

Abstract
Interleukin (IL)-6-related cytokines share gp130 as the signal-transducing protein. Cardiac myocytes produce various kinds of cytokines including IL-6 and cardiotrophin-1. Activation of gp130 transduces hypertrophic and cytoprotective signals in cardiac myocytes via JAK/STAT, MAP kinase and PI-3 kinase pathways. Besides various well-established mechanisms by which cardiac growth and myocardial remodeling are regulated, gp130 signalling may be a newly discovered mechanism that regulates these events in association with cytoprotective effect in myocardial diseases.
Keywords: cardiac myocytes, cardiotrophin-1, gp130, JAK/STAT, hypertrophy, cytoprotection, apoptosis
Background
Cytokines in cardiovascular disease
Cytokines are recognized as essential mediators of normal and pathological immune responses. It is also accepted that cytokines are involved in the cascade of events that lead to the wide range of inflammatory responses to exogenous and endogenous pathogens. Whereas heart failure is recognized as a haemodynamic disorders, more recent studies have suggested that the excessive elaboration of biologically active molecules may play an important role in the pathogenesis of heart failure by virtue of the direct effect that these molecules exert on the heart and the vasculature (Finkel et al. 1992; Packer 1992). In this regard, one of the more recent interesting and intriguing observations in clinical heart failure research is that in addition to the classic neurohormones that are elaborated in heart failure, a second portfolio of biologically active molecules, termed pro-inflammatory cytokines, are also overexpressed in heart failure (Levine et al. 1990; Matsumori et al. 1994; MacGowan et al. 1997). Given that cytokines have been shown to produce direct effect on myocardium, there has been increasing interest in studying the role that these molecules may play as biological mediators of cardiovascular diseases. Accordingly, a number of experimental and clinical studies have begun to examine the levels of cytokines and cytokine receptors in various cardiovascular diseases. Currently, circulating level of tumour necrosis factor (TNF)α and soluble TNFα receptors are most widely accepted cytokines those are recognized to implicate in the development and progression of heart failure (Ferrari et al. 1995; Torre-Amione et al. 1996; Nozaki et al. 1997). In addition, circulating level of interleukin (IL)-6 and soluble gp130 is reported to elevate in the patients with congestive heart failure and myocardial infarction (Miyao et al. 1993; MacGowan et al. 1997; Aukrust et al. 1999).
Inflammation plays an important role in the healing of tissue after injury. However, there is evidence that the accelerated inflammatory response may also extended tissue injury. Recent studies have indicated the additional importance of cytokines in the pathogenesis of myocardial injury in viral myocarditis (Shioi et al. 1996). Although anti-viral effects of inflammatory cytokines such as IL-1, IL-2, IL-6 and TNFα, have been studied in vitro (Weissenbach et al. 1980; Rook et al. 1983; Mestan et al. 1986), neither IL-1 nor TNFα had a beneficial effect on a murine model of viral myocarditis in vivo (Huber et al. 1994; Yamada et al. 1994). Whereas IL-1 and TNFα promote inflammation and are involved in the pathogenesis of clinical disorders such as septic shock, IL-6 produces a restorative effect by inducing the activity of immunoregulatory cells in murine model of viral myocarditis (Kanda et al. 1996). Experimental studies showed that the inflammatory response to myocardial infarction is associated with the induction of cytokines such as TNFα, IL-1β and IL-6 (Ono et al. 1998). Especially, recent studies have demonstrated the induction of IL-6 in the myocardium of a canine model of ischemia and reperfusion (Gwechenberger et al. 1999; Chandrasekar et al. 1999). These studies demonstrated that IL-6 synthesis is an integral part of the reaction to injury resulting from ischemia and reperfusion. Evidence of the cytokine production by cardiac myocytes (Yamauchi-Takihara et al. 1995a) has also contributed to the discovery of novel functions of cytokines in the regulation of cardiovascular disease.
Mechanisms inducing cardiac hypertrophy
Cardiac hypertrophy is induced by various stimuli, such as pressure or volume overload (Mann et al. 1989; Komuro & Yazaki 1993). Under these conditions, this hypertrophic response is assumed to be an important compensation to maintain mechanical function, however, recent clinical studies have recognized that cardiac hypertrophy is an independent risk factor of cardiac morbidity and mortality (Levy et al. 1990). Well-known stimuli of cardiac myocyte hypertrophy are mechanical stretch (Komuro et al. 1990) and various factors utilizing G protein-coupled receptors, including norepinephrine (NE) (Simpson 1985), endothelin-1 (ET-1) (Ito et al. 1991) and angiotensin II (Ang II) (Baker & Aceto 1990). Moreover, it has been reported that mechanical stress stimulates autocrine secretion of Ang II and ET-1 in cardiac myocytes (Sadoshima et al. 1993; Yamazaki et al. 1996). Recent studies have indicated that in addition to above classical neurohumoral factors, growth factors and cytokines are also implicated in myocardial hypertrophy (Duerr et al. 1995; Thaik et al. 1995; Tanaka et al. 1998).
Recently, a novel IL-6 related cytokine, cardiotrophin-1 (CT-1), was cloned and characterized as a cardiac myocyte hypertrophy-inducing factor (Pennica et al. 1995a). CT-1 activates several features of cardiac myocyte hypertrophy including sarcomeric organization and embryonic gene expression through the signal transducing receptor gp130 (Pennica et al. 1995b). Although cardiac hypertrophy is associated with marked changes in cardiac gene expression, some of which are not observed with hypertrophy induced by growth factors and cytokines (Cittadini et al. 1996). gp130 has been identified as a signal transducing protein of IL-6-related cytokines (IL-6, IL-11, leukaemia inhibitory factor (LIF), oncostatin M(OSM), ciliary neurotrophic factor (CNTF) and CT-1), and is widely expressed in various organs including the heart (Saito et al. 1992; Kishimoto et al. 1994; Kishimoto et al. 1995) (Figure 1).
Figure 1.
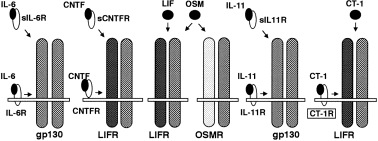
Receptor complexes sharing gp130 as a signal transducer. IL-6, when bound to either a membrane-anchored or soluble form of its receptor (IL-6R or sIL-6R), induces the formation of gp130/gp130 homodimer. Heterodimer of LIFR and gp130 is induced by the CNTF-CNTFR complex, CNTF-sCNTFR complex, LIF, OSM or CT-1. OSM also induces another heterodimer composed of OSM-specific receptor (OSMR) and 130. The IL-11-IL-11R and IL-11-sIL-11R complex is considered to induce gp130/gp130 homodimer.
Many reports have suggested that the mitogen-activated protein kinase (MAPK) family plays an important role in the differentiation and proliferation of many types of the cells (Davis 1993). One member of the MAPK family, extracellular signal-regulated protein kinase (ERK), has been reported to be required for NE-induced expression of specific genes such as atrial natriuretic factor (ANF), c-fos, and myosin light chain 2 genes (Thorburn et al. 1994; Thorburn et al. 1995). In cardiac myocytes, the MAPK pathway is activated by many factors, such as NE, ET-1 and mechanical loading, and is thought to be an important pathway for the induction of hypertrophy (Sadoshima & Izumo 1993; Yamazaki et al. 1995). However, recent studies demonstrated that the activation of ERKs is not sufficient to fully promote cardiac myocyte hypertrophy (Thorburn et al. 1995; Post et al. 1996) and the activation of JAK/STAT pathway is observed in cardiac myocytes subjected to mechanical stretch (Pan et al. 1999). This activation of JAK/STAT pathway is partially dependent on autocrine/paracrine-secreted Ang II and is mainly dependent on the IL-6 family of cytokines.
Signal transducing pathway through gp130
Many findings concerning the structure and function of IL-6 family of cytokines and their receptors have greatly contributed to the establishment of a variety of concepts about cytokines: the establishment of pleiotropy and redundancy as properties of cytokine function, the cytokine receptor superfamily, and the sharing of a signal transducing receptor subunit among several cytokine receptors. In fact, the IL-6 cytokine family plays pivotal roles in a wide variety of tissues and organs, including the immune, haematopoietic, nervous and cardiovascular systems (Hilton 1992; Patterson & Nawa 1993; Yang 1993; Kishimoto et al. 1995; Ip & Yancopoulos 1996). gp130 is a signal-transducing subunit shared by the receptors for the IL-6 family of cytokines. The binding of a ligand to its receptor induces the dimerization of gp130, leading to the activation of Janus kinase (JAK) and tyrosine phosphorylation of gp130 (Murakami et al. 1993). These events lead to the activation of multiple signal-transducing pathways, such as the STAT (signal transducer and activator of transcription), Ras-MAPK and phosphatidylinositol (PI)-3 kinase pathways whose activation is controlled by distinct regions of gp130 (Hirano et al. 1997). Three members of the JAK family, JAK1, JAK2 and Tyk2, are known to associate with gp130 prior to stimulation and to be activated when gp130 is stimulated (Kishimoto et al. 1994). Phosphorylated JAKs in turn phosphorylate a latent cytoplasmic transcription factor, STAT3, allowing it to bind to a responsive element of the target genes (Figure 2). It is now known that JAKs and STATs seem to play essential roles in cytokine function (Hirano et al. 1997). However, it is also true that the JAK-STAT pathway alone cannot explain all the events induced through cytokine receptors, and it is clear that other pathways involving src-family tyrosine kinases, RAS, MAPK, PI-3 kinase, and as yet unidentified components participate, and that the interplay among them is critically involved in the biological activities of cytokines.
Figure 2.
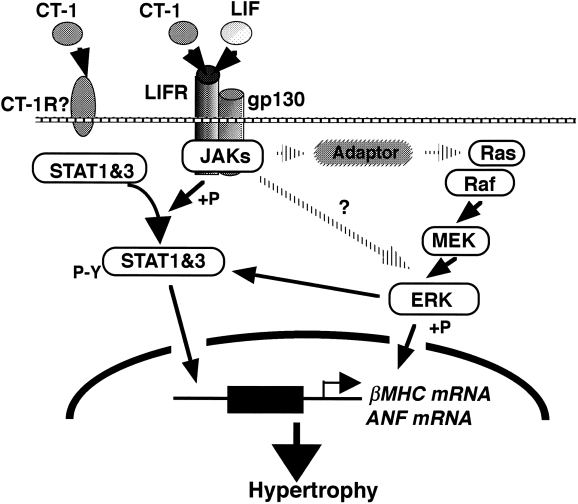
Two pathways from gp130 to hypertrophic gene expression. Heterodimerization of gp130 and LIFR activates the STAT pathway as well as the MAPK cascade.
In this review, we described the induction of IL-6 and CT-1 with various stimulations in cardiac myocytes and functional significance of the activation of JAK/STAT, MAPK and PI-3 kinase pathways in association with gp130 activation. We further discuss recent experimental data obtained in vivo as well as in transgenic animals that suggest an important role for gp130 dependent signalling pathway in cardiovascular disease.
Expression of IL-6 and CT-1 in cardiac myocytes
The expression of growing number of genes is induced by hypoxic stimulation in cardiac myocytes and vascular endothelial cells (Webster et al. 1993; Karakurum et al. 1994; Yamauchi-Takihara et al. 1995a; Yan et al. 1995). However, the molecular basis for the induction of these genes has not been fully elucidated in cardiac myocytes. In addition to NE and IL-1β stimulation, hypoxic stimulation induces the expression of IL-6 and CT-1 mRNAs in cultured neonatal rat cardiac myocytes (Yamauchi-Takihara et al. 1995a; Hishinuma et al. 1999). Subsequent reoxygenation after hypoxic stress significantly augmented the production of these cytokines (Figure 3).
Figure 3.
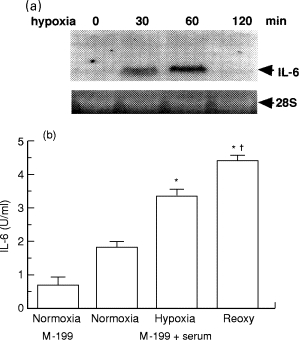
Expression of IL-6 in hypoxic cardiac myocytes. a, Northern blot analysis of IL-6 gene expression in cultured neonatal rat cardiac myocytes. b, Effect of hypoxia and reoxygenation on the production of IL-6 by cardiac myocytes. Culture medium was isolated after 4 h of incubation under normoxia (95% air-5% CO2) or hypoxia (95% N2-5% CO2) or after 2 h of hypoxia followed by 2 h of normoxia (Reoxy). IL-6 activity in the culture medium was measured by bioassay using the MH-60. BSF2 cell line. *P < 0.05 vs. Normoxia. †P < 0.05 vs. Hypoxia
The transcriptional activation of the IL-6 gene has been reported to depend on the 14-bp palindromic sequence, located at position −150, and nuclear factor (NF)-IL6 bound to this site (Isshiki et al. 1990). NF-IL6 activates the IL-6 gene and is also responsible for the regulation of the genes encoding other inflammatory cytokines, acute-phase proteins, albumin, c-fos and viral proteins (Akira et al. 1990). Moreover, the promoter region of the IL-6 gene has a binding site for the transcription factor, NF-κB, another well-characterized transcription factor that plays an important role in inflammatory responses and cell growth regulation, and is considered to be involved in the expression of many genes that encode cytokines and acute-phase proteins (Lenardo & Baltimore 1989). Transcription factors, NF-IL6 and NF-κB, are known to synergistically activate the transcription of inflammatory cytokines (Matsusaka et al. 1993).
Different lengths of IL-6 promoter-luciferase reporter plasmids were conducted and transfected into cardiac myocytes. After hypoxic stimulation, luciferase activity increased by 5.7-fold, whereas either the NF-κB or NF-IL6 binding site was disrupted, luciferase activity after hypoxic stimulation decreased by about 77% or 37%, respectively. Moreover, when both sites were disrupted, luciferase activity after hypoxic stimulation decreased by about 89%. Electrophoretic mobility shift assays (EMSAs), using oligonucleotides containing the NF-κB-or NF-IL6-binding site as a probe, demonstrated enhanced binding activity in nuclear extracts from hypoxic cardiac myocytes. The enhanced binding complex obtained with EMSA using the NF-κB probe displayed a supershift with antibody to its subunit, p50 or p65, and another enhanced binding complex obtained using the NF-IL6 probe displayed a supershift with antibody to CCAAT-enhancer-binding protein-β (C/EBP-β) (Matsui et al. 1999) (Figure 4). Taken together, although hypoxic stimulation induced transcription factors both NF-κB and NF-IL6 in cardiac myocytes, NF-κB may be the primary positive regulator of transcriptional activation of the IL-6 gene in the context of hypoxia.
Figure 4.
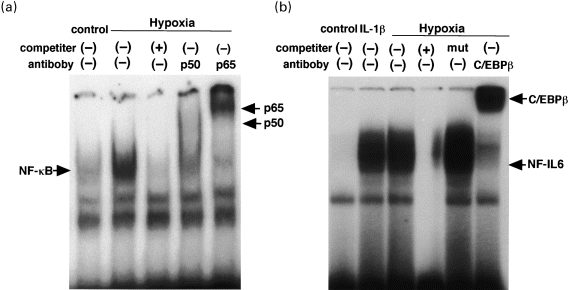
Hypoxia-mediated enhancement of nuclear binding activity for (a) NF-κB or (b) NF-IL6 motif in cardiac myocytes. Cardiac myocytes were stimulated with hypoxia or 1ng/ml IL-1β for 1 hour.
Activation of gp130 transduces hypertrophic signal in cardiac myocyte
Activation of gp130 transduces hypertrophic signals
IL-6 stimulation induces homodimerization of gp130, whereas stimulation with CT-1 or LIF leads to the formation of heterodimers composed of gp130 and LIF receptor (Pennica et al. 1995b). Evidence that IL-6 generates distinct signal only in the combination with IL-6 receptor in cardiac myocyte is based on the low expression level of IL-6 receptor (Saito et al. 1992; Yamauchi-Takihara et al. 1995b). Studies of gp130 mediated hypertrophic signalling have been conducted by using CT-1 or LIF, because LIF receptor was abundantly detected in cardiac myocytes (Matsui et al. 1996). LIF stimulation increases the size, induces the expression of c-fos, β-myosin heavy chain (MHC) and ANF mRNAs and stimulates [3H] leucine incorporation in cardiac myocytes (Matsui et al. 1996) (Figure 5). The hypertrophic signalling induced by cytokine signalling through gp130 is known to be distinct from the hypertrophic response seen after stimulation of G protein coupled receptors, both on a morphological and a molecular level. CT-1 and LIF induce a predominant increase in myocardial cell length with the addition of new sarcomeric units in series but no concomitant increase in cell width (Wollert et al. 1996). The changes in cardiac myocyte morphology induced by G protein vs. gp130-dependent signalling pathways are reminiscent of the changes observed in cardiac myocytes isolated from pressure and volume overloaded hearts, respectively (Anversa et al. 1986). It must be pointed out however, that the relative importance of G protein vs. gp130-dependent signalling pathways, in pressure and volume overload-induced hypertrophy remains to be clarified.
Figure 5.
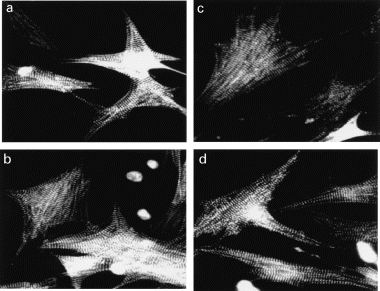
Myocardial hypertrophy induced by norepinephrine, (NE) leukaemia inhibitory factor (LIF) or IL-6. Cultured neonatal rat cardiac myocytes were stimulated for 36 hours with (a) vehicle, (b) 2 μm NE, (c) 103 U/ml LIF, (d) the combination of 103 U/ml IL-6 and soluble IL-6 receptor (0.5 μg/ml). The cells were fixed with 3.7% formaldehyde, permeablized and incubated with anti-sarcomeric α-actinin antibody and FIT conjugated anti-mouse IgG.
To evaluate the molecular mechanisms involved in LIF induced cardiac myocyte hypertrophy, the activation of the gp130 dependent signalling pathway was examined. LIF stimulation rapidly tyrosine phosphorylates gp130 and subsequently activates both JAK1 and JAK2 within a few minutes, followed within 5 min by tyrosine phosphorylation of STAT1 and STAT3 in cardiac myocytes (Kunisada et al. 1996). LIF also activates MAP kinase in a characteristic way, i.e. the magnitude of activation is half of and the duration is shorter than that of NE-induced MAPK activation. This distinct kinetic pattern of MAPK activation observed after gp130 activation might be associated with a different physiological function in the gp130-dependent signalling pathway (Kunisada et al. 1996). Intravenous administration of LIF activates JAK/STAT and MAPK pathways in the hearts, which is almost identical time course of the STAT activation observed in the liver. In addition, it has been also reported that acute pressure overload activates JAK1, JAK2 and Tyk2 kinases, as well as STAT1, 2 and 3 in the heart (Pan et al. 1997). To summarize, the JAK/STAT and MAPK pathways are located at downstream of gp130 in cardiac myocyte and are rapidly activated both in vivo and in vitro.
IL-6 and IL-6 receptor double transgenic mouse demonstrates myocardial hypertrophy
To investigate the physiological role of gp130 in vivo and to determine the pathological consequence of abnormal activation of gp130, transgenic mice having continuously activated gp130 were generated. Although an earlier study demonstrated abundant expression of gp130, it failed to detect IL-6 receptor expression in the myocardium (Saito et al. 1992). Transgenic mice (TG) were carried out by mating mice from IL-6 and IL-6 receptor trangenic lines. Double TG overexpressing both IL-6 and IL-6 receptor showed constitutive tyrosine phosphorylation of gp130 and STAT3 in the hearts. The distinguishing feature of the double TG is a dramatic increase in cardiac weight, with an increased left ventricular wall thickness and cardiac myocyte size that became apparent at 5 month of age (Hirota et al. 1995) (Figure 6). Importantly, TG overexpessing either IL-6 or IL-6 receptor alone did not show detectable myocardial abnormalities (Suematsu et al. 1989). Likewise, only a combination of IL-6 and soluble IL-6 receptor induces gp130 activation and hypertrophy of cardiac myocytes in vitro. Conceivably, cardiac myocytes derived IL-6 may associate with circulating soluble IL-6 receptor, and the IL-6/IL-6 receptor complex may then act on gp130 in an autocrine/paracrine manner. In addition, the expression of CT-1 in the adult myocardium implies CT-1 as a candidate cytokine to activate a hypertrophic response in vivo (Pennica et al. 1996b; Hishinuma et al. 1999). Ishikawa et al. (1996) reported that CT-1 mRNA was highly expressed in the hypertrophied ventricles of spontaneously hypertensive rats-stroke prone/IZM at a stage of established hypertension.
Figure 6.
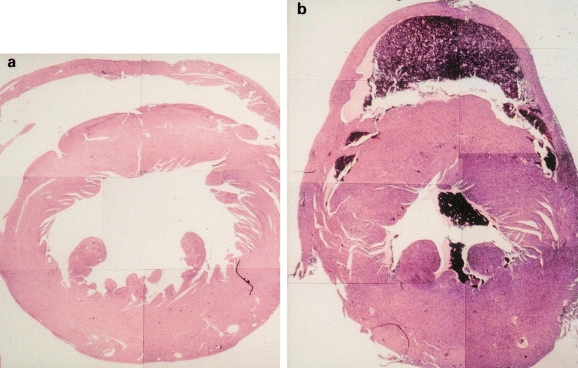
Transverse sections of the hearts from a 5-month-old control mouse (a) and IL-6 and IL-6R double transgenic mouse (b).
Role of STAT3 in gp130-mediated cardiac myocyte hypertrophy
Studies focusing on the significance of STAT3 in inducing cardiac myocyte hypertrophy, an adenovirus vector was used to overexpress wild-type STAT3 or dominant negative-type (DN) STAT3 in cultured cardiac myocytes. Although STAT3 phosphorylation was not observed in wild-type STAT3 before LIF stimulation, augmented phosphorylation was observed after the stimulation. In contrast, phosphorylation of STAT3 was not detected in DN STAT3 either with or without LIF stimulation. Pretreatment with PD98059, a MAPK kinase inhibitor, did not affect the level of tyrosine phosphorylation of STAT3 in cardiac myocytes. These results indicate that the activation of the JAK/STAT pathway, especially the STAT3-dependent pathway, is enhanced in STAT3 transfected, but not fully activated in DN STAT3 transfected cardiac myocytes after LIF stimulation, and that it is independent of the status of MAPK activation (Kunisada et al. 1998). Indeed, LIF-induced c-fos and ANF mRNA expressions were significantly enhanced with STAT3 overexpression and almost completely inhibited with DN STAT3 in cardiac myocytes (Figure 7). Although the expression of these mRNAs was reduced after PD98059 treatment, there were substantial differences in the expression level between wild-type STAT3 and DN STAT3 transfected cells (Kunisada et al. 1998).
Figure 7.
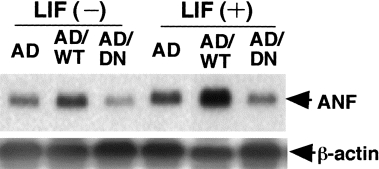
Atrial natriuretic factor (ANF) mRNA expression in cardiac myocytes after LIF stimulation. Cardiac myocytes were transfected with control adenovirus vector (AD), recombinant adenovirus carrying wild-type STAT3 (AD/WT) or dominant negative-type STAT3 (AD/DN). They were stimulated with LIF (103 U/ml) for 24 hours.
To summarize, the induction of cardiac myocyte hypertrophy and c-fos and ANF mRNAs expression by LIF was amplified by STAT3 overexpression, whereas it was attenuated under the condition with inhibited STAT3 signalling. Furthermore, when MAPK activity was inhibited, gene expression and protein synthesis were significantly suppressed even in the cells that overexpressed STAT3. This would be explained by the crosstalk between JAK/STAT and MAPK cascades, both of which are necessary for maximal transcriptional upexpression. Without serine phosphorylation, which is induced by activated MAPK, the transcriptional activity of tyrosine-phosphorylated STAT is reduced (Zhang et al. 1995). In addition, gene activation by STAT3, which obligatorily requires tyrosine phosphorylation to become active, is reported to depend for maximal activation on serine phosphorylation (Wen et al. 1995).
Role of PI-3 kinase in gp130 mediated cardiac myocyte hypertrophy
PI-3 kinase is activated by cytokine stimulation through different types of receptors to transduce intracellular responses. Several growth factor receptors, cytokine receptors, and G-protein-coupled receptors are able to stimulate PI-3 kinase activity (Shubeita et al. 1990; Chung et al. 1994; Gold et al. 1994; Foncea et al. 1997; Saward & Zahradka 1997). Furthermore, recent studies have revealed that PI-3 kinase plays an important role in the activation of p70 S6 kinase (Chung et al. 1994) and the prevention of apoptosis (Yao & Cooper 1995). Recently, PI-3 kinase is reported to be involved in the regulation of gp130-dependent hypertrophic signalling in cardiac myocytes (Oh et al. 1998a). Following the activation of gp130, a substantial increase in PI-3 kinase activity was observed in cardiac myocytes, with a rapid increase at 10 min and a decline at 60 min. JAK1 and JAK2 bound to PI-3 kinase and LIF stimulation increased the PI-3 kinase activity in both immunoprecipitates. A specific PI-3 kinase inhibitor, wortmannin, completely inhibited LIF induced activation of p70 S6 kinase and protein synthesis and partially inhibited MAPK activation. Therefore, the maximal stimulation of the protein kinase cascade by LIF requires the activation of PI-3 kinase, which may thus be an important mediator in LIF-induced transduction of hypertrophic signal.
Angiotensin II interferes with LIF induced STAT3 activation
Ang II is also known to activate STATs and induce hypertrophy and apoptosis in cardiac myocytes (Kajstura et al. 1997; Kodama et al. 1998). Recently, potential interactions between gp130 dependent and Ang II signalling pathways have been reported (Bhat et al. 1996; McWhinney et al. 1997). Although LIF attenuated the DNA fragmentation induced by serum depletion, Ang II augmented the DNA fragmentation and inhibited LIF-mediated cytoprotective effect in cardiac myocytes (Tone et al. 1998). LIF is known to tyrosine phosphorylate STAT3 in cardiac myocytes rapidly and transiently, however, it is not observed by Ang II stimulation. Ang II pretreatment inhibited LIF-induced phosphorylation of STAT3 in a dose dependent manner, and this inhibitory effect was blocked by the Ang II type I (AT1) receptor antagonist CV11974. These results demonstrate that negative crosstalk between gp130 and AT1 receptor dependent signalling exists in cardiac myocytes. This crosstalk may contribute to the modulation of pathophysiological process in myocardial disease.
Expression of the SSI family of the genes is increased in gp130 transgenic hearts
Several clinical studies have demonstrated that abnormalities in the cytokine network are observed in patients with severe congestive heart failure (Levine et al. 1990; Torre-Amione et al. 1996; Kelly & Smith 1997), and that several immunological parameters, particularly soluble gp130, correlate with variables reflecting deranged haemodynamic status (Aukrust et al. 1999). Recently, the feedback mechanism responsible for switching off the cytokine signalling has been described by several groups. A new family of negative regulators of cytokine signalling was isolated and characterized, namely CIS (cytokine-inducible SH2 protein) (Yoshimura et al. 1995), SOCS (suppressor of cytokine signalling) (Starr et al. 1997), JAB (JAK2 binding protein) (Endo-Takaho et al. 1997) or SSI (STAT-induced STAT inhibitor) (Naka et al. 1997). Because IL-6 and IL-6 receptor double transgenic mice represent a constitutively activated model of gp130 in vivo (Hirota et al. 1995), as described previously, it is not clear whether signalling pathways through gp130 are enhanced by exogenous stimuli when gp130 is overexpressed in vivo.
gp130 TG were generated under the control of cytomegalovirus (CMV)-enhancer and chicken β-actin promoter, and a cardiac selective overexpressing line was used to examine the activation of signaling pathways downstream of gp130. The expression of gp130 in TG hearts increased 8-fold compared to those of wild-type littermates (WT), and the tyrosine phosphorylation of gp130 was augmented at basal level, and the activity of other signal transducing molecules, including STAT1, STAT3 and ERK1/2, was also potentiated in TG. Enhanced activation of gp130 after LIF stimulation was observed in both TG and WT hearts. However, the activation of STAT1, STAT3 and ERK1/2 was not augmented and enhanced β-MHC mRNA expression was not observed after LIF stimulation in TG. In this regard, STAT-induced STAT inhibitor (SSI) family mRNA expression was examined and demonstrated that SSI-1 and SSI-3 mRNA expressions increased after LIF stimulation and were significantly augmented in TG. These results indicate that overexpressed gp130 does not always enhance downstream signals in the hearts. Importantly, the increased expression of gp130 upregulated the SSI family of the genes and inhibited downstream signals of gp130 in the TG hearts.
Activation of gp130 transduces cytoprotective signals in cardiac myocytes
Cytoprotective function of IL-6 related cytokines
Cardiac myocyte survival is of central importance in the maintenance of cardiac function, as well as in the development of a variety of cardiac diseases. Adult cardiac myocytes are terminally differentiated and have lost their proliferative capacity. As a result, cardiac myocyte survival is critical for the maintenance of normal cardiac function. Although a wide variety of survival factors have been identified for neural cells and several other terminally differentiated cell types, relatively little is known about the specific growth factors or cytokines that are required for the maintenance of cardiac myocyte survival. Two members of the IL-6 family of cytokines, CNTF and LIF, have been shown to play an important role in maintaining the viability of motoneurones in long-term culture (Sendtner et al. 1990; Vejsada et al. 1995). Recent studies have demonstrated that CT-1 is highly expressed in embryonic limb buds and has a protective effect on axotomy-induced degeneration in neural cells (Pennica et al. 1996a). These results suggest that CT-1 can be expected to participate in normal motoneurone development and the protection of human degenerative motoneurone disease. Therefore, the ligands that activate gp130 may play an important physiological role in regulating survival of terminally differentiated cell types.
In terms of a molecular basis of the cytoprotective effects of IL-6 related cytokines, IL-6 prevented the apoptosis induced by IL-6 depletion with the upregulation of bcl-xL in an IL-6-dependent myeloma cell line (Schwarze & Hawley 1995). Bcl-x was identified as a bcl-2-related gene that functions as a regulator of apoptotic cell death (Boise et al. 1993). Bcl-x has two isoforms, bcl-xL, a preventive isoform against apoptosis, and bcl-xS, a promotive isoform.
LIF induces bcl-xL in cardiac myocytes for an antiapoptotic effect
A recent study has provided direct evidence that one of the mechanisms by which LIF can promote the survival of cardiac myocytes was via activation leading to inhibition of the apoptotic signalling pathway. The study deployed an in vitro assay system whereby cardiac myocytes enter the apoptotic signalling pathway after the deprivation of serum. LIF significantly improved cardiac myocyte survival and was associated with increased expression of bcl-x mRNA but not of bcl-2 mRNA. The isoform induced by LIF was identified as bcl-xL by Western blotting and RT-PCR using specific primers. The antisense oligonucleotide against bcl-x mRNA inhibited the protective effect of LIF in conjunction with a reduction in bcl-xL protein (Fujio et al. 1997) (Figure 8).
Figure 8.
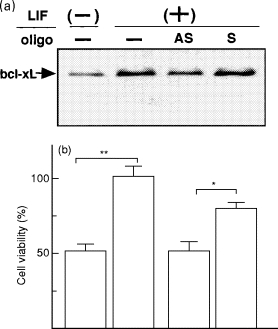
Protective effect of LIF is mediated by bcl-xL in cardiac myocytes. a, Neonatal rat cardiac myocytes were treated with LIF (103 U/ml) in the presence of 5 μm antisense (AS) or sense (S) oligonucleotides for 24 hours. Cells were directly lysed with SDS-PAGE and Western blotted with anti-bcl-x antibody. b, Cardiac myocytes were treated with LIF (103 U/ml) in the presence of 5 μm antisense or sense oligonucleotides for 72 hours. Cells were harvested with collagenase/trypsin and counted. Results are given as a percentage of basal cell number under LIF-stimulated condition without oligonucleotides. *P < 0.05; **P < 0.01.
Studies focusing on the LIF induced transcriptional regulation of the bcl-x gene were pursued using the bcl-x promoter-luciferase reporter gene and EMSAs. The location of the LIF-responsive cis-element was determined from −161 to + 10 of the promoter region of the bcl-x gene, which contains a GAS motif, TTCGGAGAA, at position −41. This motif bound to STAT1, not to STAT3, and site-directed mutagenesis revealed that this motif was essential for LIF-responsive promoter activity (Fujio et al. 1997). These findings taken together indicate that LIF induces bcl-x mRNA via the STAT1 binding cis-element in cardiac myocytes resulting in cytoprotective effects.
LIF activates Akt via PI-3 kinase in cardiac myocytes
The Akt encodes a serine threonine protein kinase that is activated by several growth factor-generated signals that are transduced via PI-3 kinase (Burgering & Coffer 1995; Kohn et al. 1995; Reif et al. 1997). Activation of Akt is known to deliver a survival signal that inhibits the apoptosis induced by growth factor withdrawal in neural cells and fibroblasts (Dudek et al. 1997; Kulik et al. 1997). In addition, activation of Akt ultimately leads to inhibition of caspase activity and protection from apoptotic cell death (Yang et al. 1995). Bad is a distant member of the bcl-2 family that promotes cell death. The active but not the inactive forms of Akt were found to phosphorylate Bad, and thus act as an antiapoptotic (Peso et al. 1997; Scheid & Duronio 1998). A recent study has further demonstrated that IL-3 induced Bad phosphorylation is inhibited by PI-3 kinase specific inhibitors (Peso et al. 1997).
To confirm the essential role of the PI-3 kinase cascade in the gp130-mediated survival function, Akt activation was examined in LIF stimulated cardiac myocytes. LIF induced a rapid activation of Akt and Bad, and these were completely inhibited by wortmannin but not by rapamycin (Oh et al. 1998a; Oh et al. 1998b). Thus, LIF regulation of Akt and Bad employs PI-3 kinase in cardiac myocytes. Further study was conducted to provide gp130-dependent survival effect in cardiac myocytes by using doxorubicin (Dox), one of the most effective and widely used groups of anticancer drugs and well known to present myocardial cell death (Buzdar et al. 1985). Cardiac myocyte apoptosis significantly increased in Dox-treated cells as determined by TUNEL assay and DNA fragmentation, which was significantly reduced by LIF pretreatment (Figure 9). Dox-induced caspase-3 activation and decrease in Bcl-xL abundance were completely inhibited by LIF. LIF phosphorylated Bad through PI 3-kinase and reduced the heterodimerization of Bad with Bcl-xL. LIF protected cardiac myocytes from Dox-induced cell death via PI 3-kinase/Akt pathway associated with the induction of Bcl-xL and Bad phosphorylation (Oh et al. 1998b).
Figure 9.
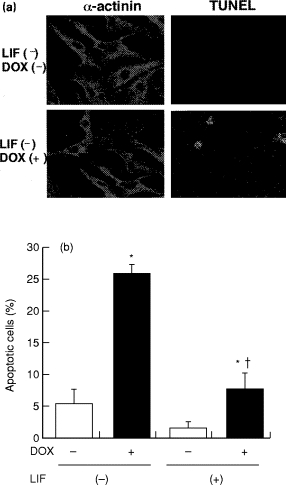
Protective effect of LIF on doxorubicin (Dox) induced cardiac myocyte apoptosis. a, In situ labelling of DNA fragmentation. Cardiac myocytes were depicted by red fluorescence using anti α-actinin antibody. Green fluorescence presents the TdT labelling of nuclei. The cells were incubated with (LIF - Dox +) or without (LIF - Dox - ) 0.5 μm Dox for 16 hours. b, Quantitative analysis of the percentage of cells undergoing apoptosis as measured by TUNEL technique. Cardiac myocytes were pretreated with LIF (103 U/ml) and stimulated with or without 0.5μm Dox for 16 hours. Data are expressed as mean ± SE. *P < 0.05 vs. Dox -; †P < 0.05 vs. LIF -.
Pathological role of gp130-dependent signalling pathway in the heart
Role of gp130 in the prevention of heart failure
The pleiotrophic functions of the IL-6 family of cytokines are mediated by the cytokine receptor subunit gp130 as the common signal transducer. Mice lacking individual component of this cytokine family or their receptors displayed milder phenotypes than expected. Not surprisingly, when receptor component used by several members of the gp130 family is inactivated, the consequences are more severe. Thus, mice lacking LIF receptor, a shared component of the receptor complexes of LIF, CNTF and CT-1, are not viable and die perinatally, exhibiting placental, skeletal, neural defect, and loss of motor neurones (Li et al. 1995; Ware et al. 1995). Animals that harbor a complete deficiency in the common signal transducer gp130 display embryonic lethality and defects in diverse embryonic compartments (Yoshida et al. 1996). To assess the physiological role of this cytokine family in adult animal, gp130 was inducibly inactivated via Cre-loxP-mediated recombination in vivo. Postnatally induced inactivation of gp130 also results in neurological, cardiac, haematopoietic, immunological, hepatic, and pulmonary defects in mice, demonstrating the widespread importance of gp130-dependent cytokines (Betz et al. 1998).
A growing body of evidence suggests the possibility that cytokines which operate via gp130 pathways might play a critical role in the onset of cardiac failure (Hirota et al. 1996; Wollert & Chien 1997). Recently, unequivocal evidence for a critical role of a gp130-dependent myocyte survival pathway was demonstrated by using Cre-loxP technique to achieve a ventricular chamber-specific knockout of gp130. This conditional mutant animal present normal cardiac structure and function. However, during acute pressure overload to the hearts from these mice display rapid onset of dilated cardiomyopathy and massive induction of myocyte apoptosis vs. the control mice that exhibit compensatory hypertrophy (Hirota et al. 1999). This study identified that cardiac myocyte apoptosis is a critical in the transition between compensatory cardiac hypertrophy and heart failure and suggested that ligands operating via gp130 may represent important role in the prevention of ventricular dilatation. Accordingly, identifying the downstream pathway by which gp130-dependent ligands can promote cardiac myocyte survival becomes of critical interest. The STAT-dependent induction of bcl-xL and PI-3 kinase mediated Akt activation may represent a part of mechanisms by which gp130-dependent cytokines suppress apoptosis in cardiac myocytes. In addition, the effect of IL-6 family of cytokines in embryonic cardiac myocyte survival suggest that deregulation of apoptosis may contribute to the ventricular chamber hypoplasia observed in gp130 knock-out mice (Yoshida et al. 1996)
Role of JAK/STAT pathway in acute myocardial infarction
In case of acute myocardial infarction, a large number of cardiac myocytes are known to die as a result of apoptosis as well as of necrosis (Gottlieb et al. 1994; Itoh et al. 1995; Kajstura et al. 1996; Olivetti et al. 1996; Bialik et al. 1997). The heart from acute myocardial infarction is a complex of various stressful states characterized by a reduction in oxygen supply followed by the inflammatory responses and mechanical stretching. This implies two main cellular events inducing apoptosis, hypoxia and mechanical loading (Tanaka et al. 1994; Teiger et al. 1996). Recent in vivo study has demonstrated that acute myocardial infarction activates JAK/STAT pathway mainly in the border area of the myocardium in rat hearts (Negoro et al. 1999). AG-490, a specific JAK2 inhibitor, was found to block the tyrosine phosphorylation of JAK2 and thus present the potential to inhibit the JAK/STAT pathway. AG-490 pretreatment significantly increased the caspase-3 activity and Bax expression in viable myocardium after the infarction. In addition, while few apoptotic myocytes were identified by means of TUNEL assay in the border area of the infarcted myocardium, a significant increase in apoptotic cells was observed when AG-490 was pretreated. The administration of JAK2 inhibitor resulted in a deterioration of myocardial viability in rat acute myocardial infarction. Therefore, the activation of JAK/STAT pathway in acute myocardial infarction heart may exerts a pivotal role in the remodeling of the diseased hearts.
Role of STAT3 in the prevention of heart failure
More recently, a direct evidence for the cytoprotective role of STAT3 mediated signalling pathway was also demonstrated in vivo. Transgenic mice with cardiac specific overexpression of the STAT3 gene (STAT3-TG) were generated and treated intraperitoneally with Dox. After 10 days, only 25% of the control mice survived compared with 80% of the STAT3-TG (Figure 10). The dead mice were found to suffer massive bilateral pleural effusion and ascites, and identified as congestive heart failure. The molecular mechanisms of the protective effects of STAT3 on Dox-treated mice were further examined. Expression of the cardiac α-actin gene was significantly reduced in control hearts after Dox treatment and this was compatible with previously published data (Ito et al. 1990; Jeyaseelan et al. 1997). However, a robust expression of transcript corresponding to cardiac α–actin was observed in STAT3-TG hearts. In addition, control hearts showed increased β–MHC and ANF gene expression after Dox treatment, however, the expression did not change after Dox treatment in STAT3-TG hearts. The expression of ANF and β–MHC genes is induced not only in myocardial hypertrophy but also in heart failure (Drexler et al. 1989; Feldman et al. 1993). The augmented expression of the ANF and β–MHC genes observed in control hearts is probably a compensatory mechanism to prevent progression of heart failure. In STAT3-TG hearts, on the other hand, the expressions of these genes were increased, but they were not further elevated after Dox treatment. These results suggested that the STAT3-TG did not suffer from severe congestive heart failure.
Figure 10.
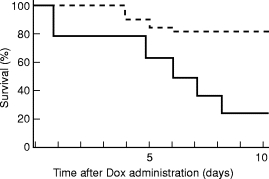
Survival rates for the mice treated with doxorubicin (Dox). Three-month-old STAT3-TG (n = 20, - - - - -) and WT (n = 20, —-) mice were administered intraperitoneally with Dox at a single dose of 15 mgjak/kg and bred under normal conditions. Survival curves represent significantly better survival in STAT3-TG than WT.
In conclusion, the present study demonstrates that STAT3-TG survive significantly longer after Dox treatment and that the overexpression of STAT3 in the hearts reduces the deteriorative effects of Dox (Kunisada et al. 1999). Conceivably, STAT3 plays a crucial role in a cardiac myocyte survival, resulting in the prevention of progression in heart failure.
Conclusions
In conclusion, the JAK/STAT, MAPK and PI-3 kinase pathways are all necessary for full activation of gp130-mediated hypertrophic and cytoprotective signals in cardiac myocytes. As discussed earlier, investigations of signalling pathways through gp130 in cardiac myocytes should uncover novel mechanisms of cardiac myocyte growth and survival. Although recent investigations have addressed the participation of cytokines in various cardiovascular diseases, we have but a limited understanding of their underlying functions and mechanisms. Our investigations of hypertrophic and cytoprotective functions of IL-6 related cytokines are expected to provide new insights into the pathophysiological significance of cytokines in various myocardial diseases. Clarification of the regulation of these cascades in normal and diseased human hearts may well lead to a novel therapeutic approach to myocardial diseases.
Acknowledgments
This study was supported by a Grant-in Aid for Scientific Research from the Ministry of Education, Science Sports and Culture of Japan and grants from the Ministry of Health and Welfare of Japan and the study group of Molecular Cardiology. We thank C. Kusunoki for excellent secretarial assistance.
References
- Akira S, Isshiki H, Sugita T, et al. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anversa P, Ricci R, Olivetti G. Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy. J. Am. Coll. Cardiol. 1986;7:1140–1149. doi: 10.1016/s0735-1097(86)80236-4. [DOI] [PubMed] [Google Scholar]
- Aukrust P, Ueland T, Lien E, et al. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1999;83:376–382. doi: 10.1016/s0002-9149(98)00872-8. [DOI] [PubMed] [Google Scholar]
- Baker KM, Aceto JF. Angiotensin II stimulation of protein synthesis and cell growth in chick heart cells. Am. J. Physiol. 1990;259:H610–H618. doi: 10.1152/ajpheart.1990.259.2.H610. [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Abraham ST, Baker KM. Angiotensin II interferes with interleukin 6-induced Stat3 signaling by a pathway involving mitogen-activated protein kinase kinase 1. J. Biol. Chem. 1996;271:22447–22452. doi: 10.1074/jbc.271.37.22447. [DOI] [PubMed] [Google Scholar]
- Bialik S, Geenen DL, Sasson IE, et al. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J. Clin. Invest. 1997;100:1363–1372. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidyl-inositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Buzdar AU, Marcus C, Smith TL, Blumenschein GR. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer. 1985;55:2761–2765. doi: 10.1002/1097-0142(19850615)55:12<2761::aid-cncr2820551206>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Betz UAK, Bloch W, Broek M, et al. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J. Exp. Med. 1998;188:1955–1965. doi: 10.1084/jem.188.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regulation of CCAAT/Enhancer binding protein, interleukin-6, interleukin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99:427–433. doi: 10.1161/01.cir.99.3.427. [DOI] [PubMed] [Google Scholar]
- Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. PDGF- and insulin-dependent p70 S6K activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- Cittadini A, Stromer H, Katz SE, et al. Differential cardiac effects of growth hormone and insulin-like growth factor-1 in the rat. A combined in vivo and in vitro evaluation. Circulation. 1996;93:800–809. doi: 10.1161/01.cir.93.4.800. [DOI] [PubMed] [Google Scholar]
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- Drexler H, Hanze J, Finckh M, Lu W, Just H, Lang RE. Atrial natriuretic peptide in a rat model of cardiac failure. Circulation. 1989;79:620–633. doi: 10.1161/01.cir.79.3.620. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–664. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Duerr RL, Huang S, Miraliakbar HR, Clark R, Chien KR, Ross J. Insulin-like growth factor-1 enhances ventricular hypertrophy and function during the onset of experimental cardiac failure. J. Clin. Invest. 1995;95:619–627. doi: 10.1172/JCI117706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo-Takaho A, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ. Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Bachetti T, Confortini R, et al. Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation. 1995;92:1479–1486. doi: 10.1161/01.cir.92.6.1479. [DOI] [PubMed] [Google Scholar]
- Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- Foncea R, Andersson M, Ketterman A, et al. Insulin-like growth factor-I rapidly activates multiple signal transduction pathways in cultured rat cardiac myocytes. J. Biol. Chem. 1997;272:19115–19124. doi: 10.1074/jbc.272.31.19115. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T. Signals through gp130 upregulate bcl-x gene expression via STAT1-binding cis-element in cardiac myocytes. J. Clin. Invest. 1997;99:2898–2905. doi: 10.1172/JCI119484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MR, Duronio V, Saxena SP, Schrader JW, Aebersold R. Multiple cytokines activate phosphatidylinositol 3-kinase in hemopoietic cells. J. Biol. Chem. 1994;269:5403–5412. [PubMed] [Google Scholar]
- Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- Hilton DJ. LIF: lots of interesting functions. Trends Biochem. Sci. 1992;17:72–76. doi: 10.1016/0968-0004(92)90505-4. [DOI] [PubMed] [Google Scholar]
- Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 1997;8:241–252. doi: 10.1016/s1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- Hirota H, Chen J, Betz UA, et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc. Natl. Acad. Sci. USA. 1995;92:4862–4866. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Yoshida K, Taga T, Kishimoto T. gp130 signaling pathways: recent advances and implications for cardiovascular disease. Trends Cardiovasc. Med. 1996;6:109–115. doi: 10.1016/1050-1738(96)00037-0. [DOI] [PubMed] [Google Scholar]
- Hishinuma S, Funamoto M, Fujio Y, Kunisada K, Yamauchi-Takihara K. Hypoxic strress induces cardiotrophin-1 expression in cardiac myocytes. Biochem. Biophys. Res. Commun. 1999;264:436–440. doi: 10.1006/bbrc.1999.1535. [DOI] [PubMed] [Google Scholar]
- Huber SA, Polgar J, Schultheiss P, Schwimmbeck P. Augmentation of pathogenesis of coxsackievirus B3 infections in mice by exogenous administration of interleukin-1 and interleukin-2. J. Virol. 1994;68:195–206. doi: 10.1128/jvi.68.1.195-206.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip NY, Yancopoulos GD. The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Annu. Rev. Neurosci. 1996;19:491–515. doi: 10.1146/annurev.ne.19.030196.002423. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Saito Y, Miyamoto Y, et al. cDNA cloning of rat cardiotrophin-1 (CT-1): augmented expression of CT-1 gene in ventricle of genetically hypertensive rats. Biochem. Biophys. Res. Commun. 1996;219:377–381. doi: 10.1006/bbrc.1996.0241. [DOI] [PubMed] [Google Scholar]
- Isshiki H, Akira S, Tanabe O, et al. Constitutive and interleukin-1 (IL-1)-inducible factors interact with the IL-1-responsive element in the IL-6 gene. Mol. Cell Biol. 1990;10:2757–2764. doi: 10.1128/mcb.10.6.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Hirata Y, Hiroe M, et al. Endothelin-1 induces hypertrophy with enhanced expression of muscle-specific genes in cultured neonatal rat cardiomyocytes. Circ. Res. 1991;69:209–215. doi: 10.1161/01.res.69.1.209. [DOI] [PubMed] [Google Scholar]
- Ito H, Miller SC, Billingham ME, et al. Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc. Natl. Acad. Sci. USA. 1990;87:4275–4279. doi: 10.1073/pnas.87.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh G, Tamura J, Suzuki M, et al. DNA fragmentation of human infarcted myocardial cells demonstrated by the nick end labeling method and DNA agarose gel electrophoresis. Am. J. Patholl. 1995;146:1325–1331. [PMC free article] [PubMed] [Google Scholar]
- Jeyaseelan R, Poizat C, Wu HY, Kedes L. Molecular mechanisms of doxorubicin-induced cardiomyopathy. J. Biol. Chem. 1997;272:5828–5832. doi: 10.1074/jbc.272.9.5828. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cheng W, Reiss K, et al. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- Kajstura J, Cigola E, Malhotra A, et al. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J. Mol. Cell. Cardiol. 1997;29:859–870. doi: 10.1006/jmcc.1996.0333. [DOI] [PubMed] [Google Scholar]
- Kanda T, McManus JE, Nagai R, et al. Modification of viral myocarditis in mice by interleukin-6. Circ. Res. 1996;78:848–856. doi: 10.1161/01.res.78.5.848. [DOI] [PubMed] [Google Scholar]
- Karakurum M, Shreeniwas R, Chen J, et al. Hypoxic induction of interleukin-8 gene expression in human endothelial cells. J. Clin. Invest. 1994;93:1564–1570. doi: 10.1172/JCI117135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RA, Smith TW. Cytokines and cardiac contractile function. Circulation. 1997;95:778–781. doi: 10.1161/01.cir.95.4.778. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fukuda K, Pan J, et al. Biphasic activation of the JAK/STAT pathway by angiotensin II in rat cardiomyocytes. Circ. Res. 1998;82:244–250. doi: 10.1161/01.res.82.2.244. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Kovacina KS, Roth RA. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J. 1995;14:4288–4295. doi: 10.1002/j.1460-2075.1995.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro I, Kaida T, Shibazaki Y, et al. Stretching cardiac myocytes stimulates protooncogene expression. J. Biol. Chem. 1990;265:3595–3598. [PubMed] [Google Scholar]
- Komuro I, Yazaki Y. Control of cardiac gene expression by mechanical stress. Annu. Rev. Physiol. 1993;55:55–75. doi: 10.1146/annurev.ph.55.030193.000415. [DOI] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell. Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisada K, Hirota H, Fujio Y, et al. Activation of JAK-STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation. 1996;94:2626–2632. doi: 10.1161/01.cir.94.10.2626. [DOI] [PubMed] [Google Scholar]
- Kunisada K, Negoro S, Tone E, Funamoto M, Yamauchi-Takihara K. STAT3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci. USA. 2000;97:315–319. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisada K, Tone E, Fujio Y, Matsui H, Yamauchi-Takihara K, Kishimoto T. Activation of gp130 transduces hypertrophic signals via STAT3 in cardiac myocytes. Circulation. 1998;98:346–352. doi: 10.1161/01.cir.98.4.346. [DOI] [PubMed] [Google Scholar]
- Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Li M, Sendtner M, Smith A. Essential function of LIF receptor in motor neurons. Nature. 1995;378:724–727. doi: 10.1038/378724a0. [DOI] [PubMed] [Google Scholar]
- MacGowan GA, Mann DL, Kormos RL, Feldman AM, Murali S. Circulating interleukin-6 in severe heart failure. Am. J. Cardiol. 1997;79:1128–1131. doi: 10.1016/s0002-9149(96)00063-x. [DOI] [PubMed] [Google Scholar]
- Mann DL, Kent RL, Cooper GT. Load regulation of the properties of adult feline cardiocytes: growth induction by cellular deformation. Circ. Res. 1989;64:1079–1090. doi: 10.1161/01.res.64.6.1079. [DOI] [PubMed] [Google Scholar]
- Matsui H, Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K. Leukemia inhibitory factor induces a hypertrophic response mediated by gp130 in murine cardiac myocytes. Res. Commun. Mol. Pathol. Pharmacoll. 1996;93:149–162. [PubMed] [Google Scholar]
- Matsui H, Ihara Y, Fujio Y, et al. Induction of interleukin (IL) -6 by hypoxia is mediated by nuclear factor (NF) -kappa B and NF-IL6 in cardiac myocytes. Cardiovasc. Res. 1999;42:104–112. doi: 10.1016/s0008-6363(98)00285-5. [DOI] [PubMed] [Google Scholar]
- Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br. Heart J. 1994;72:561–566. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaka T, Fujikawa K, Nishio Y, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA. 1993;90:10193–10197. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney CD, Hunt RA, Conrad KM, Dostal DE, Baker KM. The type I angiotensin II receptor couples to Stat1 and Stat3 activation through Jak2 kinase in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 1997;29:2513–2524. doi: 10.1006/jmcc.1997.0489. [DOI] [PubMed] [Google Scholar]
- Mestan J, Digel W, Mittnacht S, et al. Antiviral effects of recombinant tumour necrosis factor in vitro. Nature. 1986;323:816–819. doi: 10.1038/323816a0. [DOI] [PubMed] [Google Scholar]
- Miyao Y, Yasue H, Ogawa H, et al. Elevated plasma interleukin-6 levels in patients with acute myocardial infarction. Am. Heart J. 1993;126:1299–1304. doi: 10.1016/0002-8703(93)90526-f. [DOI] [PubMed] [Google Scholar]
- Murakami M, Hibi M, Nakagawa N, et al. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–1810. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Negoro S, Kunisada K, Tone E, Funamoto M, Yamauchi-Takihara K. Activation of JAK/STAT pathway transduces cytoprotective signals in rat acute myocardial infarction. Circulation. 1999;100:I–491. doi: 10.1016/s0008-6363(00)00138-3. [DOI] [PubMed] [Google Scholar]
- Nozaki N, Yamaguchi S, Yamaoka M, Okuyama M, Nakamura H, Tomoike H. Soluble tumor necrosis factor soluble receptors are elevated in relation to severity of congestive heart failure. Jpn. Circ. J. 1997;61:657–664. doi: 10.1253/jcj.61.657. [DOI] [PubMed] [Google Scholar]
- Oh H, Fujio Y, Kunisada K, et al. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70, S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem. 1998a;273:9703–9710. doi: 10.1074/jbc.273.16.9703. [DOI] [PubMed] [Google Scholar]
- Oh H, Kunisada K, Funamoto M, Negoro S, Yamauchi-Takihara K. Activation of gp130 inhibits doxorubicin induced cell death by bcl-xL/caspase–3 interaction and PI3-kinase/Akt pathway in cardiac myocytes. Circulation. 1998b. pp. I–462.
- Olivetti G, Quaini F, Sala R, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol. Cell. Cardiol. 1996;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts. Circulation. 1998;98:149–156. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992;20:248–254. doi: 10.1016/0735-1097(92)90167-l. [DOI] [PubMed] [Google Scholar]
- Pan J, Fukuda K, Kodama H, et al. Role of angiotensin II in activation of the JAK/STAT pathway induced by acute pressure overload in the rat heart. Circ. Res. 1997;81:611–617. doi: 10.1161/01.res.81.4.611. [DOI] [PubMed] [Google Scholar]
- Pan J, Fukuda K, Saito M, et al. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ. Res. 1999;84:1127–1136. doi: 10.1161/01.res.84.10.1127. [DOI] [PubMed] [Google Scholar]
- Patterson PH, Nawa H. Neuronal differentiation factors/cytokines and synaptic plasticity. Cell. 1993;72:123–137. doi: 10.1016/s0092-8674(05)80032-7. [DOI] [PubMed] [Google Scholar]
- Pennica D, King KL, Shaw KJ, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc. Natl. Acad. Sci. USA. 1995a;92:1142–1146. doi: 10.1073/pnas.92.4.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennica D, Shaw KJ, Swanson TA, et al. Cardiotrophin-1: biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 1995b;270:10915–10922. doi: 10.1074/jbc.270.18.10915. [DOI] [PubMed] [Google Scholar]
- Pennica D, Arce V, Swanson TA, et al. Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron. 1996a;17:63–74. doi: 10.1016/s0896-6273(00)80281-0. [DOI] [PubMed] [Google Scholar]
- Pennica D, Swanson TA, Shaw KJ, et al. Human cardiotrophin-1: protein and gene structure, biological and binding activities, and chromosomal localization. Cytokine. 1996b;8:183–189. doi: 10.1006/cyto.1996.0026. [DOI] [PubMed] [Google Scholar]
- Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Post GR, Goldstein D, Thuerauf DJ, Glembotski CC, Brown JH. Dissociation of p44 and p42 mitogen-activated protein kinase activation from receptor-induced hypertrophy in neonatal rat ventricular myocytes. J. Biol. Chem. 1996;271:8452–8457. doi: 10.1074/jbc.271.14.8452. [DOI] [PubMed] [Google Scholar]
- Reif K, Burgering BM, Cantrell DA. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70, S6 kinase. J. Biol. Chem. 1997;272:14426–14433. doi: 10.1074/jbc.272.22.14426. [DOI] [PubMed] [Google Scholar]
- Rook AH, Masur H, Lane HC, et al. Interleukin-2 enhances the depressed natural killer and cytomegalovirus-specific cytotoxic activities of lymphocytes from patients with the acquired immune deficiency syndrome. J. Clin. Invest. 1983;72:398–403. doi: 10.1172/JCI110981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993;12:1681–1692. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- Saito M, Yoshida K, Hibi M, Taga T, Kishimoto T. Molecular cloning of a murine IL-6 receptor-associated signal transducer, gp130, and its regulated expression in vivo. J. Immunol. 1992;148:4066–4071. [PubMed] [Google Scholar]
- Saward L, Zahradka P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 1997;81:249–257. doi: 10.1161/01.res.81.2.249. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Duronio V. Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/Akt: Involvement of MEK upstream of Bad phosphorylation. Proc. Natl. Acad. Sci. USA. 1998;95:7439–7444. doi: 10.1073/pnas.95.13.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze MMK, Hawley RG. Prevention of myeloma cell apoptosis by ectopic bcl-2 expression or interleukin 6-mediated up-regulation of bcl-xL. Cancer Res. 1995;55:2262–2265. [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990;345:440–441. doi: 10.1038/345440a0. [DOI] [PubMed] [Google Scholar]
- Shioi T, Matsumori A, Sasayama S. Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Circulation. 1996;94:1930–1937. doi: 10.1161/01.cir.94.11.2930. [DOI] [PubMed] [Google Scholar]
- Shubeita HE, McDonough PM, Harris AN, et al. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. J. Biol. Chem. 1990;265:20555–20562. [PubMed] [Google Scholar]
- Simpson P. Stimulation of hypertrophy of cultured neonatal rat heart cells through an alpha 1-adrenergic receptor and induction of beating through an alpha 1- and beta 1–adrenergic receptor interaction. Circ. Res. 1985;56:884–894. doi: 10.1161/01.res.56.6.884. [DOI] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signaling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Suematsu S, Matsuda T, Aozasa K, et al. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA. 1989;86:7547–7550. doi: 10.1073/pnas.86.19.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Ito H, Adachi S, et al. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ. Res. 1994;75:426–433. doi: 10.1161/01.res.75.3.426. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ryoke T, Hongo M, et al. Effects of growth hormone and IGF-I on cardiac hypertrophy and gene expression in mice. Am. J. Physiol. 1998;275:H393–H399. doi: 10.1152/ajpheart.1998.275.2.H393. [DOI] [PubMed] [Google Scholar]
- Teiger E, Than VD, Richard L, et al. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J. Clin. Invest. 1996;97:2891–2897. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1b modulates the growth and phenotype of neonatal rat cardiac myocytes. J. Clin. Invest. 1995;96:1093–1099. doi: 10.1172/JCI118095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Carlson M, Mansour SJ, Chien KR, Ahn NG, Thorburn A. Inhibition of a signaling pathway in cardiac muscle cells by ative mitogen-activated protein kinase kinase. Mol. Biol. Cell. 1995;6:1479–1490. doi: 10.1091/mbc.6.11.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Frost JA, Thorburn A. Mitogen-activated protein kinases mediate changes in gene expression, but not cytoskeletal organization associated with cardiac muscle cell hypertrophy. J. Cell Biol. 1994;126:1565–1572. doi: 10.1083/jcb.126.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone E, Kunisada K, Fujio Y, et al. Angiotensin II interferes with leukemia inhibitory factor-induced STAT3 activation in cardiac myocytes. Biochem. Biophys. Res. Commun. 1998;253:147–150. doi: 10.1006/bbrc.1998.9767. [DOI] [PubMed] [Google Scholar]
- Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD) J. Am. Coll. Cardiol. 1996;27:1201–1206. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- Vejsada R, Sagot Y, Kato AC. Quantitative comparison of the transient rescue effects of neurotrophic factors on axotomized motoneurons in vivo. Eur. J. Neurosci. 1995;7:108–115. doi: 10.1111/j.1460-9568.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- Ware BW, Horowits MC, Renshaw BR, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121:1283–1299. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]
- Webster KA, Discher DJ, Bishopric NH. Induction and nuclear accumulation of fos and jun proto-oncogenes in hypoxic cardiac myocytes. J. Biol. Chem. 1993;268:16852–16858. [PubMed] [Google Scholar]
- Weissenbach J, Chernajovsky Y, Zeevi M, et al. Two interferon mRNAs in human fibroblasts: in vitro translation and Escherichia coli cloning studies. Proc. Natl. Acad. Sci. USA. 1980;77:7152–7156. doi: 10.1073/pnas.77.12.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Wollert KC, Taga T, Saito M, et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. J. Biol. Chem. 1996;271:9535–9345. doi: 10.1074/jbc.271.16.9535. [DOI] [PubMed] [Google Scholar]
- Wollert KC, Chien KR. Cardiotrophin-1 and the role of gp130-dependent signaling pathways in cardiac growth and development. J. Mol. Med. 1997;75:492–501. doi: 10.1007/s001090050134. [DOI] [PubMed] [Google Scholar]
- Yamada T, Matsumori A, Sasayama S. Therapeutic effect of anti-tumor necrosis factor-alpha antibody on the murine model of viral myocarditis induced by encephalomyocarditis virus. Circulation. 1994;89:846–851. doi: 10.1161/01.cir.89.2.846. [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T. Hypoxic stress induced cardiac myocyte derived interleukin-6. Circulation. 1995a;91:1520–1524. doi: 10.1161/01.cir.91.5.1520. [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K, Ihara Y, Yoshizaki K, Kishimoto T. Hypoxic stress induced cardiac myocyte derived interleukin-6. — Activation of IL-6 is mediated by NF-IL6 and NF-Kb binding sites —. In: Yazaki Y, editor. Cardiac Development and Gene Regulation. Tokyo: Excerpta Medica Ltd; 1995b. pp. 111–126. [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, et al. Angiotensin II partly mediates mechanical stress-induced cardiac hypertrophy. Circ. Res. 1995;77:258–265. doi: 10.1161/01.res.77.2.258. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, et al. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J. Biol. Chem. 1996;271:3221–3228. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- Yan SF, Tritto I, Pinsky D, et al. Induction of interleukin-6 (IL-6) by hypoxia in vascular cells. J. Biol. Chem. 1995;270:11463–11471. doi: 10.1074/jbc.270.19.11463. [DOI] [PubMed] [Google Scholar]
- Yang YC. Interleukin 11: an overview. Stem Cells. 1993;11:474–486. doi: 10.1002/stem.5530110617. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Taga T, Saito M, et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc. Natl. Acad. Sci. USA. 1996;93:407–411. doi: 10.1073/pnas.93.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A, Ohkubo T, Kiguchi T, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]