

Abstract
To determine if West Nile virus (WNV) infection of insect cells induces a protective RNAi response, Drosophila melanogaster S2 and Aedes albopictus C6/36 cells were infected with WNV, and the production of WNV-homologous small RNAs was assayed as an indicator of RNAi induction. A distinct population of ~25 nt WNV-homologous small RNAs was detected in infected S2 cells but not C6/36 cells. RNAi knockdown of Argonaute 2 in S2 cells resulted in slightly increased susceptibility to WNV infection, suggesting that some WNV-homologous small RNAs produced in infected S2 cells are functional small interfering RNAs. WNV was shown to infect adult D. melanogaster, and adult flies containing mutations in each of four different RNAi genes (Argonaute 2, spindle-E, piwi, and Dicer-2) were significantly more susceptible to WNV infection than wildtype flies. These results combined with the analysis of WNV infection of S2 and C6/36 cells support the conclusion that WNV infection of D. melanogaster, but perhaps not Ae. albopictus, induces a protective RNAi response.
Keywords: RNAi, innate immunity, West Nile virus, Drosophila
Introduction
Introduction of long double-stranded RNA (dsRNA) into the cytoplasm of cells either experimentally or by viral infection results in the suppression of gene expression via RNA interference (RNAi)(Fire et al., 1998). The long dsRNAs are degraded by an RNAse III-type endonuclease called Dicer into 21–28 nt small interfering RNAs (siRNA) (Bernstein et al., 2001; Ketting et al., 2001; Knight and Bass, 2001). Single strands of the siRNAs are then incorporated into a RNA-induced silencing complex (RISC), where they direct RISC binding to target mRNAs containing complementary sequences (Hammond et al., 2000). The nuclease activity of the argonaute-2 protein of RISC then degrades the target mRNA, silencing its expression (Liu et al., 2004; Meister et al., 2004). RNAi also includes at least two other pathways by which dsRNAs trigger the silencing of endogenous gene expression by translational suppression via microRNAs and transcriptional suppression via changes in chromatin structure (Meister and Tuschl, 2004; Mello and Conte Jr, 2004).
The siRNA-mediated RNAi pathway has been shown to provide innate immunity against viral infection in the nematode Caenorhabditis elegans (Lu et al., 2005; Schott et al., 2005; Wilkins et al., 2005), the fruit fly Drosophila melanogaster (Galiana-Arnoux et al., 2006; van Rij et al., 2006; Wang et al., 2006; Zambon et al., 2006), and the mosquito Anopheles gambiae (Keene et al., 2004; Li et al., 2004). Mutations to components of the RNAi pathway in each species have been shown to increase the susceptibility of that species to infection by one or more RNA viruses. Since the experimental introduction of long dsRNA induces RNAi across a broad spectrum of invertebrates, it is likely that RNAi has at least the capacity to provide natural antiviral immunity in many, if not all, invertebrate animals (Li and Ding, 2005; Marques and Carthew, 2007). It is unclear if RNAi provides innate immunity against viral infection in mammals. The primary response of mammalian cells to viral infection or experimentally introduced long dsRNAs is activation of the interferon response (Gantier and Williams, 2007; Takeuchi and Akira, 2007). While strong induction of the interferon response might preclude induction of the RNAi pathway, even mammalian cells lacking key components of the interferon response, fail to induce an RNAi response when exposed to long dsRNAs, suggesting that differentiated mammalian cells lack the intrinsic ability to activate the RNAi pathway in response to long dsRNAs (Sledz et al., 2003). In contrast, some mammalian viruses express proteins that can suppress the RNAi pathway, at least as assayed in invertebrate cells, which has been interpreted as evidence that mammalian viruses might normally be subject to RNAi-mediated suppression (Li et al., 2004).
Arboviruses are a large group of RNA viruses that are transmitted between hosts by arthropod vectors, primarily mosquitoes. Many are significant human pathogens, such as Dengue virus (DENV; Flaviviridae), Chikungunya virus (CHIKV; Togaviridae), and West Nile virus (WNV; Flaviviridae). The sporadic emergence and spread of arboviral diseases is a persistent public health risk in both tropical and temperate regions of the world (Gubler, 2002; Mackenzie et al., 2004; Solomon, 2004). For example, a new strain of CHIKV that emerged in Africa in 2004 has created a large-scale epidemic across South Asia, thus far causing more than a million cases of disease and hundreds of deaths (Charrel et al., 2007; Murdur, 2007), and WNV first appeared in the Western Hemisphere in New York City in 1999, and spread rapidly across North American, thus far causing more than 10,000 cases of neuroinvasive disease and 930 deaths in the United States (Beasley, 2005; Hayes and Gubler, 2006; Hayes et al., 2005; Kramer et al., 2007)(www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm).
The early demonstration that RNAi could be used to suppress gene expression in the dipteran insect D. melanogaster (Caplen et al., 2000; Kennerdell and Carthew, 1998; Misquitta and Paterson, 1999) raised the prospect of being able to use RNAi to genetically engineer mosquitoes to be resistant to arbovirus infection, and ultimately, use the resistant mosquitoes as biocontrol agents to suppress disease transmission (James, 2000). Mosquitoes clearly have a functional siRNA-mediated RNAi pathway. RNAi has been used experimentally to suppress gene expression in a variety of mosquito cell line lines (Brown et al., 2003b; Hoa et al., 2003; Konet et al., 2007; Levashina et al., 2001), as well as in adult mosquitoes in which dsRNAs have been delivered via direct injection into the hemocoel and by expression from viral vectors and germline transgenes (Attardo et al., 2003; Bian et al., 2005; Blandin et al., 2002; Brown et al., 2003a; Hoa et al., 2003). RNAi has also been used to make mosquito cells resistant to DENV infection (Adelman et al., 2002; Caplen et al., 2002), and most recently, resistance to DENV infection has been engineered into Aedes aegypti by expressing DENV-homologous hairpin RNAs from germline transgenes (Franz et al., 2006).
While it is clear from the aforementioned studies that exposing the RNAi pathway of mosquitoes to DENV-homologous dsRNAs can confer resistance to subsequent DENV infection, such studies do not address the question of whether a DENV infection itself induces a protective RNAi response that modulates infection, akin to what has been demonstrated for viral infection in D. melanogaster and C. elegans. What is the role, if any, for example, of the RNAi pathway in determining the normal susceptibility of mosquitoes to infection by different arboviruses? There is evidence that RNAi-mediated innate immunity modulates susceptibility of An. gambiae to infection by the alphavirus O’nyong-nyong (ONNV; Togaviridae)(Keene et al., 2004). Whether RNAi provides a general antiviral response for other arboviruses and other mosquito species, however, and particularly for infection of culicine mosquitoes by flaviviruses such as DENV or WNV, is not know.
To determine if WNV infection of insect cells induces an RNAi response, we infected D. melanogaster S2 and Ae. albopictus C6/36 cells with WNV and looked for WNV-homologous small RNAs as an indicator of RNAi induction. Small RNAs were detected in the S2 but not the C6/36 cells. Drosophila genetics were then used to rigorously test if WNV infection induces an RNAi response in adult D. melanogaster Adult flies containing mutations in each of four different genes of the RNAi pathway were shown to be significantly more susceptible to WNV infection than wildtype flies, supporting the conclusion that WNV infection of D. melanogaster induces a protective RNAi response.
Results
WNV siRNAs in infected S2 cells
WNV-homologous siRNAs are predicted to be produced in infected cells if WNV infection induces an RNAi response. We looked for the production of such siRNAs in 3 different cell lines: African green monkey kidney cells (Vero), Ae. albopictus mosquito cells (C6/36), and D. melanogaster fruit fly cells (S2). WNV infection would not be expected to produce siRNAs in Vero cells, since long dsRNAs do not induce the RNAi pathway in mammals (Caplen et al., 2000). This is true even in cells, like Vero cells, in which dsRNA fails to induce an interferon response, the primary pathway activated by long dsRNA in mammals (Emeny and Morgan, 1979; Sledz et al., 2003). In contrast, induction of the siRNA-mediated RNAi pathway is the primary response to long dsRNA in plants and invertebrates and has been shown to provide innate immunity against infection by RNA viruses in these organisms (Ding et al., 2004; Li and Ding, 2005). D. melanogaster S2 cells have been one of the primary resources used to both characterize and use the siRNA-mediated RNAi pathway (Boutros et al., 2004; Caplen et al., 2000), and mosquito C6/36 cells have been shown to have a functional siRNA-mediated RNAi pathway inducible by long dsRNA (Adelman et al., 2002; Caplen et al., 2002). C6/36 cells are highly susceptible to WNV infection and are routinely used for studies of WNV and other arboviruses, while D. melanogaster S2 cells have been shown previously to be susceptible to WNV infection (Hannoun and Echalier, 1971). S2 and C6/36 cells, therefore, provided the opportunity to ask whether WNV infection produces WNV-homologous small RNAs in insect cells known to have RNAi pathways inducible by long dsRNAs.
The three cell types were infected with WNV, and the production of WNV-homologous small RNAs was monitored by Northern-blot hybridization. The S2, C6/36, and Vero cells were infected at MOI’s of 5, 1, and 0.1, respectively, MOI’s inversely related to each cell type’s sensitivity to infection. Vero and C6/36 cells produced virus titers of ~108 PFU/ml 1 and 3 days after inoculation, respectively, while infected S2 cells required 6 days to produced titers of ~106 PFU/ml (Fig. 1A). Total RNA was extracted from cells at various times after infection, enriched for small RNAs, and analyzed by Northern-blot hybridization. A discrete population of WNV-homologous small RNAs ~25 nt in length were detected in infected S2 cells but not in infected C6/36 or Vero cells (Fig. 1B). Although larger than the 21- to 23-nt siRNAs characteristically produced in in vitro RNAi reactions, the WNV-homologous small RNAs, are comparable in size to repeat-associated siRNAs produced from double-stranded RNAs originating from endogenous or transgenic repeated genes in the D. melanogaster genome (Aravin et al., 2003; Aravin et al., 2001; Bernstein et al., 2001; Elbashir et al., 2001; Pal-Bhadra et al., 2002; Sarot et al., 2004; Zamore et al., 2000). The results illustrated were obtained using a single-stranded riboprobe that hybridizes to negative-sense WNV sequences. Only negative-sense siRNAs are capable of directing RISC binding to positive-sense WNV genomes, stimulating their degradation, and thereby inhibiting infection. Also, WNV negative-sense RNAs must originate from double-stranded viral replication intermediates where the negative-sense strand is synthesized. Riboprobes that hybridize to positive-sense WNV sequences also identified small RNAs in samples of S2 RNA, as would be predicted, but such RNAs would not be involved in RISC-associated suppression of infection, and from a technical stand point, are more difficult to definitively exclude as arising from degradation of abundant positive-strand viral genomes present in the cell (data not shown). The absence of small RNAs in the C6/36 cells is unlikely to be due to technical problems, as the positive results observed for S2 cells provided a positive control for the experiment. The data illustrated in Fig. 1 are from a single experiment in which RNA samples from all three cell types were processed and analyzed in parallel, and the two Northern-blot panels illustrated are from different sections of the same blot. The ~4-kb size of the riboprobe, combined with the low hybridization stringencies used to ensure stable hybridization between the probe and small RNAs, resulted in some nonspecific hybridization to bulk RNA present on the blots, including the 30-nt 2S ribosomal RNA in S2 cells (Fig. 1B). Finally, control riboprobes of comparable size and specific activity, but with homology to D. melanogaster genomic sequences unrelated to WNV, did not hybridize to any RNAs unique to infected S2 cells (data not shown).
Fig. 1.

Detection of WNV-homologous small RNAs in infected S2 cells. (A) D. melanogaster S2 cells, Ae. albopictus C6/36 cells and African green monkey kidney cells (Vero) were infected with WNV at the indicated MOI, and the cells incubated at 28°C (S2 and C6/36) or 37°C (Vero). At various times after infection, the concentration of infectious virus particles in the culture medium was determined by plaque assay. (B) On the days indicated (arrows in panel A and labels in panel B), cells were harvested, and small RNA’s were isolated and analyzed by Northern-blot hybridization. Uninfected cells were collected as a control (lane c). Day 2 for Vero cells is before widespread cytopathy is detected. 25-nt small RNAs homologous to negative-sense WNV sequences are indicated with an arrow. The positions of 30-nt 2S ribosomal RNA and 21-nt bantam microRNA are indicated. (C) The ethidium bromide-staining pattern in the region of the gel containing the 5s rRNA and tRNAs is shown as a loading control.
While the production of a discrete population of WNV-homologous small RNAs in infected S2 cells is consistent with WNV infection of these cells inducing an RNAi response, we needed to determine whether the observed WNV-homologous small RNAs were actually functional siRNAs capable of inhibiting infection. This was determined by measuring virus production in infected S2 cells in which the RNAi pathway had been suppressed by knockdown of Argonaute 2 (AGO2) expression. AGO2 encodes the primary argonaute protein in D. melanogaster responsible for siRNA-mediated RNAi (Hammond et al., 2001; Okamura et al., 2004), and both knockdown of AGO2 in S2 cells and mutation of AGO2 in flies has been shown to disrupt RNAi-mediated innate immunity against infection by RNA viruses (Li et al., 2002; Li et al., 2004; Rij van et al., 2006; Wang et al., 2006; Zambon et al., 2006). S2 cells were transfected with synthetic siRNAs against AGO2, or against bacterial lacZ sequences as a negative control, and the cells infected with WNV. At various times after infection, the titer of virus released into the culture medium was quantitated. The specific AGO2 siRNAs used were shown previously to suppress the RNAi pathway in S2 cells (Li et al., 2004). Virus was detected earlier in AGO2-treated cells compared to the lacZ controls. In 3 of 4 independent replicates of AGO2-treated cells, virus was first detected 24 hr post infection (p.i.), while in control cells, virus was not detected in 4 of 4 replicates at 24 hr p.i., but was first detected at 36 hr p.i. (Table 1; P ≤ 0.089 for 24 hr data, t-test). No differences in viral titer were observed between the AGO2 and lacZ-treated cells at 36 and 48 hr p.i. (Table 1; P ≤ 0.97 for 48 hr data, t-test). While admittedly a modest effect, the earlier production of virus in AGO2-treated cells is nonetheless consistent with at least some of the WNV-homologous small RNAs detected in infected S2 cells being functional, RISC-associated siRNAs capable of inhibiting WNV infection. The small magnitude of the effect could reflect the intrinsic weakness of the RNAi pathway’s ability to inhibit WNV infection, or alternatively, may be a consequence of technical limitations in the experimental system. It is likely, for example, that the number of individual cells in the culture that are both transfected by siRNAs and infection with virus might be quite low if, as we suspect, the efficiencies of both siRNA transfection and virus infection are low. In addition, using the RNAi pathway to knockdown a component of the RNAi pathway is inherently self-limiting, thus limiting the level of AGO2 knockdown that can be achieved by this approach.
Table 1.
Affect of AGO2 knockdown on WNV infection of S2 cellsa
| AGO2 | LacZ | |
|---|---|---|
| hours post infection | PFU/ml, each replicate | PFU/ml, each replicate |
| 18 | 0,0 | 0,0 |
| 24 | 60, 60, 10, 0 | 0, 0, 0, 0 |
| 36 | 556, 378 | 860, 650 |
| 48 | 2200, 2200, 956, 733 | 3300,1200,900,800 |
S2 cells were transfected with siRNAs directed against AGO2 or Lac Z and then infected with 5 MOI of WNV. At the indicated times after infection, the concentration of infectious virus particles in the culture medium was determined by plaque assay.
WNV Infects D. melanogaster
We reasoned that classical genetic approaches available with D. melanogaster might provide the necessary tools to rigorously demonstrate that WNV infection of D. melanogaster induces a protective RNAi response. AGO2 mutations are available in D. melanogaster, providing the opportunity to directly compare infection susceptibility in wildtype and AGO2 mutant flies, assuming, of course, that WNV can infect adult flies. Given the broad host range of WNV and the fact that WNV can infect S2 cells, it seemed reasonable to expect that WNV would be able to infect adult flies (Hannoun and Echalier, 1971; Lawrie et al., 2004; Mumcuoglu et al., 2005).
Initial attempts to infect D. melanogaster by feeding them WNV were unsuccessful, but infection could be successfully initiated by injecting virus into the hemocoel. WNV infection of D. melanogaster was characterized in relation to WNV infection (also by hemocoel injection) of Culex pipiens, the primary vector of WNV in the Northeastern U.S. (Bernard et al., 2000). First, the dose of virus at which 50% of injected animals become infected (ID50) was determined (Fig. 2). Increasing titers of WNV were injected into adult females of each species, the animals incubated at 27°C, and the titer of virus produced in individual animals was determined by plaque assay at day 7 p.i. The ID50 for D. melanogaster was ~21 PFU compared to ~1.6 PFU for Cx. pipiens (Fig. 2; calculated using program ID50 5.0). A higher ID50 in D. melanogaster is not surprising given that D. melanogaster is separated from mosquitoes by ~ 250 million years of evolution, likely reducing the efficiency of host:virus interactions the virus requires for replication (Zdobnov et al., 2002).
Fig. 2.

The ID50 for WNV injected into D. melanogaster and Cx. pipiens. The indicated number of PFU’s of WNV was injected into adult female D. melanogaster (A) and Cx. pipiens (B). After seven days at 27°C, the titer of WNV in each animal was determine by plaque assay and is indicated by a triangle. The lower limit of detection for the assays is indicated by a dashed line. All animals for which no PFU’s were detected are indicated by a single triangle below the dashed line. The total number of animals scored as infected and uninfected at each dose of WNV is indicated above and below the dashed line, respectively.
We next compared the kinetics of WNV replication during the course of infection in the two species (Fig. 3). Animals of each species were inoculated with ~10x their respective ID50 of WNV, and then incubated at 27°C for 14 days. The titer of virus in individual animals was determined by plaque assay at various times p.i. (Fig. 3A). In both species, infectious virus accumulated rapidly during the first 5–6 days (acute phase), followed by slower accumulation from days 6–14 (plateau phase). D. melanogaster had an initial lag in virus production such that the first clear evidence of viral replication was seen at day 2 p.i. compared to day 1 for Cx. pipiens. The low levels of virus detected in D. melanogaster 12 and 24 h p.i. most likely represent residual inoculum as the levels are below the amounts initally injected. The initial lag in virus production combined with a slightly slower rate of increase between days 2–5 resulted in D. melanogaster having ~10-fold lower virus titers at all time points tested.
Fig. 3.

Kinetics of infection in D. melanogaster and Cx. pipiens. Approximately 10-times the ID50 of infectious WNV particles were injected into adult female D. melanogaster (filled circles) and Cx. pipiens (open circles), and the animals incubated at 27°C for 14 days. At the times indicated, individual animals were homogenized, and the titer of WNV was determined either by measuring the number of PFUs in each lysate by plaque assay (A), or by measuring the number of WNV genomes in each lysate by 5′ nuclease real-time RT-PCR (B). The mean and standard deviation from measurements made on 6–10 animals is shown for each time point. The lower limit of detection for the plaque assay is indicated by a dashed line.
The kinetics of infection was also quantitated by measuring the accumulation of WNV genomes by 5′-nuclease real-time RT-PCR (Fig. 3B). As observed by plaque assay, there was an initial lag in the accumulation of genomes in D. melanogaster compared to Cx. pipiens, with the first evidence of viral replication in D. melanogaster being seen at day 2 p.i. compared to as early as 12 hrs for Cx. pipiens. In D. melanogaster, genomes accumulated rapidly from days 2–5 p.i. and then continued to increase slowly through day 14. In Cx. pipiens, WNV genomes accumulated rapidly to day 3 p.i., then remained essentially constant through day 14. The longer period of proliferation and continued increase in genome copy number through day 14 p.i. resulted in D. melanogaster accumulating more WNV genomes per animal than Cx. pipiens after about day 5 p.i. The total amount of RNA extracted from individual animals was roughly equivalent for the two species, so the same relative similarities and differences were observed whether genome copy number was quantitated per animal or standardized to the amount of RNA in each extract (data not shown). The greater number of viral genomes, yet lower number of infectious particles, measured in D. melanogaster compared to Cx. pipiens may suggest that some aspect of particle assembly may be rate limiting during WNV infection of flies. Alternatively, the number of WNV particles produced in flies could actually be higher than is suggested by Vero cell plaque assays if WNV particles produced in flies are intrinsically less efficient at infecting Vero cells than particles produced in mosquitoes. Finally, WNV infection of D. melanogaster caused no obvious detrimental effects on the viability, motility, fertility, fecundity, or CO2 sensitivity of infected flies up to day 30 p.i. (data not shown).
AGO2 mutations increase susceptibility to WNV infection
Having established that WNV can infect adult flies, we then measured the susceptibility of AGO2 mutant flies to WNV infection. Approximately 2 PFU of WNV was injected into wildtype and AGO2 mutant flies, and at day 7 p.i., the titer of virus in each fly was measured by plaque assay (Fig. 4). Flies were infected with a limiting inoculum of virus (2 PFU) in order to maximize the sensitivity of the assay at revealing increased susceptibility in the AGO2 mutants. AGO2414/AGO251B compound-heterozygous flies containing two different AGO2 mutant chromosomes were tested to avoid potentially confounding phenotypes that can arise from homozygosing recessive background mutations. AGO2 mutant flies were significantly more susceptible to infection than wildtype flies (Fig. 4). Only 3 of 74 wildtype flies (4%) had detectable virus compared to 32 of 86 AGO2 mutants (37%), a 9-fold increase in frequency. In addition, the titer of virus in infected flies was at least 17-fold higher in AGO2 mutants compared to wildtype (P ≤ 0.0041, t-test).
Fig. 4.
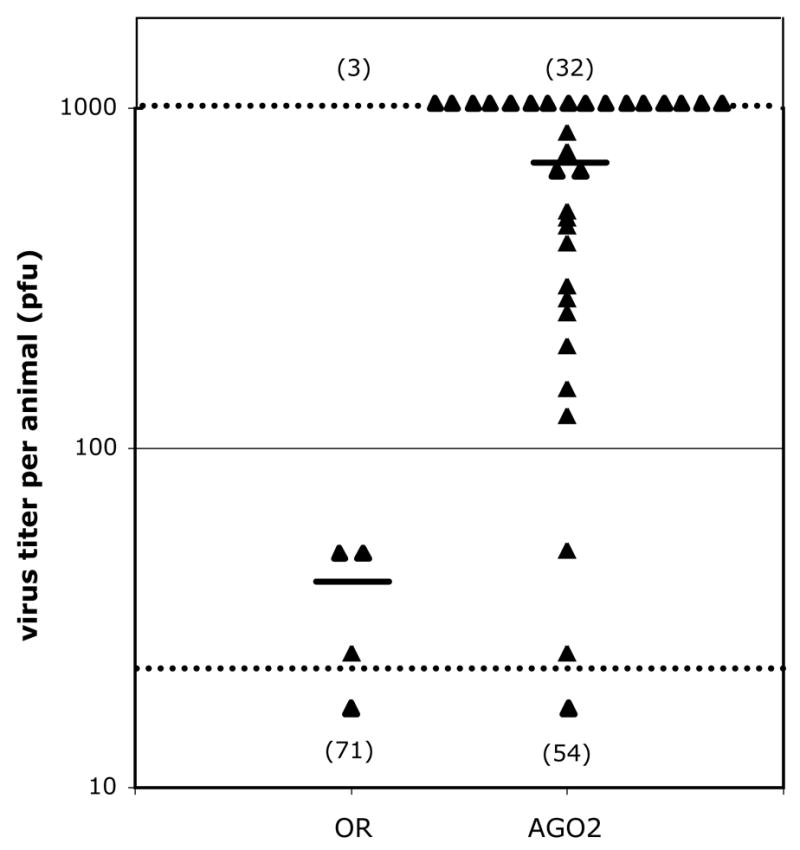
AGO2 mutant flies are more susceptible to WNV infection. 2 PFU of WNV was injected into wildtype Oregon R (OR) and mutant AGO2414/AGO251B (AGO2) flies. After 7 days at 27°C, the titer of WNV in each fly was determined by plaque assay and is indicated by a triangle. The lower and upper limits of detection for the assay are indicated by dashed lines. All flies for which no PFU’s were detected are indicated by a single triangle below the lower limit of detection, with the total number of uninfected flies indicated below this triangle. The total number of flies scored as infected is indicated above the upper dashed line. The average titer for all infected flies for each genotype is indicated by a horizontal line.
We also characterized the kinetics of infection in AGO2 mutant flies, predicting that AGO2 mutants would accumulate virus at a higher rate during infection than wildtype flies. Approximately 10x the ID50 of WNV was injected into wildtype and AGO2 mutant flies, and at various times after inoculation, the titer of WNV genomes was measured by 5′-nuclease, real-time RT-PCR. The number of genomes measured at days 3, 5, and 7 p.i. were all significantly higher in AGO2 mutants compared to wildtype (P ≤ 0.035, P ≤ 0.0012, P ≤ 0.0015, respectively; t-test), ultimately producing ~4-fold higher titer of genome copy number at days 5 and 7 p.i. (Fig. 5). So, even at high inoculums of virus, AGO2 mutants accumulated significantly more WNV genomes during the course of infection than wildtype flies. We conclude from this result, as well as the increased frequency of infection shown in Fig. 4, that mutation to AGO2 increases the susceptibility of flies to WNV infection.
Fig. 5.

AGO2 mutant flies have higher titers of WNV. Approximately 10-times the ID50 of infectious WNV particles were injected into wildtype and AGO2414/AGO251 mutant flies, and the flies cultured at 27°C. At the times indicated, individual flies were homogenized, and the titer of WNV was determined by 5′ nuclease real-time RT-PCR. The mean and standard error from measurements made on 10–11 flies is shown for each time point.
Mutation of other RNAi genes also increases susceptibility
AGO2 encodes functions in D. melanogaster beyond its role in siRNA-mediated RNAi. AGO2 has overlapping functions with AGO1 in translational repression via the microRNA pathway and with the RITS pathway that regulates formation of centromeric heterochromatin (Deshpande et al., 2005; Meyer et al., 2006). In addition, AGO2 plays a role in siRNA-independent turnover of specific transcripts (Xu et al., 2004). These alternate functions raise the possibility that the increased susceptibility to WNV infection seen in AGO2 mutations could be a consequence, directly or indirectly, of disruption to non-siRNA-mediated pathways. To address this possibility, we tested whether mutation to other RNAi genes known to contribute to innate immunity against RNA viruses would, like mutation to AGO2, increase susceptibility to WNV infection. We reasoned that if WNV infection induces a protective RNAi response, then mutation to other RNAi genes involved in this response should also increase susceptibility. Furthermore, if mutations in multiple RNAi genes increase susceptibility, then gene-specific effects on siRNA-independent pathways are less likely to be the origin of the observed increase in susceptibility.
WNV infections were characterized in flies containing mutations in three genes: spindle-E (spn-E; also called homeless [hls]), piwi, and Dicer-2 (Dcr-2). spn-E encodes a DExH-class RNA helicase, which are involved in assembly of both RISC and RITS complexes, and has been shown to influence susceptibility of flies to Drosophila X virus (DXV; Birnaviridae) (Aravin et al., 2001; Gillespie and Berg, 1995; Kennerdell et al., 2002; Pal-Bhadra et al., 2004; Stapleton et al., 2001; Zambon et al., 2006). The primary function of piwi, the founding member of the argonaute gene family, is in maintaining genome integrity in the germline, but it has also been shown to have somatic functions, including influencing the susceptibility of flies to DXV infection (Aravin et al., 2006; Cox et al., 1998; Girard et al., 2006; Klattenhoff and Theurkauf, 2008; Pal-Bhadra et al., 2002; Pal-Bhadra et al., 2004; Saito et al., 2006; Zambon et al., 2006). Dcr-2 encodes the RNAse III enzyme in D. melanogaster primarily responsible for production of siRNAs (Lee et al., 2004). Dcr-2 has been shown to play a role in the RNAi-mediated antiviral response against infection of D. melanogaster by a number of viruses including Flock House virus (FHV; Nodaviridae), Cricket Paralysis virus (CrPV; Dicistroviridae), Drosophila C virus (DCV; Dicistroviridae), and Sindbis virus (SINV; Togaviridiae) (Galiana-Arnoux et al., 2006; Rij van et al., 2006; Wang et al., 2006).
The kinetics of infection in spn-E, piwi, and Dcr-2 mutants was characterized relative to wildtype flies. Compound-heterozygous flies were created for each gene to avoid potentially confounding phenotypes arising from homozygosing recessive background mutations, as was done for AGO2. Approximately 10x the ID50 of WNV was injected into mutant and wildtype flies, and at various times after inoculation, the titer of WNV genomes was measured by 5′-nuclease, real-time RT-PCR (Fig. 6). Mutations in spn-E and piwi both produced increased rates of WNV replication from days ~2–5 p.i., resulting in significantly higher titers of WNV genomes (Fig. 6A). Higher titers were seen at days 5 and 7 in spn-E mutants (P ≤ 0.00012 and P 2.7×10−14, respectively; t-test) and at days 3, 5, and 7 for piwi mutants (P ≤ 0.00013, P ≤ 6.8×10−6, and P ≤ 7.1×10−14, respectively; t-test). The increased rates of WNV replication ultimately produced ~10-fold higher titers of WNV genomes at day 7 p.i. in both mutants (Fig. 6A). Mutation of Dcr-2 did not produce increased rates of WNV replication from days 1–5 p.i. and actually had lower titers of virus at those time points (Fig. 6B). The rate of WNV replication, however, did continue to increase from days 5–7 p.i. when replication in wildtype flies was essential flat, producing ~2-fold higher titers of WNV genomes at day 7 p.i. (P ≤ 0.042, t-test; Fig. 6B). The modest effect of the Dcr-2 mutations was not entirely unexpected. It is known that Dcr-1, which is essential for synthesis of microRNAs, can also synthesize siRNAs in Dcr-2 mutants (Lee et al., 2004). In addition, the effect of Dcr-2 mutations on infection susceptibility varies significantly for different viruses. For example, mutation of Dcr-2 increases virus titers more than 100-fold in DCV infected flies, while having little or no effect in flies infected by DXV (Rij van et al., 2006; Zambon et al., 2006).
Fig. 6.
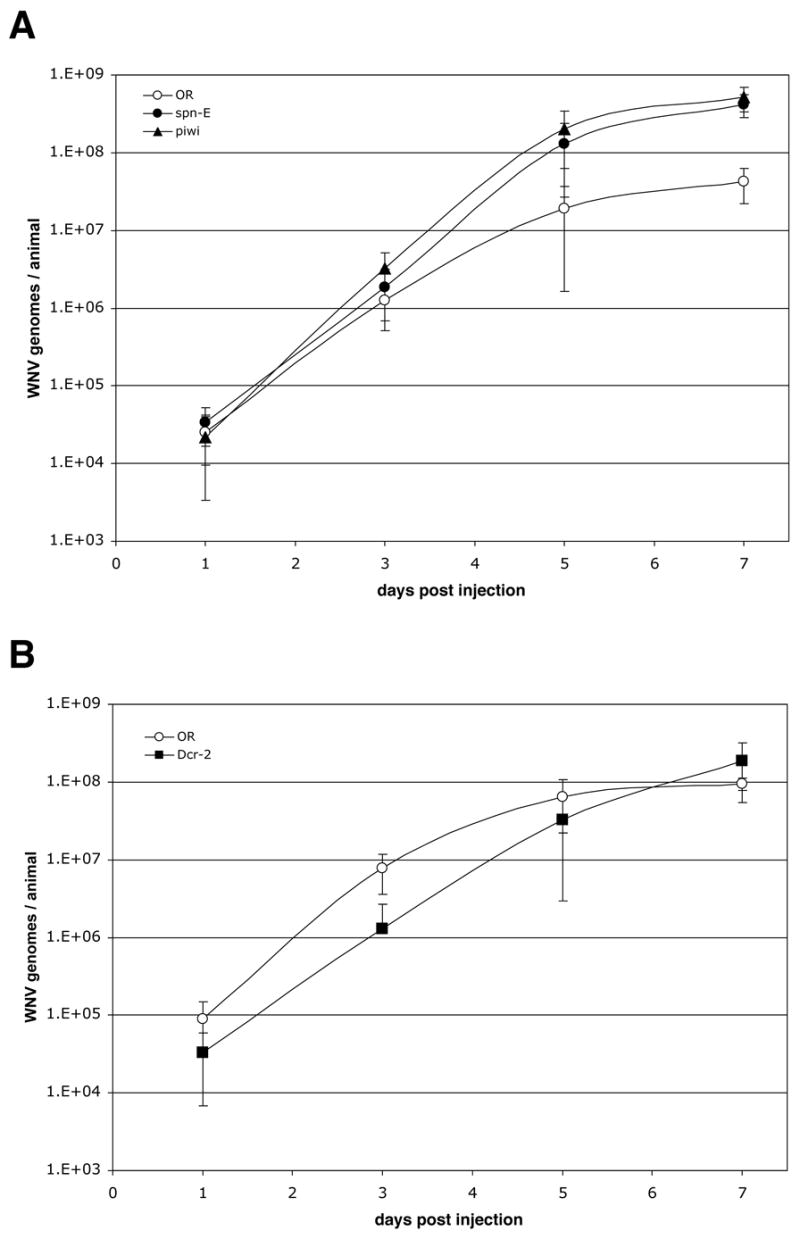
Mutation to other RNAi genes also increases titers of WNV. Approximately 10-times of infectious WNV particles was injected into wildtype, spn-EhlsΔ125/spn-EhlsE616, the ID50 piwi1/piwi2, and Dcr-2R416X/Dcr-2L811fsX mutant flies, and the flies cultured at 27°C. spn-E and piwi mutants were analyzed in one experiment (A), and Dcr-2 in another (B). At the times indicated, individual flies were homogenized, and the titer of WNV was determined by 5′ nuclease real-time RT-PCR. The mean and standard deviation from measurements made on 9–20 individual flies is shown for each time point.
The increased titers of WNV seen in the piwi and spn-E mutant flies, and to a lesser extent the Dcr-2 mutants, corroborates the increase in susceptibility seen in AGO2 mutants, consistent with mutations in all four RNAi genes increasing the susceptibility of flies to WNV infection by disrupting the siRNA-mediated RNAi pathway. These genetic results (Figs. 4–6), combined with the evidence that WNV infection of D. melanogaster S2 cells produces functional WNV-homologous siRNAs (Fig. 1 and Table 1), support the conclusion that WNV infection of D. melanogaster induces an RNAi response that inhibits infection.
Discussion
We infected D. melanogaster S2 and Ae. albopictus C6/36 cells with WNV and looked for the production of WNV-homologous small RNAs as an indicator of RNAi induction, to determine if WNV infection of insect cells induces an siRNA-mediated RNAi response. Small RNAs were detected in S2 but not in C6/36 cells, suggesting that an RNAi response is induced in cells of D. melanogaster but not Ae. albopictus (Fig. 1). RNAi knockdown of AGO2 expression resulted in S2 cells becoming more susceptible to WNV infection, suggesting that at least some of the small RNAs that are produced in S2 cells are functional siRNAs (Table 1). In order to more rigorously test whether WNV infection of D. melanogaster does, in fact, induce a protective RNAi response, adult flies containing mutations in each of four different genes of the RNAi pathway: AGO2, spn-E, piwi, and Dcr-2, were shown to be significantly more susceptible to WNV infection than wildtype flies (Figs. 4–6). We conclude from these results that WNV infection of D. melanogaster induces an siRNA-mediated RNAi response that inhibits infection.
Significant increases in infection susceptibility were seen in each of four different RNAi-mutant flies suggests that the RNAi pathway, in toto, could be a principle reason D. melanogaster is less susceptible to WNV infection than Cx. pipiens. Simultaneously mutating all components of the RNAi pathway to test this idea, however, is impossible, since some RNAi genes provide essential functions, precluding the creation of viable flies containing mutations in such genes (Lee et al., 2004).
WNV-homologous siRNAs were not detected in infected C6/36 cells, suggesting that WNV infection of Ae. albopictus cells does not induce an RNAi response or induces such a minimal response that detectable levels of siRNAs are not produced (Fig. 1). siRNAs were detected in S2 cells even though they produced titers of WNV that were 3 logs lower than titers produced by C6/36 cells (Fig. 1), further suggesting that any RNAi response induced in C6/36 cells is extremely low relative to levels of viral replication. Therefore, it is unlikely that the siRNA-mediated RNAi pathway inhibits the susceptibility of C6/36 cells to infection by WNV. Why doesn’t WNV infection of C6/36 cells induce an RNAi response, given that WNV replication produces long dsRNA replication intermediates in cells, and experimental introduction of dsRNAs into C6/36 cells clearly induces an RNAi response (Adelman et al., 2002; Caplen et al., 2002; Chu and Westaway, 1985)? Both cytological and biochemical evidence indicates that WNV replication occurs in the context of membrane associated complexes (Mackenzie et al., 1999; Uchil and Satchidanandam, 2003; Westaway et al., 1997). The structure of such complexes may hide dsRNA replication intermediates from Dicer nucleases, thus allowing the virus to evade detection by the RNAi pathway. Alternatively, WNV could express an inhibitor of the RNAi pathway, as has been demonstrated for other viruses, although experiments designed to test this possibility failed to find any evidence for general suppression of the RNAi pathway in WNV-infected cells, again suggesting that evasion, rather than suppression, is the primary mechanism by which WNV, and likely other flaviviruses, avoids RNAi-mediated innate immunity in insects (Geiss et al., 2005; Li and Ding, 2005).
The absence of an RNAi response in WNV-infected C6/36 cells, if extrapolated to adult mosquitoes, suggests that RNAi may not play a significant role in determining the susceptibility of Ae. albopictus to WNV infection. In fact, to date, there is no evidence that RNAi normally modulates susceptibility of any mosquito species to flavivirus infection. Clearly, the RNAi pathway can be experimentally activated to confer resistance to flavivirus infection. Resistance to DENV infection can be engineered into transgenic Ae. aegypti by artificially activating the RNAi pathway (Franz et al., 2006). Such experiments, however, only demonstrate that flavivirus infections can be inhibited by the RNAi pathway when RISC complexes have been “preloaded” with virus-homologous siRNAs prior to infection, not that flaviviral infections themselves induce an RNAi response that modulates susceptibility. In contrast, there is evidence that the RNAi pathway may normally modulate susceptibility of An. gambiae to infection by O’nyong-nyong virus (ONNV; Togaviridae; Alphavirus). RNAi-mediated knockdown of AGO2 expression increased the susceptibility of An. gambiae to infection by ONNV (Keene et al., 2004). It is unclear, however, the extent to which this observation can be extrapolated to flavivirus infections of culicine mosquitoes, given differences in the mechanism of replication between alphaviruses and flaviviruses. For example, RNA recombination has been observed in alphaviruses but is rare or absent in flaviviruses, consistent with alphavirus replication intermediates being more accessible than their flavivirus counterparts, at least between replication complexes and perhaps within the cytoplasm generally (Strauss and Strauss, 1997; Twiddy and Holmes, 2003). Thus, alphavirus dsRNA replication intermediates may be more readily accessible to surveillance by Dicer nucleases. Elucidating the role, if any, played by siRNA-mediated RNAi in determining the susceptibility of mosquitoes to flavivirus infection will require experiments directed specifically to this question and done in relevant host:flavivirus systems.
Why does WNV infection of D. melanogaster induce a protective RNAi response, while infection of Ae. albopictus C6/36 cells likely does not? WNV has probably evolved to evade the RNAi pathway in natural mosquito hosts by hiding its dsRNA replication intermediates within membrane-associated complexes, as discussed above. In a heterologous host like D. melanogaster, however, that evasion mechanism may be compromised. For example, host:virus interactions required for assembly of viral replication complex are likely to be suboptimal in a heterologous host due to genetic divergence of relevant host genes. dsRNA replication intermediates within such replication complexes may therefore be less well protected from Dicer surveillance, allowing activation of an RNAi response. If the genetic divergence of D. melanogaster and the resulting reduction in the fidelity of viral replication is, in fact, the underlying reason WNV infection of D. melanogaster induces a protective RNAi response, it would be interesting to determine if the same is true in different species of mosquitoes. The probability of WNV infection inducing a protective RNAi response may be a function of how genetically divergent a specific mosquito species is relative to mosquito species to which the virus is optimally adapted. In this way, the RNAi pathway might limit the host range of arboviruses, while having little or no impact on infection susceptibility in host species to which the virus is best adapted. Finally, it is also possible that the RNAi pathway in D. melanogaster is quantitatively or qualitatively different in a way that makes RNAi in flies intrinsically better at inhibiting WNV infection. Experimentally induced RNAi knockdown of endogenous genes in C6/36 cells has been reported to be weaker than the degree of knockdown that can be a achieved in S2 cells, consistent with this possibility (Caplen et al., 2002).
What ultimately inhibits WNV replication at the transition from the acute to the plateau phase of infection, is not known. The transition occurs at a discrete time, day 3 p.i. in Cx. pipiens and day 5 in D. melanogaster, as measured by WNV genomes (Fig. 3B). Understanding how WNV replication is inhibited to create this transition might provide insights into host:virus interactions that determine the overall kinetics of infection. Infection of AGO2, piwi, and spn-E mutants of D. melanogaster clearly produced higher maximum titers of WNV than infection of wildtype flies, suggesting that transition to the plateau phase of infection is not caused by viral replication depleting a limited host resource. If that were the case, infection in both mutant and wildtype flies would be expected to produce the same maximum virus titers, with the transition to the plateau phase occurring earlier in mutants than in wildtype flies. The transition, however, occurred at the same time in mutant and wildtype flies (Figs. 5–6). Analysis of the RNAi mutants also suggests that the RNAi response itself is probably not what ultimately limits infection. If it were, mutations that weaken the ability of the RNAi response to inhibit infection, like mutations in RNAi genes, would be predicted to lengthen the time required for the RNAi response to “catch up with” and eventually inhibit the acute phase of infection, but again, the transition to the plateau phase of infection occurred at the same time in mutant and wildtype flies, arguing against this idea (Figs. 5–6). If WNV infection is not inhibited by depletion of limited host resources or by the RNAi pathway, then perhaps another innate immune response activated by WNV infection plays a role. Looking at WNV infection in flies containing mutations in genes involved in other innate immune pathways would address this possibility.
WNV’s ability to infect a wide phylogenetic range of hosts, including diverse species of mammals, birds, and insects, suggests that host genetic factors that the virus needs in order to replicate are likely to be broadly conserved across species. WNV can infect adult D. melanogaster, producing viremias similar to those in the mosquito Cx. pipiens, and Drosophila genetics can be used to identify specific host genes that are important for WNV infection of flies (Figs. 2–6). Beyond the limited number of RNAi genes analyzed here, unbiased, functional genetic screens that can be done with relative ease in D. melanogaster could be used to systematically identify host genes important for WNV infection of flies. The mosquito orthologs of genes identified in D. melanogaster could then be tested for their importance for WNV infection of mosquitoes, providing a systematic way to identify mosquito genes that are important for host:virus interactions that determine infection susceptibility.
Materials and methods
Cells and virus
D. melanogaster S2 cells were grown semi-adherently in 6-well plates at 28°C in serum free medium supplemented with 2 mM L-glutamine (Invitrogen). Aedes albopictus cells (C6/36, ATCC #CRL-1660) and African green monkey kidney cells (Vero, ATCC #CCL-81) were grown in 6-well plates at 28°C and 37°C, respectively, in minimal essential medium supplemented with 10% FBS, 2 mM L-glutamine, 1.5 g/l sodium bicarbonate, 0.1 mM non-essential amino acids, 100 U/ml penicillin and streptomycin. Cells were infected with WNV by decanting medium from the well and adding 100 μl of virus at the appropriate multiplicity of infection (MOI). Following a 1-hr adsorption period, cells were rinsed once, and 3 ml of fresh medium was added, and the cells incubated at the appropriate temperature. For viral growth analysis, samples of culture supernatant were removed at appropriate times after infection and stored at −80°C. The stock virus used for infections (WNV 3356) was derived from WNV NY003356, a primary isolate from kidney tissue of an American crow collected in 2000 in Staten Island, NY (Ebel et al., 2001). The virus stock was prepared by three rounds of plaque purification in Vero cells.
Plaque Assays
Plaque assays using Vero cells were done essentially as described previously (Payne et al., 2006). Plaque assays were done on either culture supernatants from infected cells or homogenates of infected flies or mosquitoes. Infected animals were homogenized by mixer mill in 500 μl of Dulbecco’s phosphate-buffered saline supplemented with 20% FBS, 50 μg/ml penicillin and streptomycin, 50 μg/ml gentamicin, 2.5 μg/ml Fungizone, and stored at −80°C until assayed.
RNAi treatment of S2 cells
S2 cells were treated with AGO2-homologous or lacZ-homologous siRNAs once a day for 3 consecutive days, after which cells were infected with WNV at an MOI of 5. Samples of culture supernatant were collected at various times after infection and stored at −80°C until WNV titers were measured by plaque assay. On the first day of siRNA treatment, 1×106 cells were plated in the well of a 6-well plate in 860 μl of medium for each sample. 140 μl of siRNA transfection mix containing TransMessenger Transfection Reagent (Qiagen) and 7 μM of double-stranded siRNAs was added to the cells, and the cells incubated overnight. On days 2 and 3, the culture medium was removed, replaced with 860 μl of fresh medium, and the cells again treated with siRNAs as done on day 1. The siRNAs used were double-stranded 21-mers (Qiagen). For AGO2, equal molar amounts of two different siRNAs shown previously to inhibit the RNAi pathway in S2 cells were used (Li et al., 2004). lacZ siRNAs were used for the negative control (Li et al., 2004).
Northern-blot Analysis
Total RNA was isolated from infected cells using TRI Reagent following the manufacturer’s protocols (Molecular Research Center). RNA samples were enriched for small RNAs by size-selective precipitation using polyethylene glycol. Samples containing ~20 μg of size-selected RNA were fractionated on 15% polyacrylamide-8M urea gels and transferred to GeneScreen Plus membrane (New England Biolabs). Blots were probed simultaneously with two different P32-labelled riboprobes homologous to NS1 sense-strand sequences. The primer pairs used to make the riboprobe templates were GGATTGACGCCAGGGTGTACT and GCACTTGACGAGGACTCTCC (1700 nt RNA), and GGCAGTTCTGGGTGAAGTCAA and GGTGAGCCTGATGTTCCA (1900 nt RNA). D. melanogaster bantam microRNA was detected on the Northern blots as a 21-nt size marker in the lanes containing RNA from S2 cells (Brennecke et al., 2003).
Fly and Mosquito Strains and Genetics
D. melanogaster were maintained on cornmeal-brewer’s yeast-glucose medium at 23°C and 55% relative humidify. Wildtype D. melanogaster were Oregon R. AGO2414/AGO251B compound heterozygotes were created by crossing AGO2414 and AGO251B homozygous flies (Okamura et al., 2004; Xu et al., 2004). Dcr-2R416X/Dcr-2L811fsX compound heterozygotes were created by crossing Dcr-2R416X and Dcr-2L811fsX homozygous flies (Lee et al., 2004). piwi1/piwi2 compound heterozygotes were created by crossing piwi1/CyO and piwi2/CyO flies (Lin and Spradling, 1997). spn-EhlsΔ125/spn-EhlsE616 compound heterozygotes were created by crossing spn-EhlsΔ125/TM3 and spn-EhlsE616/TM3 flies (Gillespie and Berg, 1995). Cx. pipiens mosquitoes were collected from the wild in Pennsylvania, and had been colonized in the lab for ~6 months at the time of their use in the experiments described. The mosquitoes were maintained at 27°C and 85% relative humidity with a photoperiod of 16:8 hr (light:dark). Adult females were maintained in 0.5 liter cardboard cups and fed 10% sucrose ad libitum. Flies and mosquitoes were transferred to the BSL3 insectary for virus inoculation 3–7 days or ~7 after emergence, respectively.
Injections
D. melanogaster were anesthetized with triethylamine or ice and injected intra-abdominally with ~100 nl of Dulbecco’s modified eagle medium (DMEM) containing WNV at the appropriate concentration. Pre-pulled 30 μm needles were used (World Precision Instruments), and the injection volume was controlled using a pneumatic injector. Appropriate backpressure was determined empirically for each session of injection. Mutant and wildtype flies for any single experiment were always injected during the same injection session using the same injector settings and reagents, and the injected flies were always incubated together. Cx. pipiens were anesthetized with CO2 and injected intra-thoracically with ~100 nl of DMEM containing WNV at the appropriate concentration, essentially as described previously (Rosen and Gubler, 1974). After injection, both species were incubated at 27°C with a 16:8 light/dark photoperiod.
5′ Nuclease Real-Time RT-PCR
Total RNA was isolated using TRI Reagent following the manufacturer’s protocol (Molecular Research Center). RNA recovery was quantitated using Quant-iT RNA assays (Invitrogen). The amount of WNV RNA in a 10-μl sample of each extract was determined using a 5′ nuclease real-time RT-PCR assay with a primer-probe set homologous to sequences in the WNV envelope gene as described previously (Shi et al., 2001). The absolute number of WNV genomes was calculated from concurrently assayed WNV RNA standards.
Acknowledgments
We thank Haruhiko Siomi, Fen-Biao Gao, Richard Carthew, Haifan Lin, and Celeste Berg for fly strains, the Arbovirus Laboratory insectary team for mosquito rearing, and the Wadsworth Center Tissue Culture facility for Vero cultures. This work was supported by the New York State Department of Health (R.L.G. and L.D.K.) and a grant from the NIH to P.-Y.S. (AI061193).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelman ZN, Sanchez-Vargas I, Travanty EA, Carlson JO, Beaty BJ, Blair CD, Olson KE. RNA silencing of Dengue virus type 2 replication in tranformed C6/36 mosquito cells transcribing an inverted-repeat RNA derived from the virus genome. J Virol. 2002;76(24):12925–12933. doi: 10.1128/JVI.76.24.12925-12933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein JJ, Kuramochi-Miyagawa S, Nakano T, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M, Tuschl T. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol. 2001;11:1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Attardo GM, Higgs S, Klingler KA, Vanlandingham DL, Raikhel AS. RAN interference-mediated knockdown of a GATA factor reveals a link to anautogeny inn the mosquito Aedes aegypti. Proceed Natl Acad Sc, USA. 2003;100(23):13374–13379. doi: 10.1073/pnas.2235649100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley DWC. Recent advances in the molecular biology of West Nile virus. Curr Mol Med. 2005;5:835–850. doi: 10.2174/156652405774962272. [DOI] [PubMed] [Google Scholar]
- Bernard KA, Maffei JG, Jones SA, Kauffman EB, Ebel GD, Dupuis AP, II, Ngo KA, Nicholas DC, Young DM, Shi PY, Kulasekera VL, Eidson M, White DJ, Stone WB, Team NWS, Kramer LD. West Nile infection in birds and mosquitoes, New York State, 2000. Emerg Inf Dis. 2000;7(4):679–685. doi: 10.3201/eid0704.010415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–365. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Bian G, Shin SW, Cheon HM, Kokoza V, Raikhel AS. Transgenic alteration of Toll immune pathway in the female mosquito Aedes aegypti. PNAS. 2005;102(38):13568–13573. doi: 10.1073/pnas.0502815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandin S, Moita LF, Kocher T, Wilm M, Kafatos FC, Levashina EA. Reverse genetics in the mosquito Anopheles gambiae: targeted disruption of the Defensin gene. EMBO Reports. 2002;3(9):852–856. doi: 10.1093/embo-reports/kvf180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Consortium HFA, Paro R, Perrimon N. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russel RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapototic gene hid in Drosophila. Cell. 2003;113(1):25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Brown AE, Bugeon L, Crisanti A, Catteruccia F. Stable and heritable gene silencing in the malaria vector Anopheles stephensi. Nucleic Acid Res. 2003a;31(15):e85. doi: 10.1093/nar/gng085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AE, Crisanti A, Catteruccia F. Comparative analysis of DNA vectors at mediating RNAi in Anopheles mosquito cells and larvae. J Exp Biol. 2003b;206(11):1817–1823. doi: 10.1242/jeb.00360. [DOI] [PubMed] [Google Scholar]
- Caplen NJ, Fleenor J, Fire A, Morgan RA. dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene. 2000;252:95–105. doi: 10.1016/s0378-1119(00)00224-9. [DOI] [PubMed] [Google Scholar]
- Caplen NJ, Zheng Z, Falgout B, Morgan RA. Inhibition of viral gene expression and replication in mosquito cells by dsRNA-triggered RNA interference. Molec Therapy. 2002;6(2):243–251. doi: 10.1006/mthe.2002.0652. [DOI] [PubMed] [Google Scholar]
- Charrel RN, de Lamballerie X, Raoult D. Chikungunya outbreaks - the globalization of vectorborne diseases. N Engl J Med. 2007;356:769–771. doi: 10.1056/NEJMp078013. [DOI] [PubMed] [Google Scholar]
- Chu PWG, Westaway EG. Replication strategy of Kunjin virus: evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology. 1985;140:68–79. doi: 10.1016/0042-6822(85)90446-5. [DOI] [PubMed] [Google Scholar]
- Cox DN, Chao A, Baker J, Chang L, Qiao D, Lin H. A novel class of evolutionarily conserved genes defined by piwi are essential for stem cel self-renewal. Genes Dev. 1998;12:3715–3727. doi: 10.1101/gad.12.23.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, Calhoun G, Schedl P. Drosophila argonaute-2 is required early in embryogenesis for thte assembly of centric/centromeric heterochromatin, nuclear division, nuclear migration, and germ-cell formation. Genes Dev. 2005;19:1680–1685. doi: 10.1101/gad.1316805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SW, Li H, Lu R, Li F, Li WX. RNA silencing: a conserved antiviral immunity of plants and animals. Virus Res. 2004;102:109–115. doi: 10.1016/j.virusres.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Ebel GD, Dupuis AP, II, Ngo KA, Nicholas DC, Kauffman EB, Jones SA, Maffei JG, Shi PY, Bernard KA, Kramer LD. Parial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Inf Dis. 2001;7(4):650–653. doi: 10.3201/eid0704.010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- adn 22-nucleotide RNAs. Gene Devel. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol. 1979;43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Franz AWE, Sanchez-Vargas I, Adelman ZN, Blair CD, Beaty BJ, James AA, Olson KE. Engineering RNA interference-based resistance to dengue virus type 2 in genetically modified Aedes aegypti. PNAS. 2006;103(11):4198–4203. doi: 10.1073/pnas.0600479103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiana-Arnoux D, Dostert C, Schneemann A, Hoffman JA, Imler JL. Essential function in vivo for Dicer-2 in host defense against RNA viruses in drosphila. Nature Immunology. 2006;7(6):590–597. doi: 10.1038/ni1335. [DOI] [PubMed] [Google Scholar]
- Gantier MP, Williams BRG. The response of mammalian cells to double-stranded RNA. Cytok Growth Fact Rev. 2007;18(5–6):363–371. doi: 10.1016/j.cytogfr.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss BJ, Pierson TC, Diamond MS. Actively replicating West Nile virus is resistant to cytoplasmic delivery of siRNA. Virology J. 2005;5:53–66. doi: 10.1186/1743-422X-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie DE, Berg CA. homeless is required for RNA localization in Drosophila oogenesis and encodes a new member of the DE-H family of RNA-dependent ATPases. Genes Dev. 1995;9:2495–2508. doi: 10.1101/gad.9.20.2495. [DOI] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute-2, a link between genetic and biochemical analysis of RNAi. Science. 2001;293:1146–1150. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- Hannoun C, Echalier G. Arbovirus multiplication in an established diploid cell line of Drosophila melanogaster. In: Weiss E, editor. Arthropod cell cultures and their application to the study of viruses. Springer Verlag; Berlin: 1971. pp. 227–230. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Gubler DJ. West Nile Virus: Epidemiology and clinical features of an emerging epidemic in the United States. Ann Rev Med. 2006;57:181–194. doi: 10.1146/annurev.med.57.121304.131418. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Komar N, Nasci RS, Montgomery SP, O’Leary DR, Campbell GL. Epidemiology and transmission dynamics of West Nile virus disease. Emerg Inf Dis. 2005;11(8):1167–1173. doi: 10.3201/eid1108.050289a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa NT, Keene KM, Olson KE, Zheng L. Characterization of RNA interference in an Anopheles gambiae cell line. Insect Biochem Molec Biol. 2003;33(9):949–957. doi: 10.1016/s0965-1748(03)00101-2. [DOI] [PubMed] [Google Scholar]
- James AA. Control of disease transmission through genetic modification of mosquitoes. In: Handler AM, James AA, editors. Insect Transgenesis. CRC Press; 2000. pp. 319–333. [Google Scholar]
- Keene KM, Foy BD, Sanchez-Vargas I, Beaty BJ, Blair CD, Olson KE. RNA interference acts as a natural antiviral response to O’nyong-nyong virus (Alphavirus; Togaviridae) infection in Anopheles gambiae. PNAS. 2004;101:17240–17245. doi: 10.1073/pnas.0406983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerdell JR, Carthew RW. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95:1017–1026. doi: 10.1016/s0092-8674(00)81725-0. [DOI] [PubMed] [Google Scholar]
- Kennerdell JR, Yamaguchi S, Carthew RW. RNAi is activated during Drosophila oocyte maturation in a manner dependent on aubergine and spindle-E. Gene Devel. 2002;16:1884–1889. doi: 10.1101/gad.990802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterik RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- Knight SW, Bass BL. Role for the RNAseIII enzyme DCR-1 in RNA interference and germ line development in C. elegans. Science. 2001;293:2269–2271. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konet DS, Anderson J, Piper J, Akkina r, Suchman E, Carlson J. Short-hairpin RNA expression from polymerase III promoters mediates RNA interference in mosquito cells. Insect Mol Biol. 2007;16(2):199–206. doi: 10.1111/j.1365-2583.2006.00714.x. [DOI] [PubMed] [Google Scholar]
- Kramer LD, Li J, Shi PY. West Nile Virus. Lancet - Neurol. 2007;6:171–181. doi: 10.1016/S1474-4422(07)70030-3. [DOI] [PubMed] [Google Scholar]
- Lawrie CH, Uzcategui NY, Gould EA, Nuttall PA. Exodid and argasid tick species and West Nile Virus. Emerg Inf Dis. 2004;10(4):653–657. doi: 10.3201/eid1004.030517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, Carthew RW. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- Levashina EA, Moita LF, Blandin S, Vriend G, Lagueux M, Kafatos FC. Conserved role of a complement-like protein in phagocytosis revealed by dsRNA knockout in cultured cells of the mosquito Anopheles gambiae. Cell. 2001;104(5):709–718. doi: 10.1016/s0092-8674(01)00267-7. [DOI] [PubMed] [Google Scholar]
- Li H, Li WX, Ding SW. Induction and suppression of RNA silencing by an animal virus. Science. 2002;296:1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- Li HW, Ding SW. Antiviral silencing in animals. FEBS Letters. 2005;579:5965–5973. doi: 10.1016/j.febslet.2005.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX, Li H, Lu R, Li F, Dus M, Atkinson P, Brydon EWA, Johnson KL, Garcia-Sastre A, Ball LA, Palese P, Ding SW. Interferon antagonist proteins of infuenza and vaccinia viruses are suppressors of RNA silencing. PNAS. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Spradling AC. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 1997;124:2463–2476. doi: 10.1242/dev.124.12.2463. [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor J, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Jones MK, Westaway EG. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J Virol. 1999;73(11):9555–9567. doi: 10.1128/jvi.73.11.9555-9567.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nature Medicine. 2004;10(12 suppl):S98–109. doi: 10.1038/nm1144. [DOI] [PubMed] [Google Scholar]
- Marques JT, Carthew RW. A call to arms: coevolution of animal viruses ad host innate immune responses. Trends Genet. 2007;23(7):359–364. doi: 10.1016/j.tig.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Molec Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- Mello CC, Conte D., Jr Revealing the world of RNA interference. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- Meyer WJ, Schreiber S, Guo Y, Volkmann T, Welte MA, Muller HAJ. Overlapping functions of argonaute proteins in patterning and morphogenesis of Drosophila embryos. PLOS genetics. 2006;2(8):1224–1239. doi: 10.1371/journal.pgen.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misquitta L, Paterson BM. Targeted disruption of gene function in Drosophila by RNA interference (RNA-i): a role for nautilus in embryonic somatic muscle formation. Proceed Natl Acad Sc, USA. 1999;96:1451–1456. doi: 10.1073/pnas.96.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumcuoglu KY, Banet-Noach C, Malkinson M, Shalom U, Galun R. Argasid ticks as possible vectors of West Nile Virus in Israel. Vector-Borne Zoo Dis. 2005;5(1):65–71. doi: 10.1089/vbz.2005.5.65. [DOI] [PubMed] [Google Scholar]
- Murdur G. Failure to control mosquitoes has led to two fever epidemics in India. BMJ. 2007;333:773. doi: 10.1136/bmj.333.7572.773-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Ishizuka A, Siomi H, Siomi MC. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004;18:1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhadra M, Bhadra U, Birchler JA. RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Molec Cell. 2002;9:315–327. doi: 10.1016/s1097-2765(02)00440-9. [DOI] [PubMed] [Google Scholar]
- Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SCR. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science. 2004;303:669–672. doi: 10.1126/science.1092653. [DOI] [PubMed] [Google Scholar]
- Payne AF, Binduga-Gajewdka I, Kauffman EB, Kramer LD. Quantitation of flaviviruses by fluorescent focus assay. J Virol Meth. 2006;134:183–189. doi: 10.1016/j.jviromet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Rij van RP, Saleh MC, Berry B, Foo C, Houk A, Antoniewski C, Andino R. The RNA silencing endonuclease argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Gene Devel. 2006;20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen L, Gubler DJ. The use of mosquitoes to detect and propagate dengue viruses. Am J Trop Med Hyg. 1974;23(6):1153–1160. doi: 10.4269/ajtmh.1974.23.1153. [DOI] [PubMed] [Google Scholar]
- Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Gene Devel. 2006;20:2214–2222. doi: 10.1101/gad.1454806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarot E, Payen-Groschene G, Bucheton A, Pelisson A. Evidence for a piwi-dependent RNA silencing of the gypsy endogenous retrovirus by the Drosophila melanogaster flamenco gene. Genetics. 2004;166:1313–1321. doi: 10.1534/genetics.166.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott DH, Cureton DK, Whelan SP, Hunter CP. An antiviral role for the RNA interference machinery in Caenorhabditis elegans. PNAS. 2005;102(51):18420–18424. doi: 10.1073/pnas.0507123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi PY, Kauffman EB, Ren P, Felton A, Tai JH, Dupuis AP, II, Jones SA, Ngo KA, Nicholas DC, Maffei JG, Ebel GD, Bernard KA, Kramer LD. High-throughput detection of West Nile virus RNA. J Clin Microbiol. 2001;39(4):1264–1271. doi: 10.1128/JCM.39.4.1264-1271.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz CA, Holko M, deVeer MJ, Silverman RH, Williams BRG. Activation of the interferon system by short-interfering RNAs. Nature Cell Biol. 2003;5(9):834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- Solomon T. Flavivirus encephalitis. N Engl J Med. 2004;351(4):370–378. doi: 10.1056/NEJMra030476. [DOI] [PubMed] [Google Scholar]
- Stapleton W, Das S, McKee BD. A role of the Drosophila homeless gene in repression of Stellate in male meiosis. Chromosoma. 2001;110:228–240. doi: 10.1007/s004120100136. [DOI] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. Recombination in alphaviruses. Sem Virol. 1997;8:85–94. [Google Scholar]
- Takeuchi O, Akira S. Recognition of viruss by innate immunity. Immunol Rev. 2007;220:2214–224. doi: 10.1111/j.1600-065X.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- Twiddy SS, Holmes EC. The extent of homologous recombination in members of the genus flavivirus. J Gen Virol. 2003;84:429–440. doi: 10.1099/vir.0.18660-0. [DOI] [PubMed] [Google Scholar]
- Uchil PD, Satchidanandam V. Architecture of the flaviviral replication complex. J Biol Chem. 2003;278(27):24388–24398. doi: 10.1074/jbc.M301717200. [DOI] [PubMed] [Google Scholar]
- van Rij RP, Saleh MC, Berry B, Foo C, Houk A, Antoniewski C, Andino R. The RNA silencing endonuclease Argonaute 2 mediates specific aniviral immunity in Drosophila melanogaster. Gene Devel. 2006;20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Aliyari R, Li WX, Li HW, Kim K, Carthew R, Atkinson P, Ding SW. RNA interference directs innate immunity against viruses in adult Drosophila. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol. 1997;71(9):6650–6661. doi: 10.1128/jvi.71.9.6650-6661.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins C, Dishongh R, Moore SC, Whitt MA, Chow M, Machaca K. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature. 2005;436:1044–1047. doi: 10.1038/nature03957. [DOI] [PubMed] [Google Scholar]
- Xu K, Bogert BA, Li W, Su K, Lee A, Gao FB. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr Biol. 2004;14:1025–1034. doi: 10.1016/j.cub.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Zambon RA, Vakharia VN, Wu LP. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cell Microbiol. 2006;8(5):880–889. doi: 10.1111/j.1462-5822.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: Double-stranded RNA directs ATP-dependent cleavage of mRNA at 21 to 23 nuceotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, Copley RR, Christophides GK, Thomasova D, Holt RA, Subramanian GM, Mueller HM, Dimopoulos G, Law JH, Wells MA, Birney E, Charlab R, Halpern AL, Kokoza E, Kraft CL, Lai Z, Lewis S, Louis C, Barillas-Mury C, Nusskern D, Rubin GM, Salzberg SL, Sutton GG, Topalis P, Wides R, Wincker P, Yandell M, Collins FH, Ribeiro J, Gelbart WM, Kafatos FC, Bork P. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002;298:149–159. doi: 10.1126/science.1077061. [DOI] [PubMed] [Google Scholar]