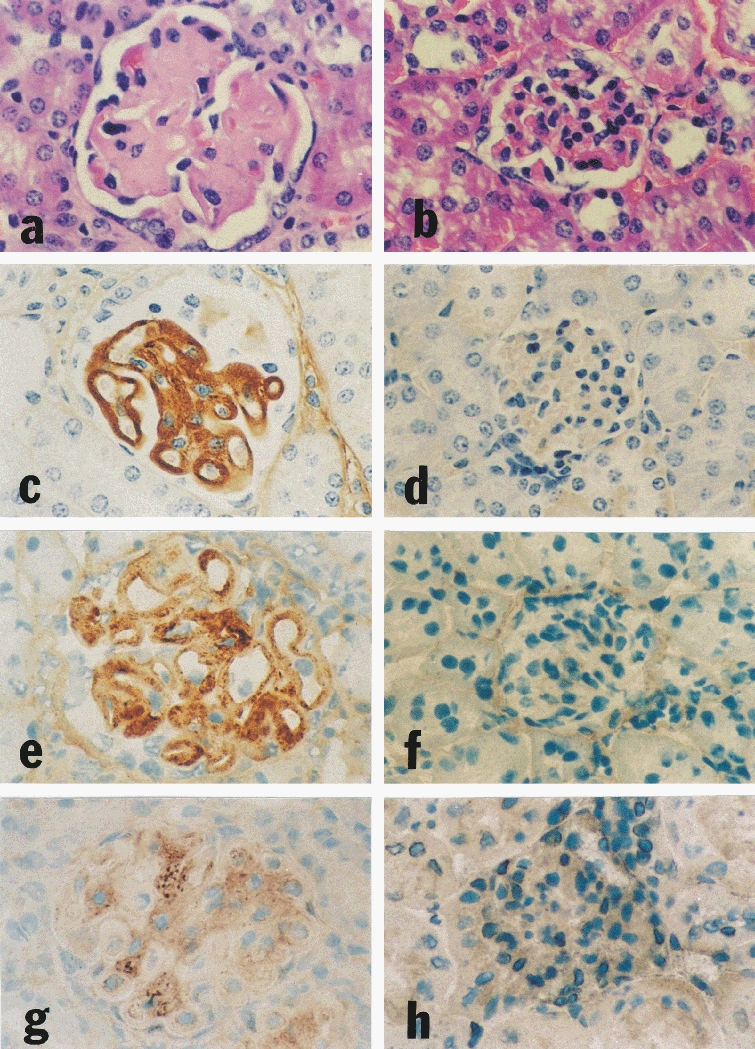
Renal histology and immunohistochemistry of eight week-old IL-4 tg and wt mice. The kidney of an IL-4 tg animal (a) shows extensive glomerular hypertrophy and hypocellularity, basement membrane thickening and mesangial accumulation of eosinophilic material compared with the kidney of a wt littermate (b). The same tg mouse exhibits heavy glomerular Ig deposition (c); the control wt mouse kidney is negative for total mouse Ig staining (d). The expanded mesangium of the tg mouse kidney contains increased collagen type I (e), compared with the normal mouse kidney of the same age (f). TGF-β1 protein is increased in glomeruli of IL-4 tg kidneys (g); the kidney of a wt littermate displays low levels of glomerular TGF-β1 protein (h). a, b (H & E stain, ×290); c-h (immunoperoxidase staining, ×290).
Figure 2.
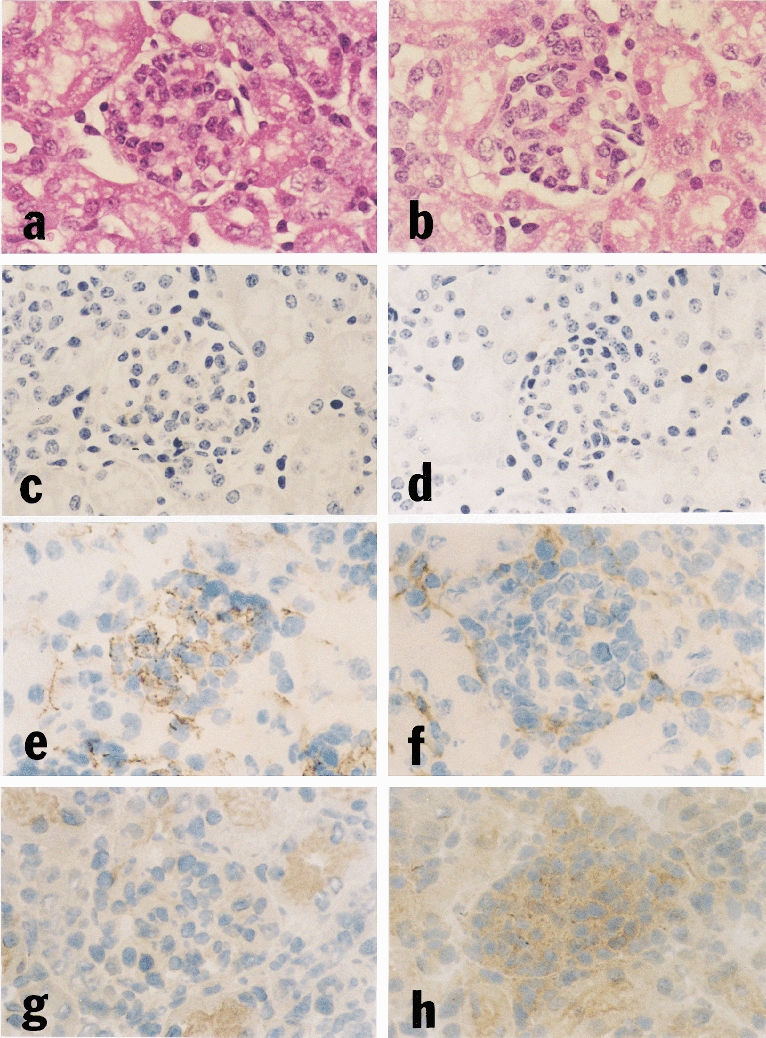
Renal histology and immunohistochemistry of seven day-old IL-4 tg and wt mice. No apparent histological difference is seen in glomeruli of (a) tg and (b) wt kidneys at this early stage. Glomerular Ig deposition is absent in both (c) tg and (d) wt kidneys, but staining for collagen type V reveals increased deposition in the mesangial area of (e) seven day-old tg mice compared with (f) wt littermates. The increased collagen deposition is not associated with increased TGF-β1 protein staining; on the contrary, glomeruli of (g) seven day-old tg mice show less TGF-β1 protein than their wt littermates (h). a, b (H & E stain, ×290); c-h (immunoperoxidase staining, ×290).
Immunohistochemistry
The glomeruli of tg mice aged between three and 12 weeks showed Ig deposits (Figure 1c), while those of wt control mice were negative (Figure 1d). In tg mice, the more severe the histological changes, the greater the glomerular Ig deposits. Extensive glomerular immunoglobulin deposition was accompanied by positive complement 3 staining (data not shown). However, complement was either undetectable or only minimal in the less severely affected glomeruli. In kidneys of seven day-old tg mice staining for Ig deposits was negative (Figure 2c).
There was no difference in the number of infiltrating macrophages/monocytes in tg animals compared with wt control mice (wt (n = 17): 0.31macs/gcs ± 0.05 vs tg (n = 28): 0.27macs/gcs ± 0.06; n.s.). Immunolabelling of kidney sections with antibodies to T cells (CD4 and CD8 subsets) was negligible in both wt and tg mice suggesting that these cells were not directly involved in the progression of disease (data not shown).
Immunohistochemistry for glomerular extracellular matrix (ECM) components revealed increased staining for collagen types I (Figure 1e,f), III, IV and V (data not shown) in the mesangium of tg glomeruli compared with wt glomeruli. Even at seven days of age some of the tg mice showed a slight but definite increase in mesangial ECM components (Figure 2e).
The mesangial ECM accumulation in tg mouse kidneys was accompanied by a marked increase in glomerular TGF-β1 protein expression (Figure 1g). Interestingly, this increase was not detected in the kidneys of seven day-old tg mice in which matrix expansion was already evident. On the contrary, staining for glomerular TGF-β1 protein in these mice was reduced (Figure 2g,h).
RT-PCR
The kidneys of MHC-I/IL-4 tg mice expressed mRNA for IL-4; wt kidneys were negative (Figure 3a,b). Consistent with the increased glomerular staining for TGF-β1 protein in three to 12 week-old tg mice was the increased renal mRNA for this cytokine (Figure 3a). The upregulation of TGF-β1 in MHC-I/IL-4 tg mouse kidneys therefore was both at the transcriptional and translational level. On the other hand, the aberrant renal IL-4 expression in seven day-old tg mice was accompanied by decreased transcription levels for TGF-β1 (Figure 3b).
Figure 3.
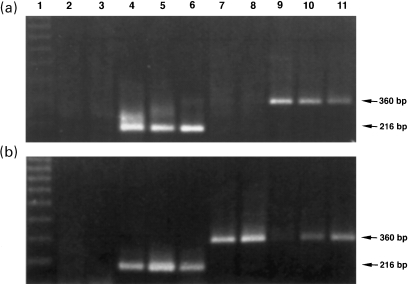
IL-4 and TGF-β1 expression in the kidneys of (a) eight week-old and (b) seven day-old tg and wt mice. RNA samples from wt (a and b: lanes 2,3 and 7,8) and tg (a and b: lanes 4,5,6 and 9,10,11) kidneys, equalized with γGCS, were subjected to RT-PCR for IL-4 (a and b: lanes 2–6) and TGF-β1 (a and b: lanes 7–11). Only tg kidneys show IL-4 expression, as demonstrated by the 216 bp-band (a and b: lanes 4,5,6). Wt kidneys do not express IL-4 mRNA (a and b: lanes 2,3). Renal TGF-β1 expression is upregulated in eight week-old tg mice, as shown by the 360 bp-band (a: lanes 9,10,11); the TGF-β1 expression in adult wt kidneys is negligible (a: lanes 7,8). In contrast, the renal TGF-β1 expression in seven day-old wt mice (b: lanes 7,8) is stronger compared with tg littermates, which show only weak 360 bp-bands (b: lanes 9,10,11) indicating decreased TGF-β1 mRNA (ethidium bromide-stained agarose gels).
Renal function
As an indicator of renal function, urinary protein and serum urea levels were examined in tg and wt mice. MHC-I/IL-4 tg mice developed oliguria and proteinuria (tg (n = 11): 2.61 mg/ml ± 0.65 vs wt (n = 7): 0.11 mg/ml ± 0.02; P < 0.005). They had significantly higher serum urea levels compared with wt animals indicating impaired kidney function (tg (n = 16): 17.0 mm ± 3.3 vs wt (n = 10): 5.9 mm ± 0.7; P < 0.005). The serum urea level (BUN) increase correlated positively with the degree of glomerulosclerosis (data not shown).
Serum IL-4 and TGF-β1 levels
As expected the sera of MHC-I/IL-4 tg mice exhibited high IL-4 levels (tg (n = 16): 76,6 pg/ml ± 7.1). The circulating IL-4 levels of wt controls (n = 10) were below detection level (< 3 pg/ml). In contrast, serum TGF-β1 levels in MHC-I/IL-4 tg mice were significantly diminished compared with those in wt mice (wt (n = 10): 38.7 ng/ml ± 2.9 vs tg (n = 16): 18.9 ng/ml ± 4.1; P < 0.005). Neither the IL-4-nor the TGF-β1-serum concentrations of tg mice showed an age-dependent pattern.
The high serum IL-4 concentrations did not correlate with the elevated serum urea in MHC-I/IL-4 tg mice, but there was a negative correlation between serum urea and serum TGF-β1 levels (correlation coefficient: −0.578; P < 0.01). Furthermore, there was an even more significant positive correlation between serum urea levels and the ratio of serum IL-4 to serum TGF-β1 in IL-4 tg mice (Figure 4a). Not surprisingly, the IL-4/TGF-β1 ratio also correlated positively with the progressive glomerulosclerotic changes in the kidneys of tg mice (Figure 4b).
Figure 4.
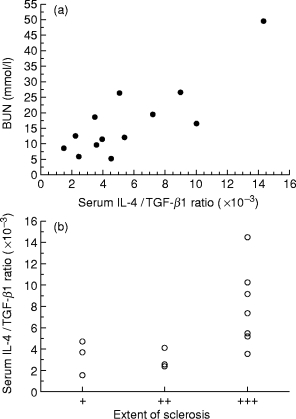
Relationship of serum IL-4/TGF-β1 ratio to renal dysfunction (a) Correlation between serum urea and IL-4/TGF-β1 serum ratio in MHC-I/IL-4 mice (n = 13; correlation coefficient: 0.822; P < 0.001). High serum urea levels are associated with high IL-4 serum levels together with decreased TGF-β1 serum concentrations. (b) Relationship of the serum IL-4/TGF-β1 ratio to the degree of glomerular scarring in MHC-I/IL-4 mice. The extent of glomerulosclerosis was evaluated semiquantitatively in a double-blind fashion. Severity of glomerulosclerosis was scored, as follows: + mild mesangial expansion and basement membrane thickening; + + mild glomerular hypertrophy and mesangial matrix accumulation; + + + extensive glomerular hypertrophy and hypocellularity, complete glomerulosclerosis.
Discussion
Recently we have reported that constitutive IL-4 expression under a MHC class I promoter leads to autoimmune-type disorders with severe renal manifestations in mice (Erb et al. 1997). This study has shown that the renal lesions of tg animals consist of glomerular hypertrophy, mesangial matrix expansion and Ig deposition resulting in severe progressive glomerulosclerosis and renal failure. The glomerular Ig deposition in these mice is accompanied by increased renal production of TGF-β1.
Kidneys from seven day-old MHC-I/IL-4 tg mice show early fibrotic changes in glomeruli with accumulation of interstitial collagen types I, III, IV and especially V, but without evidence of glomerular Ig deposition or TGF-β1 upregulation. Since these mice express the IL-4 transgene in the kidney, this suggests that the local renal IL-4 expression/production may directly mediate glomerular ECM accumulation. In a recent study it has been shown that intrinsic glomerular cells of normal human kidney express low levels of IL-4 and IL-4 receptor, and that their expression is increased in several types of glomerulonephritis raising the possibility of an IL-4-mediated pathway of fibrosis (Furusu et al. 1997). IL-4 can mediate fibrotic events directly through fibroblast activation and upregulation of collagen gene expression and/or indirectly by down-regulation of antifibrotic factors. For example, IL-4 receptor is expressed on murine lung fibroblasts suggesting that elevated levels of IL-4 at a site of injury could result in development of fibrosis (Sempowski et al. 1994), and human fibroblasts have been shown to synthesize elevated levels of ECM upon IL-4 stimulation (Postlethwaite et al. 1992). Moreover, IL-4 can modulate the collagen synthesis by human mesangial cells in a type-specific manner (Nakazato et al. 1996). Transgenic mice expressing IL-4 under the control of the human insulin promoter develop a localized host response resulting in excessive generation of ECM in the pancreas (Mueller et al. 1997). Hence, as kidneys of seven day-old MHC-I/IL-4 tg mice show signs of ECM accumulation without TGF-β involvement, it is possible that renal IL-4 expression has a direct fibrogenic effect.
In the IL-4 tg mouse model studied here increased expression of TGF-β1 occurs only after immunological insult to the kidney, and this leads to excessive matrix accumulation and accelerates the disease development. Several studies have established the role of TGF-β1 as a powerful promoter of fibrosis (Border et al. 1991; Bruijn et al. 1994; Sharma & Ziyadeh 1994), and it has been shown that the glomerular expression of TGF-β1 is increased in several types of renal disease (Wardle 1996). In vitro studies have demonstrated that the exposure of mesangial cells to IgA and IgG complexes triggers the formation of ECM, and that this is mediated through the autocrine synthesis of TGF-β (López-Armada et al. 1996). Interestingly, the renal lesions in TGF-β transgenic mice (Kopp et al. 1996), which have elevated plasma levels of TGF-β1, resemble the pathological changes of kidneys from MHC-I/IL-4 tg mice. Therefore it was initially surprising to us that TGF-β1 levels in the sera of MHC-I/IL-4 tg mice are significantly decreased when compared with wt controls. This difference in circulating TGF-β1 concentrations is unlikely to be due to lysed platelets, since MHC-I/IL-4 tg mice and wt controls had similar platelet counts (B. Rüger, unpublished observation).
As well as playing an important role in ECM accumulation, TGF-β also has powerful anti-inflammatory and immunosuppressive properties (McCartney-Francis & Wahl 1994). Several recent studies have shown a protective effect of TGF-β1 preventing or delaying the onset of various Th1 and Th2 mediated autoimmune disorders (Kuruvilla et al. 1991; Chen et al. 1994; Bridoux et al. 1997). In this context, the low TGF-β1 serum levels in MHC-I/IL-4 tg mice are consistent with the observed autoimmune disorders characterized by elevated autoantibody titres to nuclear antigens and upregulated MHC class II expression (Erb et al. 1997). Apart from the decreased serum TGF-β1 levels, MHC-I/IL-4 tg mice exhibit dramatically elevated IL-4 serum levels. It has been shown previously that MHC-I/IL-4 tg mice have increased serum IgE levels (Erb et al. 1994), however, the antibodies present in the kidneys of tg animals were of the IgM, IgG1, IgG2a and IgA isotypes, whereas IgE was not detected (Erb et al. 1997), demonstrating that glomerular IgE deposition is not involved in the disease development. Animals with the highest IL-4 serum levels did not show the most impaired kidney function. However, circulating TGF-β1 seems to play a crucial role in the development of the immune-mediated renal disorders in this model, with an apparent protective effect of higher concentrations. Moreover, the strong correlation between the IL-4/TGF-β1 serum ratio and the degree of glomerular scarring suggests that the cytokine balance is more important than absolute concentrations of single cytokines. Machold et al. have shown that TGF-β is able to inhibit IL-4 induced production of IgM, IgG, IgA and IgE in vitro (Machold et al. 1993). This is consistent with our finding in MHC-I/IL-4 tg mice that higher serum TGF-β levels are associated with fewer glomerular Ig deposits and less severe renal histological changes. However, there is no correlation between serum TGF-β concentrations and total serum IgG in MHC-I/IL-4 tg mice (unpublished observation) suggesting the immunosuppressive effect of TGF-β is on autoreactive antibodies rather than on general serum Ig levels. This is supported by a report showing that intramuscular injection of TGF-β expression vectors into lupus prone mice (MRL/lpr/lpr) decreased autoantibodies to chromatin (Raz et al. 1993). It is therefore not surprising that TGF-β deficient mice show similar autoimmune-features (Yaswen et al. 1996) as MHC-I/IL-4 tg mice. These TGF-β1 deficient mice are not able to produce TGF-β1 by themselves, but they are not ‘protein knockouts’, since TGF-β1 has been detected in several tissues, and has been shown to be derived from maternal sources through placenta and lactation (Letterio et al. 1994). Although maternal protection does provide short-term survival of these mice, the levels of maternally derived circulating TGF-β1 levels are not sufficient to prevent the onset of disease (Letterio et al. 1994).
It is of interest that despite glomerular Ig deposition being a common feature both in MHC-I/IL-4 tg mice as well as in TGF-β1 knockout mice (Yaswen et al. 1996), only the MHC-I/IL-4 tg mice show structural glomerular changes and develop glomerulosclerosis with a dramatic local increase of both renal TGF-β1 mRNA and protein. One possible explanation for the apparent discrepancies is the shorter survival time of the TGF-β deficient mice (they die by three to four weeks of age). However, this is unlikely, since some of the IL-4 tg mice have already developed extensive glomerular fibrosis by three weeks of age. Another alternative is that, although TGF-β levels are low in both the IL-4 tg mice and the TGF-β1 knockout mice, local renal production of TGF-β could be induced by Ig deposition only in MHC-I/IL-4 tg mice and this leads to fibrosis. In contrast, TGF-β deficient mice are unable to actively produce TGF-β, and therefore ECM accumulation does not occur.
The involvement in this transgenic animal model of other cytokines which are known to play a role in renal diseases, such as PDGF (Floege et al. 1992) and IL-6 (Horii et al. 1993), cannot be excluded. However, there is indirect evidence that PDGF is not involved here, as it induces marked mesangial proliferation (Isaka et al. 1993), and this is not a feature in MHC-I/IL-4 tg mice at any stage of disease development. Similarly, in tg mice expressing the human IL-6 gene under the human Ig heavy chain promoter the renal pathology is characterized by a striking proliferation of mesangial cells (Suematsu et al. 1989).
It is noteworthy that other IL-4 tg models show elevated serum autoantibody titres similar to those in the model studied here (Foote et al. 1996), but they do not develop progressive glomerular disease. As a result of the use of a MHC-class I promoter the tg mice in this study exhibit renal IL-4 expression which leads to increased mesangial matrix production prior to the immunological insult to the kidney. Possibly, this change in the glomerular matrix microenvironment promotes Ig deposition, TGF-β upregulation and further fibrosis. The other IL-4 tg models lack renal transgene expression, and show no pathological changes in their kidneys. This further supports the contention that renal IL-4 expression is a crucial factor for the development of the glomerular lesions.
In summary, this study has shown that aberrant multiorgan expression of IL-4 including the kidney causes progressive glomerulosclerosis leading to end-stage renal failure. Renal IL-4 expression leads to increased ECM production and alters the glomerular structure possibly promoting Ig deposition. TGF-β1 appears to modulate the disease progression in two ways. Firstly, the downregulation of circulating serum TGF-β1 levels is associated with glomerular Ig deposition and the progression of the renal manifestations, and secondly the upregulation of renal TGF-β1 levels following Ig deposition leads to excessive matrix accumulation and exacerbation of the disease development. As well, this model re-emphasizes that both IL-4 and TGF-β are multifunctional cytokines, which exert their actions — systemically or locally — in a context-dependent fashion.
Acknowledgments
The authors wish to thank Mrs. Jennabeth Fuge and Mrs. Margaret Coles for their help with the animal work. Aspects of this work were supported by the Health Research Council of New Zealand, the Lottery Health Research of New Zealand, the Wellington Medical Research Foundation, the Deutsche Forschungsgemeinschaft (SFB 165), and the Bundesministerium für Forschung und Technologie, Germany.
References
- 1.Border WA, Okuda S, Nakamura T, Languino LR, Ruoslahti E. Role of TGF-β1 in experimental glomerulonephritis. Ciba Found. Symp. 1991;157:178–193. [PubMed] [Google Scholar]
- 2.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 3.Bridoux F, Badou A, Saoudi A, et al. Transforming Growth Factor β (TGF-β)-dependent Inhibition of T Helper Cell 2 (Th2)-induced Autoimmunity by Self-Major Histocompatibility Complex (MHC) Class II-specific, Regulatory CD4+ T Cell Lines. J. Exp. Med. 1997;185:1769–1775. doi: 10.1084/jem.185.10.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruijn JA, Roos A, De Geus B, De Heer E. Transforming growth factor-β and the glomerular extracellular matrix in renal pathology. J. Lab. Clin. Med. 1994;123:34–47. [PubMed] [Google Scholar]
- 5.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 6.Erb KJ, Holtschke T, Muth K, Horak I, Schimpl A. T cell subset distribution and B cell hyperreactivity in mice expressing interleukin-4 under the control of major histocompatibility complex class I regulatory sequences. Eur. J. Immunol. 1994;24:1143–1147. doi: 10.1002/eji.1830240520. [DOI] [PubMed] [Google Scholar]
- 7.Erb KJ, Rüger B, Von Brevern M, Ryffel B, Schimpl A, Rivett K. Constitutive Expression of Interleukin (IL)-4 In Vivo Causes Autoimmune-type Disorders in Mice. J. Exp. Med. 1997;185:329–339. doi: 10.1084/jem.185.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Floege J, Burns MW, Alpers CE, et al. Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int. 1992;41:297–309. doi: 10.1038/ki.1992.42. [DOI] [PubMed] [Google Scholar]
- 9.Foote LC, Howard RG, Marshak-Rothstein A, Rothstein TL. IL-4 Induces Fas Resistance in B Cells. J. Immunol. 1996;157:2749–2753. [PubMed] [Google Scholar]
- 10.Furusu A, Miyazaki M, Koji T, et al. Involvement of IL-4 in human glomerulonephritis — an in situ hybridization study of IL-4 mRNA and IL-4 receptor mRNA. JASN. 1997;8:730–741. doi: 10.1681/ASN.V85730. [DOI] [PubMed] [Google Scholar]
- 11.Hasan Q, Dunbar PR, Murray-McIntosh RP, Neale TJ. Transforming Growth Factor β Isoforms in Human Glomerulopathies. Nephrol. 1998;4:353–359. [Google Scholar]
- 12.Horii Y, Iwano M, Hirata E, et al. Role of interleukin-6 in the progression of mesangial proliferative glomerulonephritis. Kidney Int. 1993;39:S71–S75. [PubMed] [Google Scholar]
- 13.Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, Imai E. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-β or platelet-derived growth factor gene into rat kidney. J. Clin. Invest. 1993;92:2597–2601. doi: 10.1172/JCI116874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isbel NM, Atkins R. Cytokines: the kidney as a model for their role in tissue injury and repair. Aust. NZ J. Med. 1996;26:636–639. doi: 10.1111/j.1445-5994.1996.tb02932.x. [DOI] [PubMed] [Google Scholar]
- 15.Kopp JB, Factor VM, Mozes M, et al. Transgenic mice with increased plasma levels of TGF-β 1 develop progressive renal disease. Lab. Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- 16.Kuruvilla AP, Shah R, Hochwald GM, Liggitt HD, Palladino MA, Thorbecke GJ. Protective effect of transforming growth factor β 1 on experimental autoimmune diseases in mice. Proc. Natl. Acad. Sci. USA. 1991;88:2918–2921. doi: 10.1073/pnas.88.7.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Letterio JJ, Geiser AG, Kulkarni AB, Roche NS, Sporn MB, Roberts AB. Maternal Rescue of Transforming Growth Factor-β 1 Null Mice. Science. 1994;264:1936–1938. doi: 10.1126/science.8009224. [DOI] [PubMed] [Google Scholar]
- 18.Lewis DB, Yu CC, Forbush KA, et al. Interleukin 4 expressed in situ selectively alters thymocyte development. J. Exp Med. 1991;173:89–100. doi: 10.1084/jem.173.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López-Armada MJ, Gómez-Guerrero C, Egido J. Receptors for Immune Complexes Activate Gene Expression and Synthesis of Matrix Proteins in Cultured Rat and Human Mesangial Cells. J. Immunol. 1996;157:2136–2142. [PubMed] [Google Scholar]
- 20.Lowenthal JW, Castle BE, Christiansen J, et al. Expression of high affinity receptors for murine interleukin 4 (BSF-1) on hemopoietic and non-hemopoietic cells. J. Immunol. 1988;140:456–464. [PubMed] [Google Scholar]
- 21.Machold KP, Carson DA, Lotz M. Transforming Growth Factor-β (TGF-β) Inhibition of Epstein-Barr Virus (EBV) -and Interleukin-4 (IL-4)-Induced Immunoglobulin Production in Human B Lymphocytes. J. Clin. Immunol. 1993;13:219–227. doi: 10.1007/BF00919975. [DOI] [PubMed] [Google Scholar]
- 22.McCartney-Francis NL, Wahl S. Transforming growth factor β: a matter of life and death (Review) J. Leukoc Biol. 1994;55:401–409. doi: 10.1002/jlb.55.3.401. [DOI] [PubMed] [Google Scholar]
- 23.Mueller R, Krahl T, Sarvetnick N. Tissue-Specific Expression of Interleukin-4 Induces Extracellular Matrix Accumulation and Extravasation of B Cells. Lab Invest. 1997;76:117–128. [PubMed] [Google Scholar]
- 24.Müller GA, Schettler V, Müller CA, Strutz F. Prevention of progression of renal fibrosis: How far are we? Kidney Int. 1996;49:S75–S82. [PubMed] [Google Scholar]
- 25.Müller W, Kühn R, Rajewski K. Major histocompatibility complex class II hyperexpression on B cells in interleukin 4-transgenic mice does not lead to B cell proliferation and hypergammaglobulinemia. Eur J. Immunol. 1991;21:921–925. doi: 10.1002/eji.1830210410. [DOI] [PubMed] [Google Scholar]
- 26.Nakajima A, Hirose S, Yagita H, Okumura K. Roles of IL-4 and IL-12 in the Development of Lupus in NZB/W F1 Mice. J. Immunol. 1997;158:1466–1472. [PubMed] [Google Scholar]
- 27.Nakazato Y, Okada H, Tajima S, et al. Interleukin-4 modulates collagen synthesis by human mesangial cells in a type-specific manner. Am. J. Physiol. 1996;270:F447–F453. doi: 10.1152/ajprenal.1996.270.3.F447. [DOI] [PubMed] [Google Scholar]
- 28.Neale TJ, Rüger BM, MacAulay H, et al. Tumor Necrosis Factor-α Is Expressed by Glomerular Visceral Epithelial Cells in Human Membranous Nephropathy. Am. J. Pathol. 1995;146:1444–1454. [PMC free article] [PubMed] [Google Scholar]
- 29.Paul WE. Interleukin-4: a Prototypic Immunoregulatory Lymphokine (Review Article) Blood. 1991;77:1859–1870. [PubMed] [Google Scholar]
- 30.Postlethwaite AE, Holness MA, Katai H, Raghow R. Human Fibroblasts Synthesize Elevated Levels of Extracellular Matrix Proteins in Response to Interleukin 4. J. Clin. Invest. 1992;90:1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raz E, Watanabe A, Baird SM, et al. Systemic immunological effects of cytokine genes injected into skeletal muscle. Proc. Natl. Acad. Sci. USA. 1993;90:4523–4527. doi: 10.1073/pnas.90.10.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rüger B, Dunbar PR, Hasan Q, et al. Human mast cells produce type VIII collagen In Vivo. Int J. Exp Path. 1994;75:397–404. [PMC free article] [PubMed] [Google Scholar]
- 33.Rüger BM, Hasan Q, Greenhill NS, Davis PF, Dunbar PR, Neale TJ. Mast cells and type VIII collagen in human diabetic nephropathy. Diabetologia. 1996;39:1215–1222. doi: 10.1007/BF02658509. [DOI] [PubMed] [Google Scholar]
- 34.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of Murine Lung Fibroblasts Express Membrane-Bound and Soluble IL-4 Receptors. J. Immunol. 1994;152:3606–3614. [PubMed] [Google Scholar]
- 35.Sharma K, Ziyadeh F. The emerging role of transforming growth factor-β in kidney diseases. Am. J. Physiol. 1994;266:F829–F842. doi: 10.1152/ajprenal.1994.266.6.F829. [DOI] [PubMed] [Google Scholar]
- 36.Suematsu S, Matsuda T, Aozasa K, et al. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA. 1989;86:7547–7551. doi: 10.1073/pnas.86.19.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takemura T, Yoshioka K, Murakami K, et al. Cellular localization of inflammatory cytokines in human glomerulonephritis. Virchows Arch. 1994;424:459–464. doi: 10.1007/BF00191429. [DOI] [PubMed] [Google Scholar]
- 38.Tepper RI, Levinson DA, Stanger BZ, Campos-Torres J, Abbas AK, Leder P. IL-4 Induces Allergic-like Inflammatory Disease and Alters T Cell Development in Transgenic Mice. Cell. 1990;62:457–467. doi: 10.1016/0092-8674(90)90011-3. [DOI] [PubMed] [Google Scholar]
- 39.Wardle N. Glomerulosclerosis: The final pathway is clarified, but can we deal with the triggers? Nephron. 1996;73:1–7. doi: 10.1159/000188990. [DOI] [PubMed] [Google Scholar]
- 40.Yan N, Meister A. Amino acid sequence of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1990;265:1588–1593. [PubMed] [Google Scholar]
- 41.Yaswen L, Kulkarni AB, Fredrickson T, et al. Autoimmune Manifestations in the Transforming Growth Factor-β 1 Knockout Mouse. Blood. 1996;87:1439–1445. [PubMed] [Google Scholar]