

Abstract
4-Alkylidene-β-lactones (hetero ketene dimers) and α-amino acids are useful precursors for total syntheses of the β-lactone containing proteasome inhibitors, salinosporamide A, cinnabaramide A, and derivatives. A key step is a nucleophile-promoted, bis-cyclization of keto acids that simultaneously generates the γ-lactam-and β-lactone of these natural products. This reaction sequence may have implications for the biosynthesis of these natural products.
Omuralide (1), derived from lactacystin (2)1 the salinosporamides e.g. salinosporamide A (3),2 and the recently disclosed cinnabaramides e.g. cinnabaramide A (4)3 are unique bicyclic β-lactone-containing natural products of bacterial origin that have recently attracted intense interest from both the synthetic and biological perspectives (Figure 1). Several synthetic efforts including numerous total syntheses of omuralide1 and three syntheses of salinosporamide A4 attest to the interest in these novel proteasome inhibitors due to their highly functionalized [3.2.0] bicyclic core and due to the validation of this therapeutic target for cancer.5 Recent crystallographic studies have elucidated fascinating details regarding inhibition of the 20S proteasome by salinosporamide A involving acylation of the active site threonine by the β-lactone with concomitant cyclization of the incipient alkoxide with the C13 chloro substituent leading to a tetrahydrofuran.6 Salinosporamide is currently in phase I human clinical studies for multiple myeloma.
Figure 1.
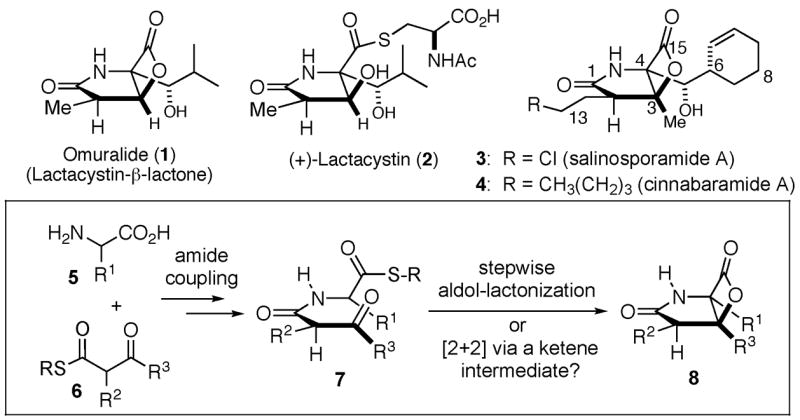
Structures of proteasome inhibitors and a possible biosynthetic origin for the γ-lactam-fused-β-lactone core.
We previously reported a catalytic, asymmetric intramolecular, nucleophile catalyzed aldol-lactonization (NCAL) process employing aldehyde acids that allows access to carbocycle-fused-β-lactones7 and this process was recently extended to keto acid substrates.8 This methodology was initially inspired by omuralide which contains such a bicyclic β-lactone core. Regarding the biosynthesis of these metabolites, one could speculate the joining of an appropriate amino acid 5 with an activated β-keto ester 6 followed by either an aldol-lactonization sequence9 or a [2+2] cycloaddition via a ketene intermediate, a mechanism commonly invoked for related bis-cyclizations (Figure 1).10
Building on our work with carbocycle-fused-β-lactones, we envisioned a concise synthetic strategy to the bicyclic core of these natural products by simultaneous formation of the C-C and C-O bonds from a keto acid precursor 10 via an intramolecular bis-cyclization process (Figure 2, 10→9). Attachment of the cyclohexenyl moiety, or other side-chains, would rely on the strategy of Corey developed in the course of their salinosporamide synthesis on simpler aldehyde γ-lactam precursors11 This would entail addition of a cyclohexenyl zinc reagent to the aldehyde derived from benzyl ether 9, however the success of this process and subsequent manipulations was not guaranteed given the presence of the β-lactone.12 The keto acid substrate 10 could be derived from coupling of an α-amino acid 11 and a ketene dimer 12, the latter serving as a suitable latent equivalent for a β-ketoester.
Figure 2.
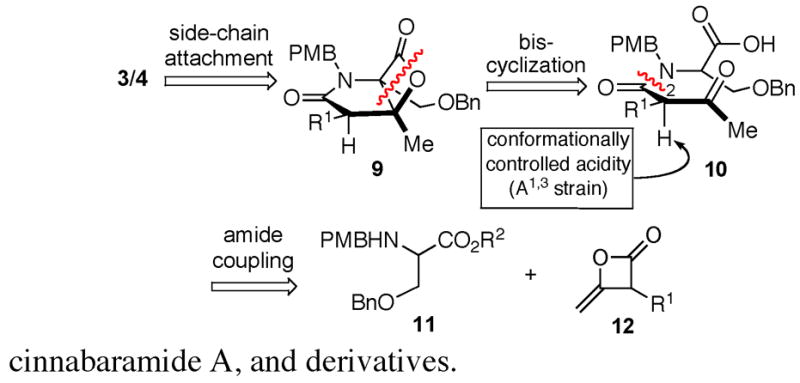
Retrosynthetic analysis of salinosporamide A, cinnabaramide A, and derivatives.
Ultimately, we sought the development of an asymmetric strategy. However, one difficulty to be overcome was the potential for enolization of the substrate β-ketoamide rendering the ketone non-electrophilic and most importantly, the possibility of rapid racemization at C2. However, due to the known conformationally controlled acidity of β-ketoamides owing to A1,3 strain,13 retention of optical activity appeared plausible. Herein we describe the implementation of the first goal of this strategy; namely, the bis-cyclization process which has led to concise total syntheses of rac-salinosporamide A (3), rac-cinnabaramide A (4), and derivatives.
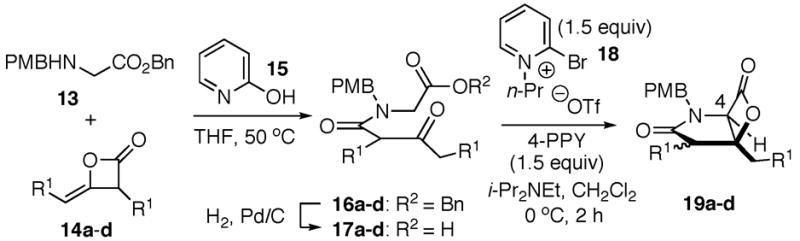
We began our studies with simple C2 unsubstituted substrates which were readily prepared by coupling of racemic ketene homodimer 14a14 with N-PMB-glycine benzyl ester (13a) by the method of Calter,15 which proceeded efficiently to provide keto acid substrate 17a following hydrogenolysis. We were pleased to find that bis-cyclization employing conditions similar to those developed for carbocycles,8 using 4-pyrrolidinopyridine (4-PPY) as nucleophilic promoter, proceeded efficiently to give bicyclic-β-lactones 19a–d (Table 1). However, the C2-unsubstituted ketoamide 17d gave only 25% yield (entry 4). Without the C2-substitutent, facile enolization of the ketoamide likely leads to diminished rates of the initial aldol step. Interestingly, increased diastereoselectivity is obtained during bis-cyclization if the reaction is performed at 25 °C for extended times (1.5 d) by selective degradation of the minor diastereomer (confirmed by 1H NMR reaction monitoring).
Table 1.
Synthesis of simplified, C4-unsubstituted salinosporamide/cinnabaramide derivatives 19a–d
 | ||||
|---|---|---|---|---|
| Entry | R1 | % yield (17)a,b | % yield (19)b | dr c |
| 1 | CyCH2 | 84 (17a) | 93 (19a) | 2.2:1 |
| 2 | nHexyl | 80 (17b) | 90 (19b) | 2.2:1 |
| 3 | PhCH2 | 72 (17c) | 85 (19c) | 2.5:1 (>19:1)d |
| 4 | H | 77 (17d) | 25 (19d) | - |
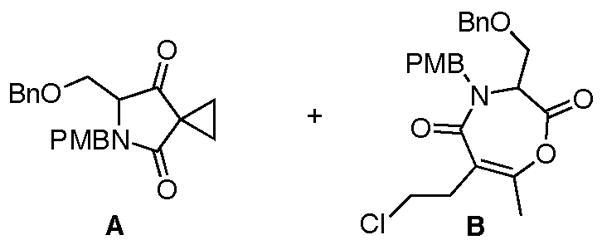
Yield is for 2 steps.
Yields refer to isolated, purified (SiO2) product.
Determined by 1H NMR analysis of crude reaction mixtures.
Observed diastereomeric ratio (dr) if reaction is allowed to proceed at 25 °C for 1.5 d (54% yield). PMB = p-methoxybenzyl, 4-PPY = 4-pyrrolidinopyridine, Cy = cyclohexyl.
A likely mechanistic pathway for this bis-cyclization building on our related work with carbocycle-fused-β-lactones8 involves initial activation of the carboxylic acid as pyridone ester 20 with modified Mukaiyama reagent 18. Following transacylation with 4-PPY, deprotonation by Hünig’s base leads to ammonium enolate 21. Subsequent net aldol-lactonization via aldolate 22 then provides the γ-lactam-fused-β-lactone 19a with concomitant regeneration of the nucleophilic promoter, 4-PPY. However, a [2+2] cycloaddition mechanism via an intermediate ketene has not been excluded at this time.16 The relative configuration of the major diastereomeric β-lactone 19a was confirmed by X-ray analysis following cleavage of the PMB group with ceric ammonium nitrate (CAN) to provide amide 23a (Scheme 1). Importantly, the relative configuration corresponds to that found in the salinosporamides and the cinnabaramides.
Scheme 1.
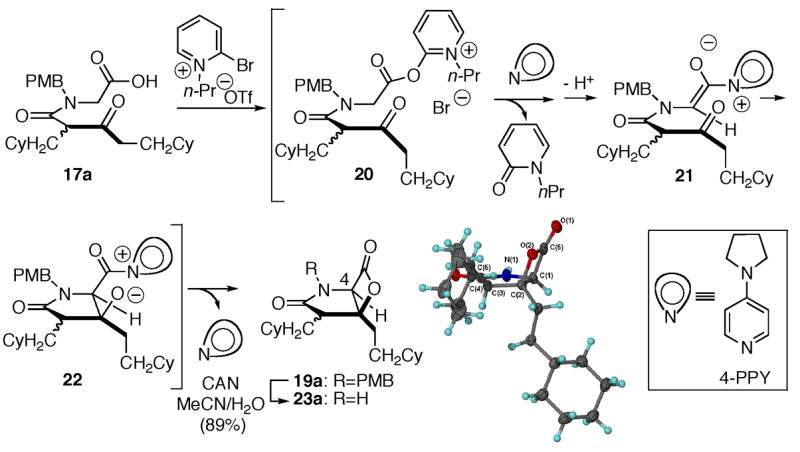
Proposed mechanism for the bis-cyclization and X-ray structure of β-lactone 23a
We next studied the impact of a C2 substituent during the bis-cyclization by targeting the synthesis of cinnabaramide A (4). The synthesis commenced by reductive amination of commercially available O-benzyl-L-serine with p-anisaldehyde (Scheme 2). Subsequent esterification provided the protected serine derivative 11a in 58% overall yield (2 steps). The required unsymmetrical ketene dimer 12a was obtained by heterodimerization of acetyl and octanoyl chlorides.14a Coupling of ketene dimer 12a with (L)-N-PMB-serine 11a gave diastereomeric esters 25 (dr, 1:1) and subsequent Sn-mediated hydrolysis17 provided acids 26 in good overall yield. The key bis-cyclization provided diastereomeric β-lactones 27/28 in 45% yield with moderate diastereoselectivity (dr 3.3:1), however the major diastereomer corresponded to that found in the natural product as verified by nOe analysis.18 Deprotection of the benzyl ether enabled separation of the major alcohol diastereomer 29 (79%, dr >19:1) and this was followed by Parikh-Doering oxidation19 to give an intermediate aldehyde which was used directly in the next step. Applying the method developed by Corey with zinc reagent 304a gave alcohols 31 in 57% yield (2 steps, dr 4.7:1). Finally oxidative cleavage of the PMB group gave rac-cinnabaramide A (4) which could be isolated diastereomerically pure in 48% yield. Spectral data for the synthetic material correlated with the published data.3 Further verification of relative configuration was accomplished by X-ray analysis.18
Scheme 2.
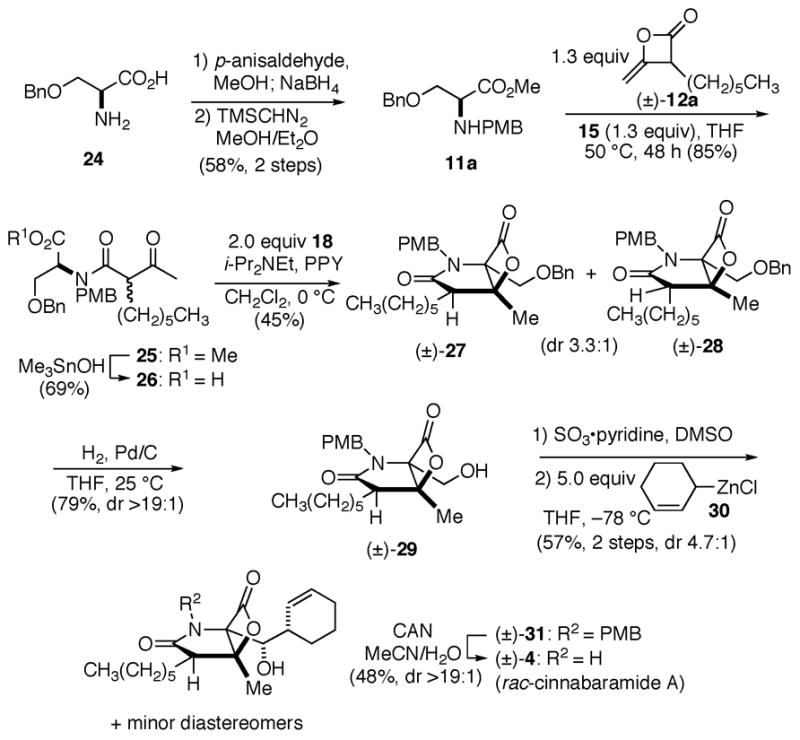
Total synthesis of rac-cinnabaramide A (4)
To further validate the mildness of this strategy, we targeted the synthesis of salinosporamide A bearing the required chloro-substituent in the keto acid substrate. In this case, N-PMB serine allyl ester 11b, available in 2 steps from serine, was utilized to enable mild ester deprotection since cyclopropane formation was observed during attempted saponification of the corresponding keto methylester (not shown, cf. 25) (Scheme 3). The amine 11b was coupled with heteroketene dimer 12b, readily available in gram quantities from heterodimerization of acetyl chloride and commercially available 4-chlorobutanoyl chloride,18 to provide keto acids 3 3 following Pd-mediated ester deprotection. Bis-cyclization provided bicyclic-β-lactones 34/35 in 25-35% yield (dr 2-3:1) favoring the relative configuration found in salinosporamide A.20 Deprotection of the benzyl ether enabled enrichment of the major diastereomer to 6-10:1 upon purification. Modified Moffatt21 oxidation using 1-(1,3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDCI)22 and dichloroacetic acid23 followed by addition of zinc reagent 30 gave predominantly two diastereomeric alcohols 37 (dr 3.5:1) in 33% yield (2 steps).24 Final deprotection of the PMB group enabled isolation of diastereomerically pure rac-salinosporamide A, which correlated with the published data and the relative configuration was further confirmed by X-ray analysis.18
Scheme 3.
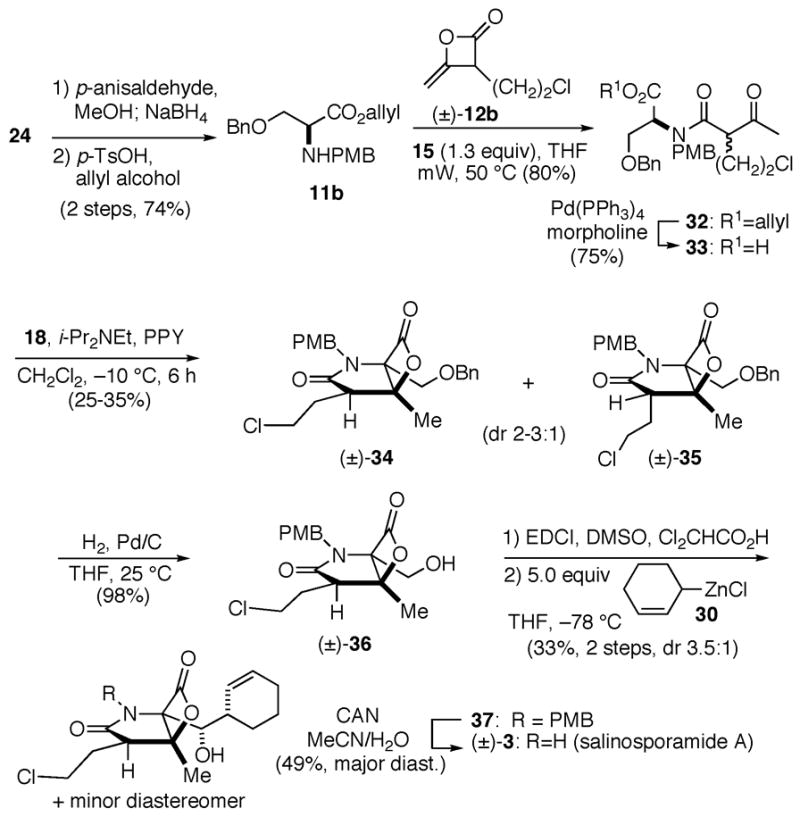
Total synthesis of rac-salinosporamide A (3)
In summary, we have developed concise synthetic routes to rac-salinosporamide A, rac-cinnabaramide A, and simplified derivatives. This strategy is unique in enabling simultaneous construction of both the γ-lactam and fused-β-lactone found in these metabolites via a bis-cyclization process. The β-lactone in these systems and the chloro-substituent in salinosporamide precursors is shown to be tolerant to several transformations which contributes to the brevity of the sequence.1c The described bis-cyclization process points to a logical biosynthetic origin for these intriguing natural products and raises the interesting question of whether such a bis-cyclization might be involved in the biosynthesis of these natural products.9 Further optimization of this process including mechanistic studies and extension to an enantioselective strategy premised on A1,3-strain in keto acid precursors, e.g. ketoacids 25 and 33, constitute our ongoing efforts in this area.
Supplementary Material
General procedures for ketene-dimerizations, bis-cyclizations and subsequent transformations with characterization data (including 1H and 13C NMR spectra) for β-lactones 3, 4, 19a–d, 23a, 27, 29, 31, 34, 36, 37 and products 11a–b, 12a–b, 14b, 16a–c, 17a–d, 25, 26, 32, 33. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the NIH (GM069784), the Welch Foundation (A-1280), and Pfizer for support of these investigations. We thank Dr. Joe Reibenspies (TAMU) for X-ray analysis and Prof. Bill Fenical (Scripps Inst. of Oceanography/UC San Diego) for a 1H NMR spectrum of natural salinosporamide A.
References
- 1.For reviews on lactacystin/omuralide syntheses, see: Corey EJ, Li WDZ. Chem Pharm Bull. 1999;47:1. doi: 10.1248/cpb.47.1.Masse CE, Morgan AJ, Adams J, Panek JS. Eur J Org Chem. 2000:2513.For a lead reference to more recent syntheses, see: Balskus EP, Jacobsen EN. J Am Chem Soc. 2006;128:6810. doi: 10.1021/ja061970a.
- 2.Isolation: Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical WF. Angew, Chem Int Ed. 2003;42:355. doi: 10.1002/anie.200390115.Analog Synthesis: Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BCM. J Med Chem. 2005;48:3684. doi: 10.1021/jm048995+.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical WF. J Org Chem. 2005;70:6196. doi: 10.1021/jo050511+.Reed KA, Manam RR, Mitchell SS, Xu J, Teisan S, Chao TH, Deyanat-Yazdi G, Neuteboom STC, Lam KS, Potts BCM. J Nat Prod. 2007;70:269. doi: 10.1021/np0603471.
- 3.Stadler M, Bitzer J, Mayer-Bartschmid A, Muller H, Benet-Buchholz J, Gantner F, Tichy HV, Reinemer P, Bacon KB. J Nat Prod. 2007;70:246. doi: 10.1021/np060162u. [DOI] [PubMed] [Google Scholar]
- 4.Reddy LR, Saravanan P, Corey EJ. J Am Chem Soc. 2004;126:6230. doi: 10.1021/ja048613p.Reddy LR, Fournier JF, Reddy BVS, Corey EJ. Org Lett. 2005;7:2699. doi: 10.1021/ol0508734.Endo A, Danishefsky SJ. J Am Chem Soc. 2005;127:8298. doi: 10.1021/ja0522783.Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2006;4:2845. doi: 10.1039/b607109k.For a study towards salinsporamide A, see: Caubert V, Langlois N. Tetrahedron Lett. 2006;47:4473.
- 5.a) Voorhees PM, Dees EC, O’Neil B, Orlowski RZ. Clin Cancer Res. 2003;9:6316. [PubMed] [Google Scholar]; b) Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J Clin Oncol. 2005;23:630. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]; c) Joazeiro CAP, Anderson KC, Hunter T. Cancer Res. 2006;66:7840. doi: 10.1158/0008-5472.CAN-06-2033. [DOI] [PubMed] [Google Scholar]
- 6.Groll M, Huber R, Potts BCM. J Am Chem Soc. 2006;128:5136. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 7.a) Cortez GS, Tennyson R, Romo D. J Am Chem Soc. 2001;123:7945. doi: 10.1021/ja016134+. [DOI] [PubMed] [Google Scholar]; (b) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]
- 8.Henry-Riyad H, Lee CS, Purohit VC, Romo D. Org Lett. 2006;8:4363. doi: 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]
- 9.While our work was in progress, a related biosynthetic pathway was proposed: Moore, B. S., International Conference on Marine Natural Products, Paris, France, Sept. 2005 and recently appeared, see: Beer LL, Moore BS. Org Lett. 2007:9, 845. doi: 10.1021/ol063102o.
- 10.For previous reports of β-lactones from keto acid derivatives via proposed [2+2] mechanisms, see: Boswell GA, Dauben WG, Ourisson G, Rull T. Bull Soc Chim Fr. 1958:1598.Kagan HB, Jacques J. Bull Soc Chim Fr. 1958:1600.Brady WT, Gu YQ. J Org Chem. 1988;53:1353.Reddy LR, Corey EJ. Org Lett. 2006;8:1717. doi: 10.1021/ol060464n.For a previous report of an aldol-lactonzation pathway, see: Merlic CA, Marlog BC. J Org Chem. 2003;68:6056. doi: 10.1021/jo034476n.
- 11.For development of a strategy for attachment of the cyclohexenyl sidechain, see ref. 4a.
- 12.Jacobsen and coworkers had previously demonstrated the stability of a related spiro-β-lactone in their studies toward omuralide (see ref. 1c).
- 13.Evans DA, Ennis MD, Le T. J Am Chem Soc. 1984;106:1154. [Google Scholar]
- 14.a) Sauer JC. J Am Chem Soc. 1947;69:2444. [Google Scholar]; b) Purohit VC, Richardson RD, Smith JW, Romo D. J Org Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]; c) Duffy RJ, Morris KA, Romo D. J Am Chem Soc. 2005;127:16754. doi: 10.1021/ja053478h. [DOI] [PubMed] [Google Scholar]
- 15.Calter MA, Orr RK, Song W. Org Lett. 2003;5:4745. doi: 10.1021/ol0359517. [DOI] [PubMed] [Google Scholar]
- 16.In addition to previous studies with carbocycle-fused β-lactones which clearly point to participation of the nucleophile in these bis-cyclization reactions, lower conversions were obtained with less nucleophilic promoters e.g. dimethylaminopyridine, suggestive of nucleophile involvement in the rate-determining or prior step.
- 17.Furlám RLE, Emesto G, Mata EG, Masearetti OA. Tetrahedron. 1998;54:13023. [Google Scholar]
- 18.See Supporting Information for experimental details.
- 19.Parikh JP, Doering WE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
-
20.Lower yields in this bis-cyclization and that leading to cinnabaramide A (26→27, 28) in comparison with C4-unsubstituted substrates (Table 1) are clearly a result of increased steric issues during the bis-cyclization. While no starting material is recovered, two major byproducts have been identified, A and B (20–30% combined yield).
- 21.Pfitzner KE, Moffatt JG. J Am Chem Soc. 1965;87:5661. [Google Scholar]
- 22.Coulton S, Southgate R. J Chem Soc Perkin Trans I. 1992:961. [Google Scholar]
- 23.Akahoshi F, Ashimori A, Sakashita H, Yoshimura T, Imada T, Nakajima M, Mitsutomi N, Kuwahara S, Ohtsuka T, Fukaya C, Miyazaki M, Nakamura N. J Med Chem. 2001;44:1286. doi: 10.1021/jm000496v. [DOI] [PubMed] [Google Scholar]
- 24.Yields in this 2 step process are lower due to incomplete oxidation and inability to purify the aldehyde due to some sensitivity of this intermediate. Diastereoselectivity is considerably lower than that reported previously (see refs. 4a–c) however this appears to be highly substrate dependent given that Danishefsky observed reduced diastereoselectivity with N-unprotected substrates (ref. 4c).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General procedures for ketene-dimerizations, bis-cyclizations and subsequent transformations with characterization data (including 1H and 13C NMR spectra) for β-lactones 3, 4, 19a–d, 23a, 27, 29, 31, 34, 36, 37 and products 11a–b, 12a–b, 14b, 16a–c, 17a–d, 25, 26, 32, 33. This material is available free of charge via the Internet at http://pubs.acs.org.