

Abstract
Background and purpose:
Two-pore-domain potassium (K2P) channels mediate potassium background (or ‘leak') currents, controlling excitability by stabilizing membrane potential below firing threshold and expediting repolarization. Inhibition of K2P currents permits membrane potential depolarization and excitation. As expected for key regulators of excitability, leak channels are under tight control from a plethora of stimuli. Recently, signalling via protein tyrosine kinases (TKs) has been implicated in ion channel modulation. The objective of this study was to investigate TK regulation of K2P channels.
Experimental approach:
The two-electrode voltage clamp technique was used to record K2P currents in Xenopus oocytes. In addition, K2P channels were studied in Chinese hamster ovary (CHO) cells using the whole-cell patch clamp technique.
Key results:
Here, we report inhibition of human K2P3.1 (TASK-1) currents by the TK antagonist, genistein, in Xenopus oocytes (IC50=10.7 μM) and in CHO cells (IC50=12.3 μM). The underlying molecular mechanism was studied in detail. hK2P3.1 was not affected by genistin, an inactive analogue of genistein. Perorthovanadate, an inhibitor of tyrosine phosphatase activity, reduced the inhibitory effect of genistein. Current reduction was voltage independent and did not require channel protonation at position H98 or phosphorylation at the single TK phosphorylation site, Y323. Among functional hK2P family members, genistein also reduced K2P6.1 (TWIK-2), K2P9.1 (TASK-3) and K2P13.1 (THIK-1) currents, respectively.
Conclusions and implications:
Modulation of K2P channels by the TK inhibitor, genistein, represents a novel molecular mechanism to alter background K+ currents.
Keywords: background potassium current, cardiac arrhythmia, cardioprotection, cellular excitability, ischaemic preconditioning, K2P channel, leak channel, membrane potential, tyrosine kinase, protein phosphorylation
Introduction
Two-pore-domain potassium (K2P) channels are recognized to be highly regulated leak pathways that control excitability, stabilizing membrane potential below firing threshold and expediting repolarization (Goldstein et al., 2001, 2005). K2P channels are identified by a unique structure of two pore-forming loop domains in each subunit. The channels assemble from two subunits to form a single ion conduction pathway. Expressed robustly throughout the cardiovascular, gastrointestinal, genitourinary and CNS, K2P channels are implicated in multiple physiological processes, including neuromodulation, neuro- and cardioprotection, regulation of cardiac rhythm, anaesthesia, apoptosis, and sensation of oxygen tension, mechanical stress, taste and temperature (Patel et al., 1998; Lauritzen et al., 2003; Heurteaux et al., 2004; Kemp et al., 2004; Richter et al., 2004; Chemin et al., 2005; Kang et al., 2005; Lalevee et al., 2006; Putzke et al., 2007). Because membrane potential is fundamental to neuronal and cardiac activity, leak current regulation is a primary and dynamic mechanism for control of cellular excitability (Goldstein et al., 2001; Patel et al., 2001; Bayliss et al., 2003).
Unravelling signal-transduction mechanisms that control excitability is critical to our understanding of cardiac and neuronal electrophysiology. Signalling via tyrosine kinases (TKs) mediates hormone- and receptor-dependent signal transduction, regulation of cell growth, differentiation, metabolism and function. Specific cardiac functions associated with TK activity include ischaemic preconditioning (Fryer et al., 1998; Benter et al., 2005) and signal transduction in angiotensin II-associated cardiac hypertrophy (Haendeler and Berk, 2000). In the brain, TKs are involved in long-term potentiation in the hippocampus (O'Dell et al., 1991). At the molecular level, TKs regulate the activity of several ion channels, including a diverse group of voltage-gated K+ channels (Hool et al., 1998; Missan et al., 2006). Although previous studies have established that K2P channels are differentially regulated by protein kinases A and C (reviewed in Goldstein et al., 2001; Bayliss et al., 2003; Mathie, 2007), there is no information on TK-related modification of K2P leak currents.
Here, K2P family members 3.1, 6.1, 9.1 and 13.1 (TASK-1, TWIK-related acid-sensitive K+ channel 1; TWIK-2, tandem of P domains in a weak inward rectifying K+ channel 2; TASK-3; and THIK-1, tandem pore domain halothane-inhibited K+ channel 1, respectively) are revealed to be inhibited by the TK inhibitor, genistein. The International Union of Pharmacology classification has accorded each K2P channel gene with an ion channel subunit product (Goldstein et al., 2005); these identifiers are used and presented with common acronyms in this study. Originally isolated from the fermentation broth of Pseudomonassp., the isoflavone compound genistein inhibits protein TKs by competing for the ATP-binding site with an IC50 of 20.4–111 μM while exhibiting little or no effects on serine/threonine kinases (Akiyama et al., 1987; Akiyama and Ogawara, 1991). Recent experimental and clinical data suggest that the phytooestrogen genistein is associated with reduced incidence of cardiovascular disease and breast, uterine and prostate cancer (Dixon and Ferreira, 2002; Park et al., 2005). Furthermore, genistein inhibits metastasis of prostate cancer in mice and enhances the efficacy of cancer therapeutics through modification of cell proliferation and survival pathways (Gescher et al., 2001; Sarker and Li, 2006; Lakshman et al., 2008).
Molecular determinants of genistein-dependent regulation of the most sensitive K2P channel, K2P3.1 (TASK-1), were studied in detail. Inhibitory effects on K2P3.1 were abolished or reduced when genistin and daidzein, inactive or less potent analogues of genistein, were applied. The phosphotyrosine phosphatase inhibitor, perorthovanadate (PVN), attenuated the effect of TK inhibition on K2P3.1. Genistein-associated blockade occurred independently of channel phosphorylation at the single TK phosphorylation site, Y323, suggesting that TK activity does not directly affect K2P3.1 channel function. Modulation of K2P channels by genistein is revealed to be a novel mechanism to alter background K+ channel function.
Methods
Molecular biology
Drug target nomenclature conforms with British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2007). Human K2P4.1 (B), K2P5.1 (B), K2P6.1 (B), K2P10.1 (B), K2P13.1 (B), K2P16.1 (P) and K2P17.1 (B) were amplified from brain (B) or pancreas (P) cDNA libraries, inserted into pCR2.1-TOPO and subcloned into pRAT, a dual-purpose expression vector, and containing a CMV promoter for mammalian expression and a T7 promoter for cRNA synthesis.
Mutations described in this study were made with a QuikChange Site-Directed Mutagenesis kit and synthetic mutant oligonucleotide primers. All cDNA constructs were confirmed by DNA sequencing. Procedures for in vitro transcription and oocyte injection were performed as published previously (Kiehn et al., 1999). Briefly, cRNAs were transcribed after vector linearization using T7 RNA polymerase and the mMessage mMachine kit. Transcripts were quantified using a spectrophotometer and by comparison with control samples separated by agarose gel electrophoresis. Stages V and VI defolliculated Xenopus oocytes were injected with 2–46 ng cRNA encoding study channels.
Tissue culture
Chinese hamster ovary (CHO) cells were cultured in minimum essential medium α (MEM α) supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin G sodium, and 100 μg mL−1 streptomycin sulphate in an atmosphere of 95% humidified air and 5% CO2 at 37 °C. Cells were passaged regularly and subcultured prior to treatment. Transient transfections of CHO cells were performed using Lipofectamine 2000 transfection reagent according to the manufacturer's instructions.
Electrophysiology
Two-electrode voltage clamp measurements were performed as described earlier (Thomas et al., 1999). This study has been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. Ovarian lobes were surgically removed in aseptic technique from female Xenopus laevis frogs anaesthetized with 1 g L−1 tricaine solution (pH=7.5). Frogs were not fed on the day of surgery to avoid emesis during anaesthesia. After surgery, the frogs were allowed to recover consciousness, followed by at least 2 months of recovery period. Oocyte collection was alternated between left and right ovaries, and no more than three surgeries were performed on one individual frog. After the final taking of oocytes, the anaesthetized frog was killed by decerebration and pithing. Following collagenase treatment, stages V and VI defolliculated oocytes were manually isolated under a stereomicroscope. Whole-cell currents were measured 1–3 days after injection with an Oocyte Clamp amplifier (Warner Instruments, Hamden, CT, USA) using pCLAMP (Axon Instruments, Foster City, CA, USA) and Origin 6 (OriginLab, Northampton, MA, USA) software for data acquisition and analysis. Data were sampled at 2 kHz and filtered at 1 kHz. Current recordings from CHO cells were performed using the whole-cell patch clamp configuration as previously reported (Thomas et al., 2001). All experiments were carried out at room temperature (20–22 °C), and no leak subtraction was done during the experiments.
Solutions and drug administration
Two-electrode voltage clamp electrodes were filled with 3 M KCl and had tip resistances of 1–5 MΩ. Recordings were performed under constant perfusion at room temperature. The standard physiological extracellular solution contained 96 mM NaCl, 4 mM KCl, 1.1 mM CaCl2, 1 mM MgCl2, 5 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid) (pH was adjusted to 7.4 with NaOH). Human K2P16.1 and K2P17.1 were activated by adjusting extracellular pH to 8.5. Currents were evoked in oocytes by step depolarization from −140 to +60 mV (500 ms) in 20-mV increments at 2-s intervals (5-s intervals for hK2P18.1) from the holding potential (−80 mV).
For whole-cell patch clamp recordings from CHO cells, electrodes were filled with the following solution (in mM): 100 K-aspartate, 20 KCl, 2.0 MgCl2, 1.0 CaCl2, 10 EGTA, 2 ATP, 10 HEPES (pH adjusted to 7.2 with KOH). The external solution for these experiments contained (in mM): 140 NaCl, 5.0 KCl, 1.0 MgCl2, 1.8 CaCl2, 10 HEPES, 10 glucose (pH adjusted to 7.4 with NaOH). Families of K2P3.1 currents were recorded during step depolarization from −120 to +80 mV (500 ms) in 20-mV increments at 5-s intervals. The holding potential was −80 mV. To quantify the inhibitory effects of genistein, currents were activated by a 500 ms test pulse to +60 mV (holding potential −80 mV). Pulses were applied in 10-s intervals during superfusion with drug solution. Current amplitudes were recorded at the end of the depolarizing test pulse once steady state had been reached (usually within 3–4 min).
Genistein, genistin and daidzein were prepared as 100 mM stock solutions in dimethylsulphoxide and stored at −20 °C. The sodium orthovanadate stock solution was prepared by adding H2O2 (30% wt wt−1 solution, 10 mM final concentration) to an aqueous solution containing 10 mM Na3VO4 and 50 mM HEPES (pH=7.4). This solution was mixed and allowed to stand at room temperature for 20 min. Excess H2O2 was eliminated by adding 200 μg mL−1 catalase (5 min incubation). The stock solution containing a mixture of vanadate and peroxovanadium complexes was made immediately before use. The orthovanadate concentration used in our experiments is based on the Na3PO4 concentration used in preparing the stock solution.
On the day of experiments, aliquots of the stock solutions were diluted to the desired concentrations with the bath solution. Human K2P3.1 currents recorded from oocytes were not significantly altered upon application of 0.6% dimethylsulphoxide (v v−1; maximum bath concentration) for 6 min (n=7; data not shown).
Materials
Human cDNA clones encoding K2P1.1-K274Q. (Rajan et al., 2005), K2P2.1 (EF165334), K2P3.1 (NM_002246), K2P3.1-H98N (Lopes et al., 2001) and K2P9.1 (NM_016601) were provided by Dr Steve Goldstein (Chicago, IL, USA). cDNA encoding human K2P18.1 (NM_181840) was generously donated by Dr C Spencer Yost (San Francisco, CA, USA). Brain (B) and pancreas (P) cDNA libraries were obtained from Clontech (Palo Alto, CA, USA), pCR2.1-TOPO from Invitrogen (Carlsbad, CA, USA) and pRAT was kindly provided by Dr Steve Goldstein (Chicago). The QuikChange Site-Directed Mutagenesis kit was from Stratagene (La Jolla, CA, USA) and the mMessage mMachine kit from Ambion (Austin, TX, USA). MEM α, fetal bovine serum and Lipofectamine 2000 transfection reagent were from Invitrogen (Karlsruhe, Germany). Genistein, genistin daidzein and Na3VO4 were all from Sigma.
Data analysis and statistics
Concentration–response relationships for drug-induced block were fit with a Hill equation of the following form: Idrug/Icontrol=1/[1+(D/IC50)n], where I indicates current, D is the drug concentration, n is the Hill coefficient and IC50 is the concentration necessary for 50% block. Data are expressed as mean±s.e.mean. We used Student's t-test (two-tailed tests) to compare the statistical significance of the results: P<0.05 was considered statistically significant. Multiple comparisons were performed using one-way ANOVA. If the hypothesis of equal means could be rejected at the 0.05 level, pairwise comparisons of groups were made and the probability values were adjusted for multiple comparisons using the Bonferroni correction.
Results
Genistein reduces K2P3.1 (TASK-1) background currents
The effects of TK inhibition on human K2P3.1 (TASK-1) leak channels were studied in X. laevis oocytes. Genistein, a broad range TK inhibitor, reduced hK2P3.1 potassium currents in a concentration-dependent manner, as displayed in Figure 1. Currents were elicited by a 500 ms depolarizing step to +20 mV from a holding potential of −80 mV. The degree of block was determined after 6 min (Figure 1a). To study the concentration dependence of hK2P3.1 inhibition by genistein, currents in the presence of the drug were normalized to their respective control values and plotted as relative current amplitudes in Figure 1b (n=4–13 cells were investigated at each concentration). The half-maximal inhibitory concentration (IC50) for block of hK2P3.1 leak channels yielded 10.7±0.8 μM with a Hill coefficient nH of 1.5±0.3. The time course is shown in Figure 1c (n=4). After a control period of 6 min with no significant changes in current amplitude, hK2P3.1 current reduction by 100 μM genistein reached steady-state conditions after 6 min. Upon washout (20 min), inhibitory effects of genistein on hK2P3.1 were partially reversible.
Figure 1.

Inhibition of human K2P3.1 (TASK-1) channels expressed in Xenopus oocytes by genistein. Representative current traces recorded from the same cell under control conditions and after superfusion with genistein (1, 10 and 100 μM, respectively) are displayed in (a). (b) Concentration–response relationships for the effect of genistein on hK2P3.1 outward currents measured at +20 mV (n=4–13 cells; mean±s.e.mean). The IC50 yielded 10.7 μM. (c) Time course of hK2P3.1 current inhibition by 100 μM genistein (n=4).
Human K2P3.1-related K2P family members K2P6.1, K2P9.1 and K2P13.1 are sensitive to genistein
Over the past 11 years, 15 different human K2P channels have been cloned. Some genes failed to produce currents (K2P7.1, K2P12.1 and K2P15.1), leaving 12 functional leak channels accessible to electrophysiological investigation. To assess specificity of genistein-induced K2P current inhibition, all functional human K2P channels cloned to date were studied (Figure 2). From a holding potential of −80 mV, depolarizing pulses were applied for 500 ms to voltages between −140 and +60 mV in 20-mV increments (0.5 Hz). This protocol was used in all experiments performed in this study, unless indicated otherwise. K2P1.1 (TWIK-1) channels, previously shown to be non-functional, were recently revealed to produce leak currents when a lysine residue is removed by mutation to glutamine (K2P1.1-K274Q; Rajan et al., 2005). This effect has been suggested to be caused by lack of covalently bound small ubiquitin-like modifier protein, SUMO. However, this hypothesis is currently being discussed controversially (Feliciangeli et al., 2007). Here, we expressed hK2P1.1-K274Q cRNA to achieve sufficient hK2P1.1 current levels in Xenopus oocytes. Both hK2P1.1-K274Q and hK2P6.1 displayed very low current levels under control conditions (0.36±0.02 μA (n=15) and 0.31±0.02 μA (n=5), respectively). However, these current amplitudes were significantly larger than mean current levels in uninjected control oocytes (0.19±0.01 μA; n=12), indicating successful channel expression. The effects of 100 μM genistein on K2P family members are summarized in Figures 2 and 3a. The most pronounced effect was observed with hK2P3.1. In addition, genistein significantly reduced hK2P6.1, hK2P9.1 and hK2P13.1 currents, respectively (Figures 2 and 3a). A phylogenetic tree illustrates that leak channels affected by TK regulation are related (Figure 3b).
Figure 2.

Synopsis of genistein effects on functional human K2P family members. Representative control measurements and recordings after application of 100 μM genistein (6 min) are shown. As wild-type hK2P1.1 (TWIK-1) produces relatively small currents, a mutant previously shown to display increased macroscopic currents in oocytes (hK2P1.1-K274Q; Rajan et al., 2005) has been used. See text for voltage protocol.
Figure 3.

Protein tyrosine kinase inhibitor genistein reduces currents of related hK2P family members. (a) Significant current reduction was observed with hK2P3.1, hK2P6.1, hK2P9.1 and hK2P13.1, respectively (**P<0.01, ***P<0.001 versus respective controls; n=5–15 cells were measured for each K2P channel). Data are given as mean±s.e.mean. (b) Phylogenetic tree of functional human K2P channels. Nucleotide sequence alignment and phylogenetic analysis were generated using ClustalW software version 1.83 (BLOSUM method) and TreeView. Human K2P channels affected by genistein are indicated.
hK2P3.1 current reduction by TK inhibition is voltage independent
Molecular mechanisms of TK-dependent regulation of the most sensitive K2P channel, hK2P3.1, were studied in detail. The effect of genistein on hK2P3.1 current–voltage (I–V) relationship was investigated under isochronal recording conditions using the protocol described in Figure 2. Families of current traces from one cell are shown for control conditions and after exposure to 100 μM genistein (6 min) in Figure 4a, revealing electrophysiological characteristics typical for a potassium-selective background leak conductance, that is, a voltage-independent portal showing Goldman–Hodgkin–Katz, or open, rectification (Figure 4b and c; Goldstein et al., 2001). Potassium channels that display open rectification pass current more readily in one direction (rectify) owing to unequal ion concentration across the membrane. Relative inhibition of hK2P3.1 currents (plotted as a function of the test pulse potential in Figure 4d) was not significantly different among the voltage steps applied (n=13 cells were studied at each potential), indicating that genistein application reduced leak currents in a voltage-independent manner.
Figure 4.

Biophysical and pharmacological determinants of genistein-induced hK2P3.1 current reduction. (a) Effects of genistein on the voltage dependence of activation. Control measurement and the effect of 100 μM genistein (6 min) are shown in one representative oocyte. (b, c) Activation curves, that is, step current amplitudes as a function of the pulse potential, recorded under isochronal conditions (b, original current amplitudes; c, values normalized to peak currents) (n=13). There was no apparent shift in the current–voltage relationship. (d) Attenuation of hK2P3.1 currents by genistein is not voltage dependent. The fraction of blocked step currents is plotted as a function of the respective test pulse potentials. Channel block did not display significant differences between −140 and +60 mV (n=13 cells). (e) The inactive analogue genistin (10 and 100 μM; n=8 and 6 cells, respectively) did not reproduce inhibitory effects of 10 and 100 μM genistein (n=22 and 12 oocytes, respectively). Similarly, 10 μM of the less potent TK inhibitor daidzein had no significant effect on hK2P3.1 currents (n=7). At higher concentrations (100 μM) daidzein weakly reduced hK2P3.1 currents (n=9). (f) Predicted topology of hK2P3.1 subunits. TM, transmembrane domain; P, pore loop domain. N and C termini are intracellular. The histidine residue that allows protons to block in the physiological pH range (H98) is indicated. Data are expressed as mean±s.e.mean. *P<0.05, ***P<0.001 versus respective controls (see text for voltage protocol).
To differentiate between TK-dependent actions of genistein and direct inhibition of hK2P3.1 channels, structurally similar analogues were applied under similar experimental conditions (Figure 4e). Currents were recorded at the end of depolarizing voltage steps to +20 mV under control conditions and after drug application for 6 min. The inactive analogue, genistin (10 and 100 μM), did not cause significant current reduction (n=8 and 6 cells, respectively), whereas genistein application reduced mean currents by 38.9±1.8% (n=22) and 81.1±1.7% (n=12) under similar conditions (10 and 100 μM, respectively). Daidzein is a different structural analogue of genistein that is commonly used as negative control similar to genistin. However, in contrast to genistin, daidzein inhibits TKs at higher drug concentrations. Accordingly, 10 μM daidzein did not induce significant hK2P3.1 current reduction (n=7), as expected, whereas 100 μM daidzein reduced outward current amplitudes by 18.2±1.3% (n=9).
TK-mediated hK2P3.1 current block does not depend on channel protonation
K2P3.1 channels are sensitive to extracellular pH variations in the physiological range, displaying a pKa value of 7.2 (Lopes et al., 2000, 2001). A single histidine residue adjacent to the potassium selectivity filter within the first pore-forming loop, H98, represents the extracellular proton-binding site in hK2P3.1 (Figure 4f). Effects of 10 μM genistein (6 min) on wild-type and mutant hK2P3.1-H98N channels insensitive to pH modulation were compared. Reduction of wild-type currents (39.2±2.4%; n=13) was not significantly different from hK2P3.1-H98N channels (33.0±1.8%; n=8), indicating that channel protonation is not required for TK regulation of hK2P3.1.
Direct TK-dependent phosphorylation of hK2P3.1 α-subunits is not involved in genistein-dependent leak channel regulation
The significance of tyrosine phosphorylation in genistein-associated hK2P3.1 current inhibition was further analysed using PVN, a membrane-permeable inhibitor of protein tyrosine phosphatases. After application of 100 μM PVN for 6 min, that is, when dephosphorylation of TK substrates was prevented, the effect of TK inhibition by 10 μM genistein (co-administered for additional 6 min) was significantly reduced compared with PVN-free conditions (Figure 5a; current reduction of 26.9±1.2 versus 38.9±1.8%, respectively; n=6 and 22 cells). The difference in current reduction by 100 μM genistein in the presence (76.4±1.1%; n=9) or absence of 100 μM PVN (81.1±1.7%; n=12) did not achieve statistical significance.
Figure 5.

Human K2P3.1 regulation by tyrosine kinases (TKs) does not require direct TK-dependent phosphorylation of the channel α-subunit. (a) Mean (±s.e.mean) relative current amplitudes in the presence or absence of the tyrosine phosphatase inhibitor, perorthovanadate (PVN; 100 μM), after application of 10 or 100 μM genistein (6 min) or compared to PVN-free control conditions (w/o PNV) for hK2P3.1 wild type (n=6–22 cells studied). Although PVN did not alter basal hK2P3.1 current amplitudes, the inhibitory effect of 10 μM genistein was significantly attenuated by simultaneous PVN treatment (*P<0.05). (b) Predicted membrane topology of hK2P3.1 subunits illustrating the location of the putative TK phosphorylation site, Y323. (c) Exchange of the tyrosine residue at position 323 for phenylalanine (to prevent phosphorylation; F; n=10) or glutamate (to mimic phosphorylation; E; n=10) did not affect basal current amplitudes under control conditions (WT, wild type; n=10). Furthermore, hK2P3.1 current reduction induced by 10 μM genistein was not altered (d; n=8–13). See text for voltage protocol.
Perorthovanadate application alone (100 μM; 6 min) did not cause significant hK2P3.1 amplitude changes (Figure 5a; n=9). The lack of response to pharmacological inhibition of tyrosine phosphatases suggests a saturated level of basal TK substrate phosphorylation under the given experimental conditions.
The antagonism of genistein-induced hK2P3.1 current reduction by PVN (Figure 5a) can be associated in the first instance with direct TK-dependent phosphorylation of the channel α-subunit at its single predicted TK consensus site, Y323 (Figure 5b). To differentiate between direct effects of a TK on the channel protein from intermediate actions within signal-transduction cascades, we performed site-directed mutagenesis to generate mutant hK2P3.1 channels that lack the consensus TK phosphorylation site (Y323F). In addition, a different mutant was generated to mimic phosphorylation at residue 323 (Y323E). Under control conditions, current amplitudes recorded from hK2P3.1-Y323F (n=10) and hK2P3.1-Y323E channels (n=10) were not significantly different from wild-type currents (Figure 5c; n=10). Furthermore, the inhibitory action of 10 μM genistein was not significantly altered by Y323 mutations compared with wild-type hK2P3.1 (Figure 5d). Taken together, these data reveal that TK regulation of hK2P3.1 channels is not mediated by direct channel phosphorylation.
Indirect actions of TK inhibition on hK2P3.1 channels are not mediated via PKA- or PKC-dependent pathways
Indirect TK-dependent actions may affect ion channel function by regulating serine/threonine kinase activity (Schröder et al., 2004; Zhou et al., 2007). To determine whether two serine/threonine kinases, protein kinases A and C, contribute to TK-dependent hK2P3.1 regulation, cells were incubated with the PKA and PKC inhibitor, staurosporine (1 μM), for 1–3 h prior to electrophysiological recordings. Figure 6a illustrates that inhibition of protein kinases A and C by staurosporine did not significantly affect hK2P3.1 current reduction by 10 μM genistein (6 min; n=11) when compared with oocytes from the same batch without staurosporine pretreatment (n=9), arguing against a significant role of PKA and/or PKC in hK2P3.1 regulation by TKs. It is noteworthy, however, that basal hK2P3.1 current amplitudes were increased by incubation with 1 μM staurosporine compared with untreated cells (mean current amplitudes at +20 mV: 7.7±0.5 versus 5.2±0.5 μA; n=11 and 9 cells, respectively). This is in line with previous studies reporting K2P3.1 current reduction by PKC-dependent mechanisms, resulting in cardiac repolarization abnormalities (Besana et al., 2004).
Figure 6.

Protein kinases A and C are not involved in tyrosine kinase (TK)-dependent hK2P3.1 regulation. (a) Oocytes were treated with 1 μM staurosporine, a serine/threonine kinase inhibitor, for 1–3 h prior to current recordings (stauro.; n=11). The effect of 10 μM genistein was then compared to control cells from the same batch without staurosporine (mock; n=9). The genistein response was not significantly altered by pretreatment with staurosporine. Predicted PKA and PKC phosphorylation sites are indicated in (b). To prevent phosphorylation, respective residues were mutated to alanine, producing hK2P3.1 ΔPKA (S392A–S393A), hK2P3.1 ΔPKC (S358A–T383A), hK2P3.1 ΔPKA ΔPKC (S358A–T383A–S392A–S393A) and hK2P3.1 ΔPKA ΔPKC ΔTK (hK2P3.1 ΔPKA ΔPKC including the Y323F mutation to prevent TK phosphorylation), respectively. Mutant channels were sensitive to 100 μM genistein similar to wild-type hK2P3.1 (c; n=4–8 cells studied). Data are given as mean±s.e.mean. See text for voltage protocol.
To further confirm that PKA- and/or PKC-dependent phosphorylation of hK2P3.1 channels is not required for TK regulation, we generated hK2P3.1 clones where all putative PKA- and/or PKC-dependent phosphorylation sites were mutated to alanine residues to prevent phosphorylation (Figure 6b). The resulting constructs were hK2P3.1 ΔPKA (S392A–S393A), hK2P3.1 ΔPKC (S358A–T383A), and hK2P3.1 ΔPKA ΔPKC (S358A–T383A–S392A–S393A), respectively. In addition, a construct lacking all putative PKA-, PKC- and TK-dependent phosphorylation sites was generated, hK2P3.1 ΔPKA ΔPKC ΔTK (hK2P3 ΔPKA ΔPKC-Y323F). When compared to wild-type hK2P3.1, mutant clones were equally sensitive to 100 μM genistein (Figure 6c; 6 min incubation; n=4–8 cells were studied), ruling out a significant role of direct phosphorylation by PKA, PKC and/or TK in genistein-mediated hK2P3.1 current reduction.
Under baseline conditions, mean hK2P3.1 wild-type current amplitudes (recorded at +20 mV) yielded 3.8±0.8 μA (n=4), whereas hK2P3.1 ΔPKC channels displayed significantly increased currents (5.5±1.4 μA; n=4). This is in accordance with our results obtained from cells following staurosporine treatment and reflects PKC-dependent inhibition of hK2P3.1 channels (Besana et al., 2004). Of note, currents recorded from cells expressing hK2P3.1 ΔPKA channels were significantly smaller (0.5±0.04 μA; n=4) compared with hK2P3.1 WT, indicating PKA-dependent activation of hK2P3.1. Mutant hK2P3.1 ΔPKA ΔPKC channels displayed an intermediate phenotype; mean current amplitudes yielded 1.6±0.3 μA (n=4).
Genistein regulates hK2P3.1 channels expressed in mammalian cells
It has been indicated that phosphorylation levels within Xenopus oocytes are highly variable (Cohen and Zilberberg, 2006). Thus, we expressed hK2P3.1 potassium channels heterologously in CHO cells to demonstrate modulation of hK2P3.1 currents in mammalian cells (Figure 7), that is, in a more stable environment. From a holding potential of −80 mV, depolarizing pulses were applied for 500 ms to voltages between −120 and +80 mV in 20-mV increments (0.2 Hz). The degree of block was determined at +60 mV after steady state had been reached (usually within 3–4 min) (Figure 7b), and the half-maximal inhibitory concentration was calculated as described in Figure 1 (Figure 7d; n=3–4 cells were investigated at each concentration). The IC50 for block of hK2P3.1 leak channels in CHO cells yielded 12.3±5.4 μM with a Hill coefficient nH of 0.83±0.21.
Figure 7.

Genistein-induced inhibition of human K2P3.1 (TASK-1) channels expressed in Chinese hamster ovary (CHO) cells. Families of current traces recorded under control conditions and after superfusion with 100 μM genistein are displayed in (a, b), respectively. (c) Representative recording from an untransfected CHO cell display little endogenous K+ conductance. (d) Concentration–response relationships for the effect of genistein on hK2P3.1 currents measured at +60 mV (n=3–4 cells; mean±s.e.mean). The IC50 yielded 12.3 μM (see text for voltage protocol).
Discussion and conclusions
K2P channels stabilize membranes of excitable cells at hyperpolarized potentials below the threshold for action potential firing. Here, we describe genistein-dependent modulation of K2P leak channels as a novel regulatory mechanism in addition to known G-protein-dependent pathways (for review, see Patel and Honore, 2001; Mathie, 2007).
K2P leak channels are modulated by genistein
Genistein reduces potassium leak currents in Xenopus oocytes and CHO cells (Figures 1 and 7). This mechanism is conserved among related K2P family members (Figures 2 and 3). In addition to hK2P3.1, the potassium leak channels hK2P6.1, hK2P9.1 and hK2P13.1 are blocked by genistein. In the present study, molecular mechanisms of genistein-dependent regulation of the most sensitive human K2P channel, hK2P3.1, were analysed in detail. The inhibitory action of genistein was voltage independent, that is, current inhibition occurred with similar potency at membrane potentials between −140 and +60 mV. K2P3.1 channels are sensitive to extracellular pH (pKa=7.2; Lopes et al., 2001). However, protein protonation is not required: mutation of a single histidine residue adjacent to the potassium selectivity filter that represents the extracellular proton-binding site in hK2P3.1, H98 (Figure 4f), did not alter genistein sensitivity of the channel.
Molecular mechanisms
Genistein may modulate ion channel and receptor function either through inhibition of TKs (Yu et al., 2004; Cho et al., 2005; Missan et al., 2006) or by direct blockade (Paillart et al., 1997; Belevych et al., 2002; Altomare et al., 2006). These pathways are not mutually exclusive, as illustrated by the work of Ogata et al. (1997), suggesting a combination of both mechanisms.
Here, several lines of evidence support the hypothesis that hK2P3.1 current reduction is mediated by TK inhibition. First, genistin, a structurally similar analogue of genistein, did not exert inhibitory effects. Second, a different structural analogue that only weakly inhibits TKs (daidzein) did not markedly affect hK2P3. Daidzein caused weak current inhibition only at high drug concentrations, as predicted. This is in line with studies demonstrating TK-dependent effects of genistein (Yu et al., 2004; Cho et al., 2005; Missan et al., 2006). Furthermore, this hypothesis is reinforced by the observation that PVN, an inhibitor of tyrosine phosphatase activity, attenuated the inhibitory effect of 10 μM genistein on hK2P3.1 currents.
In contrast, PVN did not significantly affect hK2P3.1 inhibition by 100 μM genistein. This lack of effect argues in favour of TK-independent mechanisms (Belevych et al., 2002). Moreover, the onset of current inhibition was fast (Figure 1). Rapid onset of block is consistent with direct channel blockade from the extracellular side. However, genistein and PVN have been shown to act rapidly, within 5 min, on ion channels and receptors via TK-dependent mechanisms as well (Wischmeyer et al., 1998; Cho et al., 2005), indicating that no definitive conclusions may be drawn from this observation. Finally, the signal-transduction mechanism does not involve direct TK phosphorylation of the channel protein. Mutant hK2P3.1 channels lacking the single putative TK-dependent phosphorylation site (hK2P3.1-Y323F) or mimicking phosphorylation (hK2P3.1-Y323E) were still modulated by genistein (Figure 5). Direct TK-dependent phosphorylation can be ruled out for other genistein-sensitive K2P family members, as hK2P6.1, hK2P9.1 and hK2P13.1 do not harbour any intracellular TK phosphorylation sites.
Indirect actions may affect K2P channel function via cross-talk with protein kinase A and/or protein kinase C, as suggested previously for L-type calcium currents and voltage-gated potassium channels (Schröder et al., 2004; Zhou et al., 2007). However, inhibition of PKA and PKC by preincubation with staurosporine did not affect genistein-induced hK2P3.1 current reduction. Furthermore, hK2P3.1 proteins lacking putative PKA- and PKC-dependent phosphorylation sites were sensitive to genistein similar to wild-type channels. Thus, PKA and PKC are not essential for genistein-induced inhibition of hK2P3.1, and direct phosphorylation of hK2P3.1 protein by TK, PKA and PKC is not required.
The rapid onset of block argues against increased protein turnover and protein degradation as molecular mechanisms of action. However, cross-talk with intracellular second messengers or lipid-dependent pathways (for example, diacylglycerol, phospholipase C, PIP2 and IP3) may be involved in genistein-dependent K2P regulation. In addition, accessory β-subunits or interacting proteins such as 14-3-3, endogenously expressed in Xenopus oocytes and CHO cells, may mediate inhibitory effects of genistein. Recently, association of activated Gαq subunits with hK2P3.1 and hK2P9.1 channels has been revealed to cause leak current inhibition (Chen et al., 2006; Veale et al., 2007). We may speculate that genistein-induced TK inhibition stimulates Gαq pathways, ultimately leading to association of activated Gαq proteins with hK2P3.1 channels and current inhibition.
In summary, it is reasonable to assume that genistein modulates hK2P3.1 channels, at least in part, via TK-dependent mechanisms. These pathways include intermediate signal-transduction factors. TK-dependent phosphorylation of hK2P3.1 protein was not observed. In addition, direct inhibitory effects of genistein on hK2P3.1 currents may contribute to the inhibitory effect of genistein on K2P channels as well. Future studies including chimeric approaches and analyses of the putative drug-binding site in hK2P3.1 are required to characterize the underlying molecular mechanism in detail.
Physiological and clinical implications of TK-dependent background K+ current regulation in heart and CNS
The cardiac plateau current 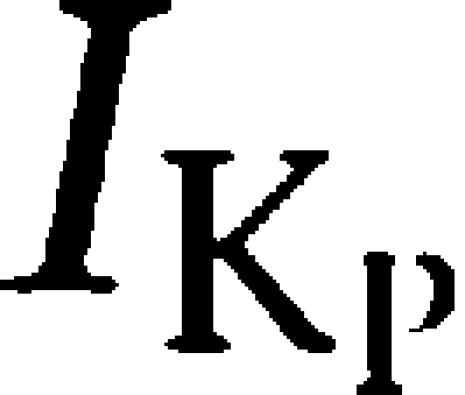 is time independent, potassium selective and influences the amplitude and duration of the cardiac action potential and, consequently, the duration of myocardial contraction (Marban, 2002). On the basis of common distribution and biophysical attributes, it has been suggested that K2P3.1 channels contribute to
is time independent, potassium selective and influences the amplitude and duration of the cardiac action potential and, consequently, the duration of myocardial contraction (Marban, 2002). On the basis of common distribution and biophysical attributes, it has been suggested that K2P3.1 channels contribute to 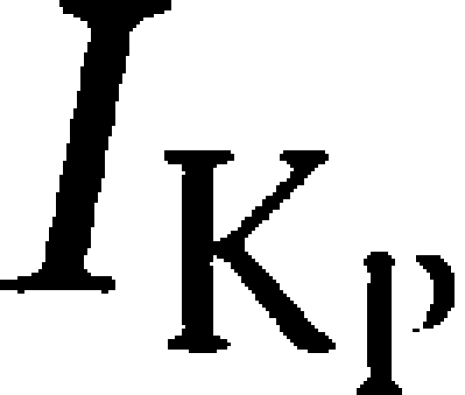 (Lopes et al., 2000). In mouse, K2P3.1 is expressed throughout the heart with prominence in the ventricles. K2P3.1 mRNA and protein have been demonstrated in rabbit heart in both atrial and ventricular cardiomyocytes. Endogenous K2P3.1-like currents have been recorded in rat cardiomyocytes, and inhibition of these currents has been proposed to mediate proarrhythmic effects. Recently, cardiac K2P channels have been associated with ischaemic preconditioning and cardioprotection (Lu et al., 2007). TK inhibition reduces beneficial effects of cardiac ischaemic preconditioning (Imagawa et al., 1997; Fatehi-Hassanabad and Parratt, 1997; Fryer et al., 1998). Thus, it is tempting to hypothesize that reduction of cardioprotective hK2P3.1 currents by TK inhibition attenuates ischaemic preconditioning in the presence of TK antagonists. Moreover, TK-related block of cardiac K2P leak currents may cause prolonged repolarization and, perhaps, dysregulation of cardiac pacemaker activity (Barbuti et al., 2002; Lalevee et al., 2006; Putzke et al., 2007).
(Lopes et al., 2000). In mouse, K2P3.1 is expressed throughout the heart with prominence in the ventricles. K2P3.1 mRNA and protein have been demonstrated in rabbit heart in both atrial and ventricular cardiomyocytes. Endogenous K2P3.1-like currents have been recorded in rat cardiomyocytes, and inhibition of these currents has been proposed to mediate proarrhythmic effects. Recently, cardiac K2P channels have been associated with ischaemic preconditioning and cardioprotection (Lu et al., 2007). TK inhibition reduces beneficial effects of cardiac ischaemic preconditioning (Imagawa et al., 1997; Fatehi-Hassanabad and Parratt, 1997; Fryer et al., 1998). Thus, it is tempting to hypothesize that reduction of cardioprotective hK2P3.1 currents by TK inhibition attenuates ischaemic preconditioning in the presence of TK antagonists. Moreover, TK-related block of cardiac K2P leak currents may cause prolonged repolarization and, perhaps, dysregulation of cardiac pacemaker activity (Barbuti et al., 2002; Lalevee et al., 2006; Putzke et al., 2007).
TK-sensitive K2P channels are strongly expressed in the CNS. In particular, K2P3.1 and K2P9.1 (with K2P1.1 and K2P10.1) are believed to contribute to the potassium standing outward current (IK(SO)) important in cerebellar granule neurons (Millar et al., 2000; Han et al., 2002; Clarke et al., 2004). Furthermore, K2P3.1 is responsive to volatile anaesthetics and has been implicated in oxygen sensation in both the brain and the carotid body. Inhibition of background potassium conductances in multiple areas of the CNS (including brainstem aminergic neurons, cerebellar granule neurons, cortex, thalamus, hippocampus and hypothalamus) is a significant mechanism by which hormones and neurotransmitters may enhance excitability and contribute to neuronal plasticity (Patel and Honore, 2001; Mathie, 2007). TK-related inhibition of K2P currents in the CNS may lead to increased excitability at the cellular level, ultimately translating into differential regulation of central nervous function.
Acknowledgments
We thank Dr Bettina Thomas for insightful comments on the paper. We are grateful to Dr C Spencer Yost for donating human K2P18.1 cDNA and to Dr Steve Goldstein for providing the pRAT vector and human cDNA clones encoding hK2P1.1-K274Q, hK2P2.1, hK2P3.1, hK2P3.1-H98N and hK2P9.1. This study was supported by research grants from the German Research Foundation (to CAK), from the Deutsche Stiftung für Herzforschung (to DT), from the German Cardiac Society (to DT), and from the National Institutes of Health (HL71789 to EF).
Abbreviations
- K2P
two-pore-domain K+ channel
- TASK
TWIK-related acid sensitive K+ channel
- THIK
tandem pore domain halothane inhibited K+ channel
- TK
tyrosine kinase
- TWIK
tandem of P domains in a weak inward rectifying K+ channel
Conflict of interest
The authors state no conflict of interest.
References
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- Akiyama T, Ogawara H. Use and specificity of genistein as inhibitor of protein-tyrosine kinases. Methods Enzymol. 1991;201:362–370. doi: 10.1016/0076-6879(91)01032-w. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels. Br J Pharmacol. 2007;150 Suppl 1:S1–S168. doi: 10.1038/sj.bjp.0707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomare C, Tognati A, Bescond J, Ferroni A, Baruscotti M. Direct inhibition of the pacemaker (If) current in rabbit sinoatrial node cells by genistein. Br J Pharmacol. 2006;147:36–44. doi: 10.1038/sj.bjp.0706433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuti A, Ishii S, Shimizu T, Robinson RB, Feinmark SJ. Block of background K+ channel TASK-1 contributes to arrhythmogenic effects of platelet-activating factor. Am J Physiol. 2002;282:H2024–H2030. doi: 10.1152/ajpheart.00956.2001. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Sirois JE, Talley EM. The TASK family: two-pore domain background K+ channels. Mol Interv. 2003;4:205–219. doi: 10.1124/mi.3.4.205. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Warrier S, Harvey RD. Genistein inhibits cardiac L-type Ca2+ channel activity by a tyrosine kinase-independent mechanism. Mol Pharmacol. 2002;62:554–565. doi: 10.1124/mol.62.3.554. [DOI] [PubMed] [Google Scholar]
- Benter IF, Juggi JS, Khan I, Yousif MH, Canatan H, Akhtar S. Signal transduction mechanisms involved in cardiac preconditioning: role of Ras-GTPase, Ca2+/calmodulin-dependent protein kinase II and epidermal growth factor receptor. Mol Cell Biochem. 2005;268:175–183. doi: 10.1007/s11010-005-3895-1. [DOI] [PubMed] [Google Scholar]
- Besana A, Barbuti A, Tateyama MA, Symes AJ, Robinson RB, Feinmark SJ. Activation of protein kinase Cɛ inhibits the two-pore domain K+ channel, TASK-1, inducing repolarization abnormalities in cardiac ventricular myocytes. J Biol Chem. 2004;279:33154–33160. doi: 10.1074/jbc.M403525200. [DOI] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honore E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, et al. Inhibition of a bachground K+ channel by Gq protein α-subunits. Proc Natl Acad Sci USA. 2006;103:3422–3427. doi: 10.1073/pnas.0507710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CH, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, et al. Rapid upregulation of α7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci. 2005;25:3712–3723. doi: 10.1523/JNEUROSCI.5389-03.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CE, Veale EL, Green PJ, Meadows HJ, Mathie A. Selective block of the human 2-P domain potassium channel, TASK-3, and the native leak potassium current, IKSO, by zinc. J Physiol. 2004;560:51–62. doi: 10.1113/jphysiol.2004.070292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Zilberberg N. Fluctuations in Xenopus oocytes protein phosphorylation levels during two-electrode voltage clamp measurements. J Neurosci Methods. 2006;153:62–70. doi: 10.1016/j.jneumeth.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Ferreira D. Genistein. Phytochemistry. 2002;60:205–211. doi: 10.1016/s0031-9422(02)00116-4. [DOI] [PubMed] [Google Scholar]
- Fatehi-Hassanabad Z, Parratt JR. Genistein, an inhibitor of tyrosine kinase, prevents the antiarrhythmic effects of preconditioning. Eur J Pharmacol. 1997;338:67–70. doi: 10.1016/s0014-2999(97)01299-5. [DOI] [PubMed] [Google Scholar]
- Feliciangeli S, Bendahhou S, Sandoz G, Gounon P, Reichold M, Warth R, et al. Does sumoylation control K2P1/TWIK1 background K+ channels. Cell. 2007;130:563–569. doi: 10.1016/j.cell.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Schultz JE, Hsu AK, Gross GJ. Pretreatment with tyrosine kinase inhibitors partially attenuates ischemic preconditioning in rat hearts. Am J Physiol. 1998;275:H2009–H2015. doi: 10.1152/ajpheart.1998.275.6.H2009. [DOI] [PubMed] [Google Scholar]
- Gescher AJ, Sharma RA, Steward WP. Cancer chemoprotection by dietary constituents: a tale of failure and promise. Lancet Oncol. 2001;2:371–379. doi: 10.1016/S1470-2045(00)00392-2. [DOI] [PubMed] [Google Scholar]
- Goldstein SAN, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. 2005;57:527–540. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- Goldstein SAN, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Berk BC. Angiotensin II mediated signal transduction. Important role of tyrosine kinases. Regul Pept. 2000;95:1–7. doi: 10.1016/s0167-0115(00)00133-6. [DOI] [PubMed] [Google Scholar]
- Han J, Truell J, Gnatenco C, Kim D. Characterization of four types of background potassium channels in rat cerebellar granule neurons. J Physiol. 2002;542:431–444. doi: 10.1113/jphysiol.2002.017590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004;23:2684–2695. doi: 10.1038/sj.emboj.7600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC, Middleton LM, Harvey RD. Genistein increases the sensitivity of cardiac ion channels to β-adrenergic receptor stimulation. Circ Res. 1998;83:33–42. doi: 10.1161/01.res.83.1.33. [DOI] [PubMed] [Google Scholar]
- Imagawa J, Baxter GF, Yellon DM. Genistein, a tyrosine kinase inhibitor, blocks the ‘second window of protection' 48 h after ischemic preconditioning in the rabbit. J Mol Cell Cardiol. 1997;29:1885–1893. doi: 10.1006/jmcc.1997.0428. [DOI] [PubMed] [Google Scholar]
- Kang D, Choe C, Kim D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol. 2005;564:103–116. doi: 10.1113/jphysiol.2004.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp PJ, Peers C, Lewis A, Miller P. Regulation of recombinant human brain tandem P domain K+ channels by hypoxia: a role for O2 in the control of neuronal excitability. J Cell Mol Med. 2004;8:38–44. doi: 10.1111/j.1582-4934.2004.tb00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn J, Thomas D, Karle CA, Schöls W, Kübler W. Inhibitory effects of the class III antiarrhythmic drug amiodarone on cloned HERG potassium channels. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:212–219. doi: 10.1007/pl00005344. [DOI] [PubMed] [Google Scholar]
- Lakshman M, Xu L, Ananthanarayanan V, Cooper J, Takimoto CH, Helenowski I, et al. Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Res. 2008;68:2024–2032. doi: 10.1158/0008-5472.CAN-07-1246. [DOI] [PubMed] [Google Scholar]
- Lalevee N, Monier B, Senatore S, Perrin L, Semeriva M. Control of cardiac rhythm by ORK1, a Drosophila two-pore domain potassium channel. Curr Biol. 2006;16:1502–1508. doi: 10.1016/j.cub.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Zanzouri M, Honore E, Duprat F, Ehrengruber MU, Lazdunski M, et al. K+-dependent cerebellar granule neuron apoptosis. Role of TASK leak K+ channels. J Biol Chem. 2003;278:32068–32076. doi: 10.1074/jbc.M302631200. [DOI] [PubMed] [Google Scholar]
- Lopes CMB, Gallagher PG, Buck ME, Butler MH, Goldstein SAN. Proton block and voltage-gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- Lopes CMB, Zilberberg N, Goldstein SAN. Block of Kcnk3 by protons: evidence that 2-P-domain potassium channel subunits function as homodimers. J Biol Chem. 2001;276:24449–24452. doi: 10.1074/jbc.C100184200. [DOI] [PubMed] [Google Scholar]
- Lu Z, Gao J, Zuckerman J, Mathias RT, Gaudette G, Krukenkamp I, et al. Two-pore K(+) channels, NO and metabolic inhibition. Biochem Biophys Res Commun. 2007;363:194–196. doi: 10.1016/j.bbrc.2007.08.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban E. Cardiac channelopathies. Nature. 2002;415:213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- Mathie A. Neuronal two pore domain potassium channels and their regulation by G protein coupled receptors. J Physiol. 2007;578:377–385. doi: 10.1113/jphysiol.2006.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JA, Barratt L, Southan AP, Page KM, Fyffe REW, Robertson B, et al. A functional role for the two-pore domain potassium channel TASK-1 in cerebellar granule neurons. Proc Natl Acad Sci USA. 2000;97:3614–3618. doi: 10.1073/pnas.050012597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missan S, Lindsell P, McDonald TF. Tyrosine kinase and phosphatase regulation of slow delayed-rectifier K+ current in guinea-pig ventricular myocytes. J Physiol. 2006;573:469–482. doi: 10.1113/jphysiol.2005.104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER, Grant SG. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- Ogata R, Kitamura K, Ito Y, Nakano H. Inhibitory effects of genistein on ATP-sensitive K+ channels in rabbit portal vein smooth muscle. Br J Pharmacol. 1997;122:1395–1404. doi: 10.1038/sj.bjp.0701532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart C, Carlier E, Guedin D, Dargent B, Couraud F. Direct block of voltage-sensitive sodium channels by genistein, a tyrosine kinase inhibitor. J Pharmacol Exp Ther. 1997;280:521–526. [PubMed] [Google Scholar]
- Park D, Huang T, Frishman WH. Phytoestrogens as cardioprotective agents. Cardiol Rev. 2005;13:13–17. doi: 10.1097/01.crd.0000126084.68791.32. [DOI] [PubMed] [Google Scholar]
- Patel A, Honore E. Properties and regulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel A, Lazdunski M, Honore E. Lipid and mechano-gated 2P domain K+ channels. Curr Opin Cell Biol. 2001;13:422–427. doi: 10.1016/s0955-0674(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, et al. A mammlian two pore domain mechanogated S-type K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzke C, Wemhöner K, Sachse FB, Rinne S, Schlichthörl G, Li XT, et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. 2007;75:59–68. doi: 10.1016/j.cardiores.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SAN. Sumoylation silences the plasma membrane leak channel K2P1. Cell. 2005;121:37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Richter TA, Dvoryanchikov GA, Chaudhari N, Roper SD. Acid-sensitive two-pore domain (K2P) channels in mouse taste buds. J Neurophysiol. 2004;92:1928–1936. doi: 10.1152/jn.00273.2004. [DOI] [PubMed] [Google Scholar]
- Sarker FH, Li Y. Using chemoprotective agents to enhance the efficacy of cancer therapy. Cancer Res. 2006;66:3347–3350. doi: 10.1158/0008-5472.CAN-05-4526. [DOI] [PubMed] [Google Scholar]
- Schröder F, Klein G, Frank T, Bastein M, Indris S, Karck M, et al. Src family tyrosine kinases inhibit single L-type Ca2+ channel activity in human atrial myocytes. J Mol Cell Cardiol. 2004;37:735–745. doi: 10.1016/j.yjmcc.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Thomas D, Wendt-Nordahl G, Röckl K, Ficker E, Brown AM, Kiehn J. High affinity blockade of HERG human cardiac potassium channels by the novel antiarrhythmic drug BRL-32872. J Pharmacol Exp Ther. 2001;297:735–761. [PubMed] [Google Scholar]
- Thomas D, Zhang W, Karle CA, Kathöfer S, Schöls W, Kübler W, et al. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J Biol Chem. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- Veale EL, Kennard LE, Sutton GL, MacKenzie G, Sandu C, Mathie A. Gαq-mediated regulation of TASK3 two-pore domain potassium channels: The role of protein kinase C. Mol Pharmacol. 2007;71:1666–1675. doi: 10.1124/mol.106.033241. [DOI] [PubMed] [Google Scholar]
- Wischmeyer E, Döring F, Karschin A. Acute suppression of inwardly rectifying Kir2.1 channels by direct tyrosine kinase phosphorylation. J Biol Chem. 1998;273:34063–34068. doi: 10.1074/jbc.273.51.34063. [DOI] [PubMed] [Google Scholar]
- Yu HG, Lu Z, Pan Z, Cohen IS. Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflugers Arch. 2004;447:392–400. doi: 10.1007/s00424-003-1204-y. [DOI] [PubMed] [Google Scholar]
- Zhou SS, Zhang LB, Sun WP, Xiao FC, Zhou YM, Li YJ, et al. Effects of monocarboxylic acid-derived Cl−channel blockers on depolarization-activated potassium currents in rat ventricular myocytes. Exp Physiol. 2007;92:549–559. doi: 10.1113/expphysiol.2007.037069. [DOI] [PubMed] [Google Scholar]