

Abstract
With the emergence of drug resistance and the genomic revolution there has been a renewed interest in the genes that are responsible for the generation of bioactive natural products. Secondary metabolites of one major class are biosynthesized at one or more sites by ultra large enzymes that carry covalent intermediates on phosphopantetheine arms. Because such intermediates are difficult to characterize in vitro, we have developed a new approach for streamlined detection of substrates, intermediates and products attached to a phosphopantetheinyl arm of the carrier site. During vibrational activation of gas phase carrier domains, facile elimination occurs in benchtop and Fourier-Transform mass spectrometers alike. Phosphopantetheinyl ejections quickly reduce >100 kDa megaenzymes to <1000 Da ions for structural assignment of intermediates at <0.007 Da mass accuracy without proteolytic digestion. This “Top Down” approach quickly illuminated diverse acyl-intermediates on the carrier domains of the nonribosomal peptide synthetases (NRPSs) or polyketide synthases (PKSs) found in the biosynthetic pathways of prodigiosin, pyoluteorin, mycosubtilin, nikkomycin, enterobactin, gramicidin and several proteins from the orphan pksX gene cluster from Bacillus subtilis. By focusing on just those regions undergoing covalent chemistry, the method delivered clean proof for the reversible dehydration of hydroxymethylglutaryl-S-PksL via incorporation of 2H or 18O from the buffer. The facile nature of this revised assay will allow diverse laboratories to spearhead their NRPS/PKS projects with benchtop mass spectrometers.
About 50% of today's drugs and 75% of today's antimicrobials are derived from secondary metabolites (1,2). Many of those secondary metabolites are of polyketide or nonribosomal peptide origin. With the emergence of resistance and the genomic revolution there is a “renaissance” ongoing in the discovery of bioactive natural products and the characterization of the genes responsible for their production (1). Non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) are large enzymes often >>100 kDa that biosynthesize their natural products (e.g., the antibiotics penicillin, vancomycin) via covalent intermediates on phosphopantetheine arms (3). Currently, even when NRPS and PKS proteins can be overproduced their direct interrogation by mass spectrometry (MS) is difficult and time-consuming, with reports from relatively few laboratories appearing in the primary literature (4-6). Therefore it would be of great benefit for the NRPS and PKS community if new MS-based methods to characterize these proteins were developed and easier to implement. This paper describes such a method.
The purpose of this paper is four-fold: 1) the paper introduces a new and efficient method that utilizes a gas phase elimination reaction that takes place during tandem mass spectrometry (MS/MS) to quickly characterize substrates, intermediates and products that are loaded onto the phosphopantetheinyl arm on carrier domains of NRPS and PKS systems, 2) the paper provides a mechanistic rationale for the phosphopantetheinyl eliminations that are utilized in this assay, 3) the paper demonstrates that the phosphopantetheinyl ejections can be performed not only on peptides but also on intact proteins. Tandem mass spectrometry on intact proteins is also known as the “Top Down” approach (7), and 4) finally, we show that the method is readily adapted to benchtop mass spectrometers making it useful to researchers that do not have access to high end MS instrumentation.
We provide support for the robustness and generality of this assay via 33 examples as this approach was used to characterize the loading of substrates, intermediates and products of proteins involved in the biosynthesis of the antibiotic nikkomycin (8), the antifungal agent pyoluteorin (9, 10), the antibiotic prodigiosin (11, 12), the antibacterial gramicidin (13, 14), the antibacterial agent mycosubtilin (15, 16), and the siderophore enterobactin (17, 18). Further, intermediates were detected via this gas phase elimination on four proteins from the orphan orphan pksX cluster of B. subtilis that produces an unknown antibiotic (19-21). In addition, we show clear evidence for the re-hydratase activity of PksH via stable isotope incorporation from solvent, an experiment greatly streamlined by observing just that portion of the protein-substrate undergoing chemistry. PksL (28 kDa), NikP1 (74 kDa), and GrsA (127 kDa) are used to show the extensibility of this assay format to Top Down experiments that circumvent the need for proteolysis before mass spectrometry.
RESULTS and DISCUSSION
Using Electrospray Ionization Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry (ESI/FT-ICRMS), thiotemplate bound substrates and intermediates of NRPS and PKS systems have been visualized (9, 12, 16, 18, 19, 23, 24). The FT-ICRMS assay has recently been adapted into a tool for substrate screening, but the mass accuracy has been between 0.05−0.5 Da and required, in many cases, digestion and active site mapping prior to the analysis (25). Detection of key peptides carrying acyl-intermediates by peptide mapping is one of the rate limiting steps in the characterization of thiothemplate enzymes by mass spectrometry (23). Using tandem mass spectrometric methods, such as infrared multiphoton dissociation (IRMPD) (26) and collisionally activated dissociation (CAD) (27), we observed two reactions that result from the elimination of the phosphopantetheinyl functionality of carrier domains found on NRPSs and PKSs. As an example, subjecting the 12+ charge state of pyrrolyl-S-PltL to IRMPD resulted in two 11+ fragment ions of high abundance. These 11+ ions correspond to the apo protein −18 Da or apo protein +80 Da (Figure 1A), and their 1+ complementary ions were also observed. These ion pairs sum to the intact mass of the precursor ions and represent the dominant dissociation channels observed in the experiment. Thus 12+ precursor ions of an intact protein generated primarily 11+ and 1+ fragment ions, along with minor product ions from amide bond cleavage. The fragmentation, as a result of IRMPD, creates two complementary ion pairs. The observed masses for the eliminated products are 354.156 and 452.132 Da, and agree with the calculated ions to within 0.007 Da (figure 1B and C).
Figure 1.

The phosphopantetheinyl elimination fragments observed during tandem mass spectrometry. A) Pyrrolyl-S-PltL subjected to the tandem mass spectrometric method IRMPD. B) Enlargement of the mass spectral region that correspond to the smaller 1+ pantetheinyl fragment ion. C) Enlargement of the mass spectral region that correspond to the smaller 1+ phosphopantetheinyl fragment ion. D) Elimination fragment of mono-chloropyrrolyl-S-PltL E) Elimination fragment of dichloropyrrolyl-S-PltL. The protein derived ions are reported as the neutral monoisotopic mass while the PPant eliminated ions are reported as the 1+ monoisotopic mass. The remaining signals in the 450−500 m/z region are frequency spikes or amide backbone cleavage of the protein itself.
Similar results were obtained when mono-chloropyrrolyl- S-PltL, dichloropyrrolyl-S-PltL and the di-bromopyrrolyl-S-PltL (9) were subjected to tandem mass spectrometry (figures 1D, 1E respectively). These results also show that this approach can be used for the direct visualization of species that have distinctive isotopic signatures without the need for protein expression in 13C,15N double-depleted media (9). As example, the chloropyrrolyl-S-PltL as well dichloropyrrolyl-S-PltL phosphopantetheinyl ejection ion show the A+2 isotope that comes from 37chlorine (figures 1D and E) which comprises 24% of the chlorine in nature. In addition, the phosphopantetheinyl ejection from di-bromopyrrolyl-S-PltL allowed the direct observation of the +2 and +4 isotopes that come from the incorporation of one and two bromine 81's when it was subjected to tandem mass spectrometry (figure 2). The natural abundance of 81Br is 49% of the total natural bromine isotopic content while for 79Br it is 51%. The isotopic pattern observed in the di-bromopyrrolyl elimination reaction is quite similar to the predicted isotopic pattern (figure 2).
figure 2.
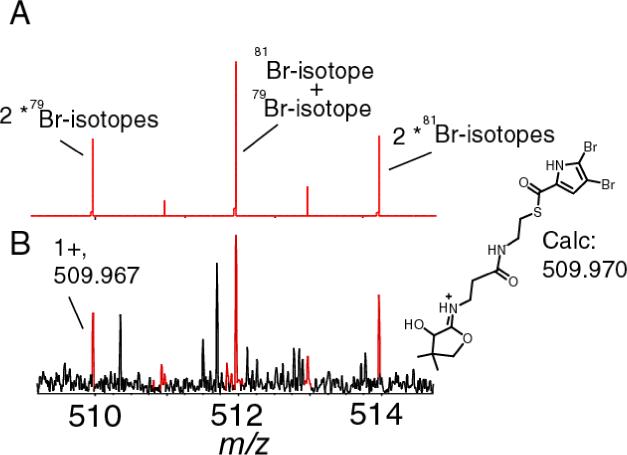
A) The theoretical isotopic distribution of the elimination ion for of di-bromopyrrolyl-S-PltL. B) Elimination ion of di-bromopyrrolyl-S-PltL.
There are two main mechanisms that can be invoked for the elimination of the phosphopantetheinyl functionality to generate a mass consistent with the apo form of the protein that has lost 18 Da and a charge. Both of these mechanisms give a product of identical mass and charge. The first mechanism involves a rearrangement in which the α-carbon of serine is deprotonated and a rearrangement takes place as shown in figure 3A. The resulting protein ions have a mass shift from the apo form of the protein with an of loss of 18 Da via the generation of a dehydroalanine and would have a reduction in charge. In this paper we refer to this 1+ ion product as PPant (figure 3A). The second proposed mechanism is analogous to the elimination of a phosphate from a perprotonated phosphorylated serine or threonine during collisionally activated dissociation as recently proposed (28). During this elimination, an oxazolium containing protein ion is produced that also has a mass shift of the apo peptide −18 Da (figure 3B).
Figure 3.

Different mechanistic proposals for the two major types of eliminations of the phosphopantetheinyl functionality during tandem mass spectrometry to generate the ejection ions with a charge of 1+. R = H or acyl substituent. z = charge. In A and B we show that the amide on the right side of the 1+ ejection ion is protonated, it is also possible that the other amide is protonated (37).
The second major type of phosphopantetheinyl elimination leaves a phosphate functionality on the protein but also involves the loss of one charge to an ejected 1+ ion. The mechanism that accounts for this is shown in figure 3C. Following the elimination reaction, a phosphate anion is left behind and the carbonyl of an amide displaces the pantetheinylate moiety forming a 5-membered ring with a protonated imine that is positively charged (figure 3C). In this paper we abbreviate this ejected ion as Pant.
To demonstrate that this elimination reaction can also be used to observe small mass changes with high mass accuracy, the ACP domain from MycA (13.4 kDa) on the mycosubtilin pathway loaded with either acetoacetate and β-aminobutyrate that was generated using an in cis aminotranferase activity was investigated (16). These two species have a calculated mass difference of 1.032 Da. Following digestion and separation of the ACP domain, the latter was subjected to MS/MS using IRMPD. Looking at the region below 600 m/z, the ions of the phosphopantetheinyl functionality loaded with β-aminobutyrate gave 1+ ion signals at 346.180 m/z and 444.154 m/z (figures 4B and D). The calculated theoretical masses for the eliminated products ions are 346.180 and 444.157 Da and are within 0.003 Da (7 ppm) of the observed values. In the case of MycA loaded with acetoacetate, the eliminated fragments that were observed correspond to the mass values of 345.151 and 443.124 Da that are within 0.003 Da of the calculated masses (345.148 and 443.125 Da, figures 4A and C). The mass difference between the acylated form and the calculated holo form was 84.021 Da. Based on the accurate mass search as described in the experimental section, the best molecular formula that could be assigned was C4H4O2 and is in agreement with the addition of the acetoacetyl substituent on the phosphopantetheinyl functionality as it matched within 5 ppm. This confirms that sufficient mass accuracy is available to assign an empirical formula to the acylated species, particularly important for discovery applications where structural elucidation of bound species is required.
Figure 4.
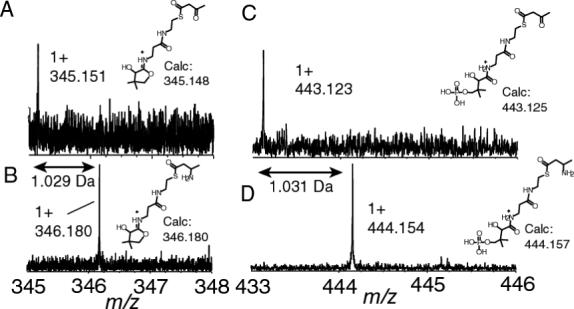
Phosphopantetheinyl fragment ions observed on the ACP2 of MycA upon incubation of acetoacetyl-S-MycA-ACP2 with Gln. A and C) the two phosphopantetheinyl elimination reactions of acetoacetyl-S-MycA-ACP2, B and D) the two phosphopantetheinyl elimination reactions of acetoacetyl-S-MycA-ACP2 treated with glutamine to form β-aminobutyryl-S-MycA-ACP2.
In this paper, the PPant and/or Pant ejection ions for 33 different acyl-S-carrier domains are provided and they are displayed in Table 1 and figure 5. In Table 1, the elimination fragments ions are shown for pyrrolyl-S-PigG, pyrrolyl-β-ketoacyl-S-PigH-ACP1 and pyrrolyl-β-ketoacyl-S-PigH-2ACP (12), the ACP and PCP domain of the 4 domain construct MycA loaded with acetoacetate, other β-ketoacids and the corresponding-β-aminobutyrates (16), pyrrolyl-S-PltL, mono-chloropyrrolyl-S-PltL, dichloropyrrolyl-S-PltL, di-bromopyrrolyl-S-PltL (9), seryl-S-PksN (25), phenylacetyl-S-PksJ (29), acetoacetyl-S-AcpK, acetyl-S-AcpK, acetoacetyl-S-PksL-2ACP, hydroxymethylglutaryl-S-PksL-2ACP, dehydromethylglutaryl-S-PksL-2ACP, 2Δ-isoprenyl-S-PksL-2ACP (19), and dihydroxybenzoyl-S-EntB(ArCP) (17, 25). To determine if larger intact enzymes could be interrogated by the Top Down approach, the 74 kDa protein NikP1 on the nikkomycin biosynthetic pathway (13,25) and the undigested 127 kDa phenylalanyl-S-GrsA or D5-deutero phenylalanyl-S-GrsA were analyzed (13, 14). In both these cases, one or both elimination products were observed with high mass accuracy. At this point, we do not know why in some of the cases one of the ejection products is observed but not the other nor do we know what influences this. Below we highlight some of the experiments to emphasize the utility of this assay.
Table 1.
The observed masses of the phosphopantetheinyl eliminated 1+ fragment ions.
| Protein Investigated | Pathway | Digestion method | Fragmentation Methodb | Ra | Elimination ion Aa (calculated) | Elimination ion Ba (calculated) |
|---|---|---|---|---|---|---|
| PigH(ACP1) | Prodigiosin | Trypsin | OCAD | A | 347.129(347.128) | ND |
| PigH(ACP1) | Prodigiosin | Trypsin | OCAD | B | 396.161(396.159) | ND |
| PigH-2ACP | Prodigiosin | not digested | IRMPD | A | 347.130(347.128) | ND |
| PigH-2ACP | Prodigiosin | not digested | IRMPD | B | 396.161(396.159) | ND |
| PigG | Prodigiosin | not digested | IRMPD | C | 354.152(354.149) | ND |
| PltL | Pyoluteorin | not digested | IRMPD | C | 354.156(354.149) | 452.132(452.126) |
| PltL | Pyoluteorin | not digested | IRMPD | D | 388.112(388.110) | ND |
| PltL | Pyoluteorin | not digested | IRMPD | E | 422.077(422.071) | 520.050(520.048) |
| PltL | Pyoluteorin | not digested | OCAD | F | 509.967(509.970) | ND |
| MycA (ACP2) | Mycosubtilin | Trypsin | OCAD | G | 345.151(345.148) | 443.124(443.125) |
| MycA (ACP2) | Mycosubtilin | Trypsin | OCAD | H | 346.180(346.180) | 444.154(444.157) |
| MycA (ACP2) | Mycosubtilin | Trypsin | OCAD | I | ND | 445.150(445.154) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | J | ND | 555.253(555.251) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | K | ND | 557.283(557.279) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | M | ND | 499.190(499.188) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | N | ND | 501.219(501.217) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | G | ND | 443.128(443.125) |
| MycA (PCP1) | Mycosubtilin | Trypsin | IRMPD | K | ND | 445.155(445.154) |
| AcpK | Orphan | not digested | IRMPD | G | 345.153(345.148) | 443.131(443.125) |
| AcpK | Orphan | not digested | IRMPD | N | ND | 401.119(499.201) |
| PksN | Orphan | Trypsin | IRMPD | O | ND | 446.143(446.136) |
| PksJ (A1) | Orphan | Trypsin | IRMPD | P | ND | 477.150(477.146) |
| PksL-2ACP | Orphan | not digested | IRMPD | G | 345.151(345.148) | 443.127(443.125)c |
| PksL-2ACP | Orphan | not digested | IRMPD | Q | 405.170(405.170) | 503.149(503.146)c |
| PksL-2ACP | Orphan | not digested | IRMPD | R | 343.171(343.170) | 441.147(441.146)c |
| PksL-2ACP | Orphan | not digested | IRMPD | S | 407.173(407.174) | ND |
| PksL-2ACP | Orphan | not digested | IRMPD | T | 406.176(407.176) | ND |
| PksL-2ACP | Orphan | not digested | IRMPD | U | 387.162(387.159) | 485.139(485.136) |
| NikP1 | Nikkomycin | CNBr | OCAD | V | ND | 496.164(496.163) |
| NikP1 | Nikkomycin | not digested | IRMPD | V | ND | 496.163(496.163) |
| EntB | Enterobactin | not digested | IRMPD | W | ND | 495.118(495.120) |
| GrsA | Gramicidin | not digested | IRMPD | X | 408.198(408.196) | 506.179(506.173) |
| GrsA | Gramicidin | not digested | IRMPD | Y | 413.231(413.227) | 511.212(511.201) |
The structures representing R on the elimination ions are shown in figure 5.
Structures of Pant and PPant are shown in figure 4 as well.
Values taken from reference 19. ND = Not detected
Figure 5.

Structures of the R groups in Table 1 of the phosphopantetheinyl 1+ fragment ions described.
Recently, we were able to observe the loaded form of the undigested NikP1 by ESI-ICRFTMS by observing the increase in mass from 74356 ± 11 to 74498 ± 01 Da upon addition of histidine (13). This is a 142 ± 11 Da increase, in agreement with the expected mass increase of 137.059 Da for histidine. However, the mass increase is not very accurate because the protein did not have resolved isotopes. Performing IRMPD on the intact protein gave rise to a 496.164 Da fragment ion corresponding to the eliminated PPant to within 2 ppm of the calculated mass. This ion is 137.060 Da larger compared to the calculated holo form (359.104 Da) and is within 0.001 Da compared to the calculated mass shift for histidine. A second intact construct that was analysed in this fashion was the 127 kDa protein GrsA. Loading of holo GrsA with phenylalanine resulted in a mass of 126,714 ± 15 Da and the protein loaded with D5-deuterated phenylalanine had a mass of 126,726 ± 12 Da and therefore would require several measurements or digestion to tell with confidence which one of the proteins was loaded with D0 or D5-phenylalanine. Subjecting the protein to IRMPD however showed the elimination fragments had a mass difference of 5.033 Da, in agreement with the 5.029 Da for the calculated difference between the two species (figure 6, right). Such use of stable isotopes to confirms the identity of a substrate or can assists in probing mechanisms.
Figure 6.

Phosphopantetheinyl elimination of D0-phenylalanyl-S-GrsA (A) compared to D5-phenylalanyl-S-GrsA (B). A, adenylation; PCP, peptidyl carrier domain; E, epimerisation domain.
In addition, this method can be used to investigate the incorporation of stable isotopes for mechanistic studies even those using D2O or H218O. This is demonstrated by a reaction of hydroxymethylglutaryl-S-PksL-2ACP (HMG-S-PksL-2ACP) with PksI or PksH. PksH and PksI are proteins from an orphan gene cluster in B. subtilis that are involved in the dehydration and decarboxylation of HMG-S-PksL-2ACP to produce 2Δ-isoprenyl-S-PksL-2ACP (Scheme in figure 7A) (16). PksH appears to be the dehydratase while PksI catalyzes the decarboxylation. Similar findings were recently obtained with the PksH and PksI homologs on the curacin system (24). Incubation of HMG-S-PksL-2ACP with PksI, resulted in no reaction. When HMG-S-PksL-2ACP was incubated with PksH, a partial dehydration product was observed. Because complete conversion to the 2Δ-isoprenyl-S-PksL-2ACP was observed when both PksI and PksH were added, it was possible that the PksH reaction was reversible. To test this hypothesis, a racemic mixture HMG-S-PksL-2ACP was prepared by incubating hydroxylmethylglutaryl-CoA with apo-PksL-2ACP and the phosphopantetheinyltransferase Sfp. This was incubated with either PksL or PksH in 70% H218O or D2O and subjected to mass spectrometry and IRMPD. If the reaction was reversible, the incorporation of a deuterium and water from the buffer was anticipated (figure 7B and C). Because a racemic mixture of HMG-S-CoA was used to prepare HMG-S-PksL-2ACP, it was anticipated that PksH would only function on one isomer and therefore a maximum of 50% of the total HMG-S-PksL-2ACP could serve as a substrate for PksH.
Figure 7.

The rehydratase activity of PksH. A) General overview of the conversion of Acac-S-PksL-2ACP to isoprenyl-S-PksL-2ACP as catalysed by PksG, PksH and PksI. For clarity the chemistry on only one of the two carrier domains is shown. B) Hypothesis for the hydroxyl exchange catalysed by PksH in 70% H218O. C) Hypothesis for the deuterium exchange catlysed by PksH in 70% D2O. D) The data showing the 18O and 2H incorporation into the elimination fragment ion of HMG-S-PksL when the reaction was carried out in 18O or 2H buffers. PksI, served as negative control and demonstrates that the reaction was specific to PksH. The starting HMG-S-PksL-2ACP is generated from racemic HMG-CoA and therefore only 50% of this protein form could serve as a substrate.
From the intact mass of HMG-S-PksL-2ACP (a 28 kDa species), it could not be unambiguously shown that partial 18O was incorporated even-though we are using FT-ICRMS to detect the intact ions. Therefore the ions were subjected to IRMPD and the PPant elimination reaction was analyzed. The elimination reaction in the presence of PksH in 18O buffer showed about 33% incorporation of 18O into HMG-S-PksL-2ACP (figure 7D). We feel quantifying the relative signals in this case is warranted, as both species should have similar ionization efficiencies and elimination reaction rates, however if the substrates on the phosphopantetheinyl functionality differ dramatically, the relative ionization efficiencies must be established when quantification is desired. The observed mass of 407.173 Da agreed well with the predicted mass of 407.174 Da. Because PksH selectively dehydrates one of the two isomers of HMG-S-PksL-2ACP, it was anticipated that we could have a maximum of 35% incorporation of 18O; therefore the 18O incorporation appears to be nearly complete. Similarly, when the PksH reaction was repeated but this time in D2O, an estimated 33% of the HMG-S-PksL-2ACP contained a deuterium as evidenced by a mass shift of 1.007 Da again in agreement with the maximum of 35% deuterium incorporation that could be obtained for this system (figure 7D). This supports the hypothesis that PksH catalyzes the dehydration as well as rehydration reaction while PksI does not recognize either of the isomers of HMG-S-PksL-2ACP as a substrate. On the other hand, when both PksI and PksH are added to HMG-S-PksL-2ACP, we see a loss of 62.002 Da (table 1) (19). These results are in agreement with the reversible dehydration reaction catalysed by PksH and decarboxylation catalyzed by PksI, results that cannot be obtained using the standard FT-ICRMS assay format with a 28 kDa protein, in particular when only partial conversion is observed and provide an example of the type of studies that can be performed using this assay format.
In this paper, we describe the use primarily of an FT-ICRMS instrument because of its superior mass accuracy and it is the main instrument in our laboratory but the assay is also applicable to non-FT-ICRMS instruments. In one of the original studies of NRPS systems by mass spectrometry, the elimination ions were observed using MS/MS of small peptides (<3 kDa) on a triple quadrupole instrument but was not utilized as an assay to observe the acylated and characterize substrates loaded onto the phosphopantetheine arm with high mass accuracy (30, 31). These results by these early observations provided an indication that this assay can be performed on smaller peptide fragments from digests with low resolution instruments but provided no indication that it could be accomplished with intact proteins rather than only digested samples. If this Top Down assay is transferable to other less expensive and more common mass spectrometric instruments, it would make it a much more general assay to study NRPS and PKS biosynthetic pathways. To demonstrate the Top Down approach on a non-FTMS instrument, the phosphopantetheinyl ejection assay was repeated using a linear ion trap (LTQ) instrument, which is a very common benchtop MS instrument. Subjecting holo AcpK to trap isolation and CAD, resulted in fairly abundant fragment ions at m/z 261.1 and 359.1. These are well in agreement with the calculated masses 261.127 and 359.104 for the phosphopantetheinyl ejections (figure 8C). Similarly, the phosphopantetheinyl ejection of the 28 kDa HMG-S-PksL-2ACP resulted in a new and very abundant peak at 405.2 m/z, in agreement with the expected mass of 405.017 Da (figure 8F). These two experiments establish that the LTQ, as a representative benchtop instrument, can be used to generate the eliminated ions from intact proteins as well.
Figure 8.
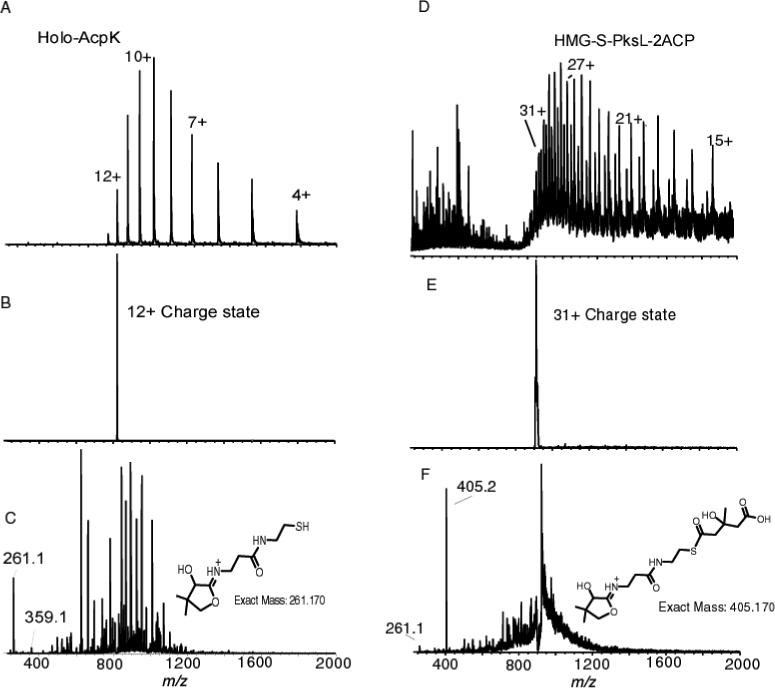
Phosphopantetheinyl ejection as observed with an ion trap instrument. A) ESI/MS of holo-AcpK (10.3 kDa). B) Trap isolation of the 12+ charge state C) CAD at 17 % of the ions isolated in B. D) Broadband spectrum of HMG-S-PksL-2ACP (28 kDa). E) Trap isolation of the 31+ charge state F) CAD at normalized collision energy of 17 % of the ions isolated in E. 405.2 m/z is HMG-Pant. 261.1 m/z is Pant.
To establish that the ion trap MS instrument could also be used to monitor small mass differences such as the 2 Da differences generated by the conversion of Acac-S-PksL-2ACP to isoprenyl-S-PksL-2ACP, HMG-S-PksL-2ACP was incubated with PksI, PksH or both PksI and H. When HMG-S-PksL-2ACP (prepared via the incubation of Ac-S-AcpK, PksG and Acac-S-PksL-2ACP) was incubated with PksI, the masses of 345.2 (Pant elimination of unreacted acetoacetyl) and 405.2 were observed (figure 9A). While upon the addition of PksH to HMG-S-PksL-2ACP, a new mass at 387.1 Da was observed (figure 9B). The observation of the low steady state dehydration using the LTQ showed a similar amount of the dehydrated species upon the addition of PksH when compared to the FT-ICRMS (figure 9B inset). This provides an indication that similar results are obtained using the LTQ when compared to the FT-ICRMS. When both PksI and PksH were added, a new mass of 343.2 Da emerged that is 2 Da smaller than the eliminated ion from acac-S-PksL-2ACP, consistent with the formation of an isoprenyl-S-PksL-2ACP product (figure. 9C). Figure 7D and F are a direct comparison between the same phosphopanteteinyl ejection ion from HMG-S-PksL-2ACP as observed by the low resolution LTQ instrument and the high resolution FT-ICRMS experiment which shows that the FT-ICRMS has a significant advantage in resolution and mass accuracy. These comparisons do not invalidate the utility of the LTQ as a tool as there are very few instances where one might need to have a mass accuracy to within 0.007 Da or less, in the majority of instances a mass accuracy of within 0.1 Da as obtained with the LTQ is sufficient. When a low instrument resolution instrument is used for this assay and the signal is in doubt, one could resort to stable isotope incorporation studies to confirm and solidify the identity of the signal.
Figure 9.
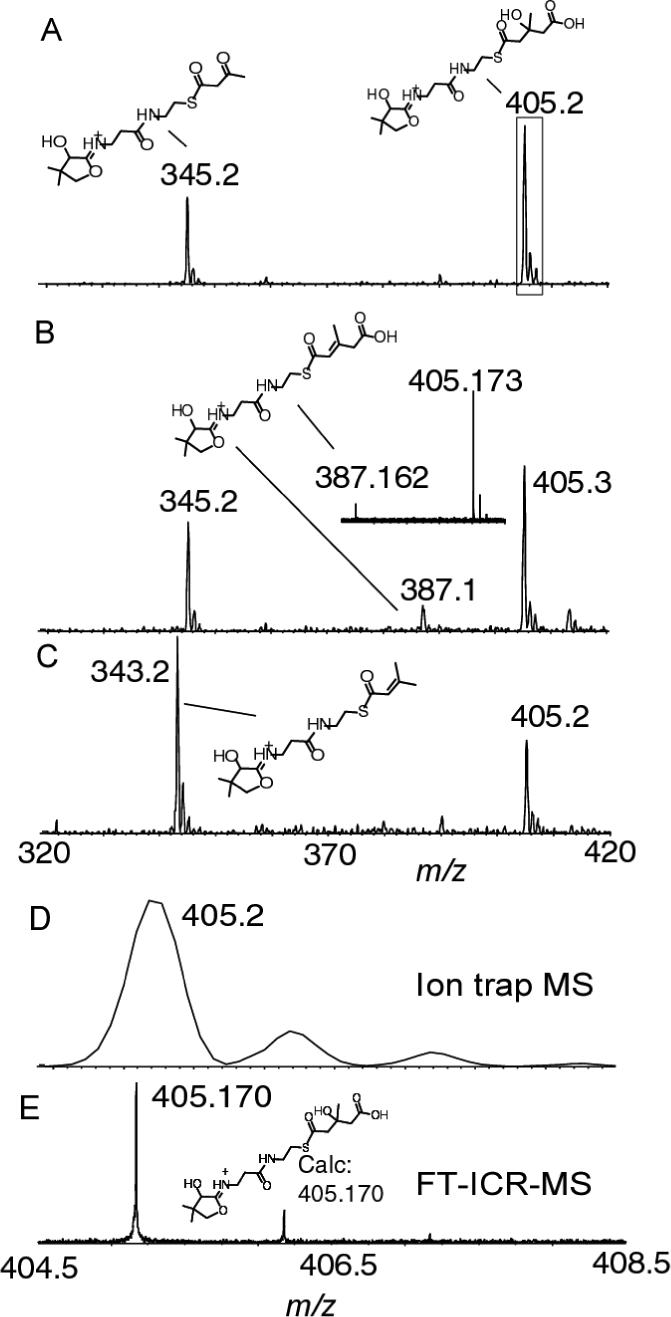
Low resolution observation of ejection ion for isoprenyl-S-PksL-2ACP as catalysed by PksH and PksI. A) Pant ejection of Acac-S-PksL-2ACP and HMG-S-PksL-2ACP following the incubation of Acac-S-PksL-2ACP with Ac-S-AcpK, PksG and PksI. For comparison with FT-ICRMS data the “boxed” data is enlarged in D. B) Pant ejection of Acac-S-PksL-2ACP and HMG-S-PksL-2ACP following the incubation of Acac-S-PksL-2ACP with Ac-S-AcpK, PksG and PksH. Inset shows the signal to show the similar level of dehydration was observed by FT-ICRMS as was observed using the LTQ C) Pant ejection of HMG-S-PksL-2ACP and isoprenyl-S-PksL-2ACP following the incubation of Acac-S-PksL-2ACP with Ac-S-AcpK, PksG and PksHI. D) The HMG-PksL-2ACP ejected ion enlarged from the boxed in ion in panel A. E) the same ion in D but generated and measured using FT-ICRMS. CAD voltages ranged from 15 to 30 %.
In summary, the direct mass spectral visualization of substrates and intermediates via a gas phase elimination presented in this paper will be very useful for studying NRPS/PKS but can also be readily extended to the study of fatty acid biosynthetic pathways. It gives the masses of the substrates at the phosphopantetheinylated active sites with very high mass accuracy and is useful for several reasons. The first major implication of this assay is that it immediately confirms that the substrate or intermediate is linked to the phosphopantetheinyl functionality or not without the need for digestion. Such a confirmation cannot be accomplished by any other assay currently available to the NRPS and PKS community. It eliminates the possibility that the substrate or intermediate is somewhere else on the protein that may be an in vitro artifact. Second, this phosphopantetheinyl ejection assay improves the speed at which activities of phosphopantetheinylated proteins can be characterized and in many cases will alleviate the need to hydrolyze the substrate or intermediate for characterization by HPLC or small molecule mass spectrometry. Third, if a substrate such as a halogen that has multiple isotopes is incorporated, the isotopic pattern can be directly verified. Fourth, it is possible to observe the direct incorporation of stable isotopes for biosynthetic studies. Finally, it is possible to confirm new activities such as the loading of phenylacetate on the first adenylation-thiolation module of PksJ. We project application of this MS/MS based method to ever larger multi-domain and even multimodular thiotemplate assembly lines >200 kDa and it is readily adapted to parent ion scanning (32, 33) and selected reaction monitoring for targeted detection of species at low levels (34).
Materials
The proteins SgcC2, SgcC1, SgcC4, Sfp, PigH, PigH-2ACP, PigJ, PigG, PigI, PigA, PltL, SsuE, PltA, GrsA, MycA(construct A4N7), AcpK, PksN, PksL-2ACP, PksI, PksH, NikP1, EntE and EntB(ArCP) were obtained as described (9, 12,16,18,19,23). The first di-domain of PksJ with an N-terminal histidine tag was overproduced in E. coli and also purified via nickel affinity chromatography (Straight, Dorrestein, Kelleher, Kolter unpublished). Racemic HMG-CoA, and all other chemicals were purchased from Sigma-Aldrich.
Digestion
When digestion was employed, it was achieved by cyanogen bromide or trypsin via protocols identical as described (9, 12,16,18,19,23).
Purification of the active sites
Both the digested proteins and the undigested proteins (except GrsA) were subjected to HPLC purification. The gradients used were water to acetonitrile gradients as described (9, 12, 16, 18, 19, 23). During the HPLC runs, the fractions containing the carrier domains were collected, frozen at −80 °C and lyophilized.
Sample preparation of GrsA for mass spectrometry
For experiments involving the protein GrsA (PheATE), the holo protein was generated by incubating 500−600 μg of protein for one hour in a reaction mixture containing 0.05 M Tris-HCl (pH 8), 0.01 M MgCl2, 0.002 M Tris(2-carboxyethyl) Phosphine (pH 6), 200 μm Coenzyme-A, and 0.5 mg/mL Sfp (4 μL). After one hour, the reaction was supplemented with 0.01 M ATP and 0.01 M amino acid (L-phenylalanine or D5-L-phenylalanine) and allowed to incubate for an additional thirty minutes to generate loaded protein. Reactions were stored at −80°C until preparation for injection into the mass spectrometer. Immediately before injection into the mass spectrometer, samples were prepared using C4 ZipTips (Millipore). To prepare each sample, the ZipTip was washed five times with 10 μL ACN and 5 times with 10 μL H2O/0.1% TFA. The sample was loaded onto the ZipTip by pipetting into the sample 10−15 times, each time injecting back into the reaction vile. The ZipTip was washed again 8 times with 10 μl H2O/0.1% TFA, and the sample eluted into 5 μL 78% ACN/2% acetic acid. To prepare the sample for injection into the mass spectrometer, 25 μL of nanospray solution (49% MeOH/ 49% H2O/ 2% formic acid) was added to the eluted sample and the sample mixed thoroughly. The values of the GrsA reported are the neutral most abundant masses and the standard deviation is a result from the calculation of the mass at each different charge state.
FT-ICR Mass spectrometry
The lyophilized fractions were resuspended in 50−400 μL of electrospray solution (78% ACN, 0.1% acetic acid or 49% methanol, 1% formic acid). Usually, the 49% methanol, 1% of formic acid was used with the larger protein/protein domains as this resulted in less unwanted fragmentation before the ions reached the cell. For mass spectrometric analysis, a custom 8.5 Tesla ESI-FTMS mass spectrometer equipped with a front-end quadrupole was utilized (35) The samples were introduced into the FTMS using a NanoMate 100 for automated nanospray (Advion Biosciences, Ithaca, NY). To introduce the samples, a back pressure of 0.45−0.8 psi and a voltage of 1.4−1.80 V was used. Typical instrument settings for the initial broadband analysis are 50−500 ms ion accumulation per scan and 10−300 scans were acquired per spectrum. While the majority of the time the quadrupole was not used to enhance the signal but when it was used, the accumulation times were changed from 50−500 ms to 500−7000 ms depending on signal intensity. For SWIFT excitation to isolate specific ions, the amplitude was set to 0.27 Vp-p and 30−50 waveform loops were used. For CAD, the ions were subjected to −10 to −40 Volts in the accumulation octupole (36). IRMPD was accomplished with a CO2 laser by irradiation on axis in regards to the cell for 80−300 ms. The instrument was externally calibrated using ubiquitin, 8560.65 Da monoisotopic Mr value (Sigma) or on the electron capture dissociated fragment ions of ubiquitin. All the values described in this manuscript were determined manually. The masses for the PPant and Pant ejection products are reported as the ion masses. The FT-ICRMS settings were as follows and ranges are indicated where appropriate; Tube lens 200−275 V, Capillary heater 3.5−4 A, Quad filter −20 V, skimmer was off, capillary offset 34 V, transfer of −10 to −80 V, transfer times 1.00 to 1.25 ms, leak gas of 3.5 to 4.5E-5 Torr. Excitation was set to 200 or 400 to 2000 m/z with a chirp rate of 6540 Hz/μsec at a 0.45 Vamplitude, For detection at low m/z, the Nyquists frequency was set such that either 204 or 408 m/z was the cutoff. All data sets were collected as 512 k or 1 Mb.
Generation of HMG-S-PksL-2ACP in 18O or deuterated buffer
100 μL of 50 mM Tris, 1 mM Tris(2-carboxyethyl) Phosphine, pH 7.6 was frozen and lyophilized. The resulting residue was redissolved in 100 μL of D2O or H218O. To generate HMG-S-PksL-2ACP, a PksL-2ACP sample (75 μL, 134 μM) was incubated with racemic HMG-CoA (10 μL, 3 mM), Sfp (3 μL, 1.2 mg/ml), and MgCl2 (1 μL, 800 mM) for 30 min., 25 μL of this solution was diluted with 70 μL of the 18O buffer or Deuterated buffer prepared above.
18O exchange of HMG-S-PksL-2ACP by PksH
To HMG-S-PksL-2ACP in 18O buffer, 5 μL of PksI (215 μM), PksH (117 μM) or both PksH and PksI were added and the reaction allowed to proceed for an additional ten minutes before the reaction was halted by the addition of 50 μL of 10% formic acid. The samples were purified by HPLC before they were analysed by mass spectrometry.
2H exchange of HMG-S-PksL-2ACP by PksH
To HMG-S-PksL-2ACP in 2H buffer, 5 μL of PksI (215 μM), PksH (117 μM) or both PksH and PksI were added and the reaction allowed to proceed for an additional ten minutes before the reaction was halted by the addition of 50 μL of 10% formic acid. The samples were purified by HPLC before they were analysed by mass spectrometry.
LTQ MS settings
The samples were introduced via direct infusion with a flow rate 5 μL/min, maximum intensity for the protein signal was obtained using automatic tuning option on the LTQ instrument (Finnigan). After the maximum signal was obtained for a given sample, the ions of interest were isolated with a 5 m/z window and the normalized collision energy for CAD was set to 15 to 30 % to induce the phosphopantetheinyl ejections.
Empirical formula calculation for the acyl substitutent
The calculated mass of the holo form (protonated, 261.127 Da) was substracted from the observed mass of 345.148 Da. This mass of 84.021 Da represents the mass difference of the acylated species compared to the holo form of the protein. This mass value was imported into Xcalibur (Thermo-Finnigan) and the mass tolerance was set to 25 ppm. Only two empirical formulas were obtained that matched to this mass difference. The first was C4H4O2 and was within 4.99 ppm from the calculated mass of an acetoacetyl loaded form, the other was C2H2N1O3 for which the mass was within 20.97 ppm and chemically does not make sense.
Acknowledgments
This work was supported in part by NIH Kirschstein NRSA postdoctoral fellowships F32-GM 073323 (PCD), F32-GM 72299 (ZDA), NSF postdoctoral Fellowship in Microbial Biology DBI-0200307 (PDS), and NIH grants GM 49338 (CTW), GM 58213 (RK), GM 67725 (NLK).
Abbreviations
- ESI/FT-ICRMS
Electrospray Ionization Fourier-Transform Ion Cycletron Resonance Mass Spectrometry
- MS
Mass Spectrometry
- MS/MS
Tandem Mass Spectrometry
- PKS
Polyketide Synthase
- NRPS
Non-ribosomal Peptide Synthetase
- IRMPD
Infrared Multiphoton Dissociation
- CAD
Collisionally Activated Dissociation
- ACP
Acyl Carrier Protein
- 2ACP
Tandem Acyl Carrier Proteins
- PCP
Peptidyl Carrier Protein
- HMG
Hydroxymethylglutaryl
- Acac
acetoacetyl
- HPLC
High Pressure Liquid Chromatography
- TFA
trifluoroacetic acid
- ACN
acetonitrile
References
- 1.Paterson I, Anderson EA. The renaissance of natural products as drug candidates. Science. 2005;310(5747):451–453. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- 2.Bode HB, Muller R. The impact of bacterial genomics on natural product research. Angew. Chem. Int. Ed. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science. 2004;303(5665):1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 4.Dorrestein PC, Kelleher NL. Dissecting Non-ribosomal and Polyketide Biosynthetic Machineries Using Electrospray Ionization Fourier-Transform MassSpectrometry. Nat. Prod. Rep. 2006 doi: 10.1039/b511400b. DOI: 10.1039. [DOI] [PubMed] [Google Scholar]
- 5.Hong H, Appleyard AN, Siskos AP, Garcia-Bernardo J, Staunton J, Leadlay PF. Chain initiation on type I modular polyketide synthases revealed by limited proteolysis and ion-trap mass spectrometry. FEBS J. 2005;272:2373. doi: 10.1111/j.1742-4658.2005.04615.x. [DOI] [PubMed] [Google Scholar]
- 6.Schnarr NA, Chen AY, Cane DE, Khosla C. Analysis of covalently bound polyketide intermediates on 6-Deoxyerythronolide B synthase by tandem proteolysis-mass spectrometry. Biochemistry. 2005;44:11836. doi: 10.1021/bi0510781. [DOI] [PubMed] [Google Scholar]
- 7.Sze SK, Ge Y, Oh H, McLafferty F. Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc. Natl. Acad. Sci. USA. 2002;99:1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Hubbard BK, O'Connor SE, Walsh CT. Formation of β-hydroxy histidine in the biosynthesis of nikkomycin antibiotics. Chem. Biol. 2002;9(1):103–112. doi: 10.1016/s1074-5521(02)00090-x. [DOI] [PubMed] [Google Scholar]
- 9.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. USA. 2005;102(39):13843–8. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowak-Thompson B, Chaney N, Wing JS, Gould SJ, Loper Joyce E. Characterization of the pyoluteorin biosynthetic gene cluster of pseudomonas fluorescens Pf-5. J. Bacteriol. 1999;181(7):2166–2174. doi: 10.1128/jb.181.7.2166-2174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson NR, et al. Biosynthesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel 2-methyl-3-n-amyl-pyrrole (MAP) assembly pathway, definition of the terminal condensing enzyme, and implications for undecylprodigiosin biosynthesis in Streptomyces. Mol. Microbiol. 2005;56:971–989. doi: 10.1111/j.1365-2958.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- 12.Garneau-Tsodikova S, Dorrestein PC, Kelleher NL, Walsh CT. Protein assembly line components in prodigiosin biosynthesis: characterization of PigA,G,H,I,J. submitted. doi: 10.1021/ja063611l. [DOI] [PubMed] [Google Scholar]
- 13.Hicks LM, Mazur MT, Miller LM, Dorrestein PC, Schnarr NA, Khosla C, Kelleher NL. Investigating Nonribosomal Peptide and Polyketide Biosynthesis by Direct Detection of Intermediates on >70 kDa Polypeptides by Using Fourier-Transform Mass Spectrometry. ChemBioChem. 2006;7:904–907. doi: 10.1002/cbic.200500416. [DOI] [PubMed] [Google Scholar]
- 14.Miller LM, et al. Parallel interrogation of covalent intermediates in the biosynthesis of gramicidin S using high-resolution mass spectrometry. Protein Science. 2005;14(10):2702–2712. doi: 10.1110/ps.051553705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duitman EH, et al. The mycosubtilin synthetase of Bacillus subtilis ATCC6633: a multifunctional hybrid between a peptide synthetase, an amino transferase, and a fatty acid synthase. Proc. Natl. Acad. Sci. USA. 1999;96(23):13294–9. doi: 10.1073/pnas.96.23.13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aron ZD, Dorrestein PC, Blackhall JR, Kelleher NL, Walsh CT. Characterization of a new tailoring domain in polyketide biogenesis: the amine transferase domain of MycA in the Mycosubtilin gene cluster. J. Am. Chem. Soc. 2005;127(43):14986–14987. doi: 10.1021/ja055247g. [DOI] [PubMed] [Google Scholar]
- 17.Lai JR, Fischbach MA, Liu DR, Walsh CT. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc. Natl. Acad. Sci. USA. 2006;103(14):5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gehring AM, Bradley KA, Walsh CT. Enterobactin Biosyntheis in Escherichia coli: Isochorismate Lyase (EntB) Is a Bifunctional Enzyme That is Phosphopantetehinylated by EntD and Then Acylated by EntE Using ATP and 2,3-Dihydroxybenzoate. Biochemistry. 1997;36:8495–8503. doi: 10.1021/bi970453p. [DOI] [PubMed] [Google Scholar]
- 19.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Convergence of isoprene and polyketide biosynthetic machinery: isoprenyl-S-carrier proteins in the pksX pathway of B. subtilis. Proc. Natl. Acad. Sci. USA. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofemeister J, et al. Genetic analysis of the biosynthesis of non-ribosomal peptide- and polyketide-like antibiotics, iron uptake and biofilm formation by Bacillus subtilis A1/3. Mol. Genet. Genomics. 2004;272(4):363–378. doi: 10.1007/s00438-004-1056-y. [DOI] [PubMed] [Google Scholar]
- 21.Stein T. Bacillus subtilis antibiotics: Structures, syntheses and specific functions. Mol. Microbiol. 2005;56(4):845–857. doi: 10.1111/j.1365-2958.2005.04587.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen X-H, et al. Structural and functional characterization of polyketide synthase gene clusters in Bacillus amyloliquefaciens FZB 42. J. Bacteriol. 2006;188:4024–4036. doi: 10.1128/JB.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLoughlin SM, et al. Chemoenzymatic approaches for streamlined detection of active site modifications on thiotemplate assembly lines using mass spectrometry. Biochemistry. 2005;44(43):14159–14169. doi: 10.1021/bi051202g. [DOI] [PubMed] [Google Scholar]
- 24.Gu L, Jia J, Liu H, Hkansson K, Gerwick WH, Sherman DH. Metabolic Coupling of Dehydration and Decarboxylation in the Curacin A Pathway: Functional Identification of a Mechanistically Diverse Enzyme Pair. J. Am. Chem. Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- 25.Dorrestein PC, et al. Activity screening of carrier domains within nonribosomal peptide synthetases using complex substrate mixtures and large molecule mass spectrometry. Biochemistry. 2006;45(6):1537–1546. doi: 10.1021/bi052333k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Little DP, Speir JP, Senko MW, O'Connor PB, McLafferty FW. Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing. Anal. Chem. 1994;66(18):2809–15. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 27.Senko MW, Speir JP, McLafferty FW. Collisional activation of large multiply charged ions using fourier transform mass spectrometry. Anal. Chem. 1994;66(18):2801–8. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 28.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicks LM, Moffitt MC, Beer LL, Moore BS, Kelleher NL. Structural characterization of in vitro and in vivo intermediates on the loading module of microcystin synthetase. ACS Chemical Biology. 2006;1(2):93–102. doi: 10.1021/cb500007v. Phenylacetyl-loaded PksJ adenylation-thiolation di-domain 1 that judged by BLAST analysis is similar to the di-domain McyG on the microcystin biosynthetic pathway which was shown, contrary to what was expected, that it did not load phenylacetate but a phenylproprionate derived substrate. PksJ (A1) could not be loaded with phenylproprionates. [DOI] [PubMed] [Google Scholar]
- 30.Stein T, et al. The multiple carrier model of nonribosomal peptide biosynthesis at modular multienzymatic templates. Journal of Biological Chemistry. 1996;271(26):15428–35. doi: 10.1074/jbc.271.26.15428. [DOI] [PubMed] [Google Scholar]
- 31.Stein T, et al. Detection of 4'-phosphopantetheine at the thioester binding site for L-valine of gramicidin S synthase 2. FEBS Letters. 1994;340:39–44. doi: 10.1016/0014-5793(94)80169-x. [DOI] [PubMed] [Google Scholar]
- 32.Lee DL, Muck W, Henion JD, Covey TR. On-line capillary zone electrophoresis-ion spray tandem mass spectrometry for the detection of dynorphins. Journal of Chromatography. 1988;485:313–321. doi: 10.1016/s0021-9673(00)90575-2. [DOI] [PubMed] [Google Scholar]
- 33.Carr SA, Huddleston MJ, Bean MF. Selective identification and differentiation of N- and O-linked oligosaccharides in glycoproteins by liquid chromatography-mass spectrometry. Protein science. 1993;2:183–196. doi: 10.1002/pro.5560020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopfgartner G, et al. Triple quadrupole linear ion trap mass spectrometer for the analysis of small molecules and macromolecules. J. Mass Spectrom. 2004;39:845–855. doi: 10.1002/jms.659. [DOI] [PubMed] [Google Scholar]
- 35.Patrie SM, et al. Construction of a hybrid quadrupole/fourier transform ion cyclotron resonance mass spectrometer for versatile MS/MS above 10 kDa. J. Am. Soc. Mass Spectrom. 2004;15:1099–1108. doi: 10.1016/j.jasms.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 36.Patrie SM, Ferguson JT, Robinson DN, Whipple D, Rother M, Metcalf WW, Kelleher NL. Top Down Mass Spectrometry of <60-kDa Proteins from Methanosarcina acetivorans Using Quadrupole FTMS with Automated Octopole Collisionally Activated Dissociation. Molecular & Cellular Proteomics. 5:14–25. doi: 10.1074/mcp.M500219-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 2000;35(12):1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]