

Abstract
Previous studies showed that Fanconi anemia (FA) murine stem cells have defective reconstitution after bone marrow (BM) transplantation. The mechanism underlying this defect is not known. Here, we report defective homing of FA patient BM progenitors transplanted into mouse models. Using cells from patients carrying mutations in FA complementation group A (FA-A), we show that when transplanted into nonobese diabetic/severe combined immunodeficiency (NOD/SCID) recipient mice, FA-A BM cells exhibited impaired homing activity. FA-A cells also showed defects in both cell-cell and cell-matrix adhesion. Complementation of FA-A deficiency by reexpression of FANCA readily restored adhesion of FA-A cells. A significant decrease in the activity of the Rho GTPase Cdc42 was found associated with these defective functions in patient-derived cells, and expression of a constitutively active Cdc42 mutant was able to rescue the adhesion defect of FA-A cells. These results provide the first evidence that FA proteins influence human BM progenitor homing and adhesion via the small GTPase Cdc42-regulated signaling pathway.
Introduction
The hematologic complications of Fanconi anemia (FA) are nearly universal among patients with FA. Progressive bone marrow (BM) failure represents a hallmark of the disease and is the leading cause of patient death.1,2 Allogeneic hematopoietic stem cell (HSC) transplantation from related and unrelated donors is the only known curative therapy for severe BM failure in patients with FA.3,4 While outcomes from related donors seem to be good,4 the use of unrelated donors as the HSC source has had limited success with poor survival due to graft rejection, graft-versus-host disease (GVHD), and treatment-related toxicities. The recent addition of fludarabine to the transplant preparative regimen for patients undergoing unrelated donor HSC transplantation has significantly reduced these problems, probably as a result of greatly enhanced immune suppression without overlapping toxicities.5,6 Gene therapy is considered a promising approach to the treatment of BM failure in FA because reconstitution of a specific FA gene may allow correction of cellular phenotypes of FA HSCs in a complementation group-specific fashion. However, several clinical gene therapy trials in patients with FA have failed to show sustained engraftment of HSC and progenitor cells transduced with the FANCA7 or FANCC8 gene in patients carrying inactivating mutations in the respective FA gene. Several studies have found very low numbers of CD34+ HSCs in the BM and granulocyte colony-stimulating factor (G-CSF)–mobilized peripheral blood (PB) in patients with FA.7,9 In addition, studies in several FA gene-targeted mouse knockout models have shown that FA HSC and progenitor cells have high rates of stress-induced apoptosis and reduced repopulating capacity.10–17 Together, these data from humans and mouse models suggest that FA HSCs/progenitor cells have greatly reduced quality and quantity.
Here, we demonstrate that hematopoietic progenitor cells from patients carrying mutations in the FA complementation group A (FA-A) are defective in cell adhesion and BM homing in xenogeneic mouse models. These defects are attributable, at least in part, to the altered Cdc42 GTPase activity that is subjected to FANCA regulation. These results provide strong evidence that in addition to maintaining genomic stability, FA proteins may function to regulate cell adhesion and homing.
Methods
Patients, cell culture, and retroviral constructs
Bone marrow (BM) cells were collected from patients with FA-A (aged 6-21 years) enrolled in the Fanconi Anemia Comprehensive Care Center at Cincinnati Children's Hospital Medical Center (CCHMC) and healthy non-FA human volunteers (aged 27-34 years) according to the Declaration of Helsinki and protocols approved by the Institutional Review Board of CCHMC. Mononuclear cells were cultured in Iscove modified Dulbecco medium (IMDM) containing 10% fetal calf serum (FCS) and cytokines (100 ng/mL stem cell factor [SCF], 50 ng/mL G-CSF, and 50 ng/mL thrombopoietin [TPO]; all from PeproTech, Rocky Hill, NJ). The human FANCA cDNA18 was cloned into the S11FAIEGnls vector with enhanced green fluorescent protein (eGFP) expression as described previously.6 The Cdc42(F28L) cDNA, a Cdc42 mutant that constitutively exchanges GDP for GTP but still hydrolyzes GTP,19 was subcloned into the BamH1 and EcoR1 sites of the MIEG3 retroviral vector.
Cell-cell adhesion and cell-matrix adhesion and migration assays
Cell-cell adhesion was performed by adding 5 × 105 BM cells from each healthy donor and patient with FA-A onto confluent stromal monolayers. The cocultures were incubated for 4 hours, and the adherent cells were subjected to colony-forming cell (CFC) assay. Progenitor adhesion was expressed as a percentage of the input CFCs. For cell-matrix adhesion assay, 105 BM cells or 5 × 105 lymphoblasts were added to non–tissue culture plates coated with 25 μg/mL recombinant fibronectin fragment CH-296 (TakaraBio, Otsu, Japan). After a 2-hour incubation, adherent cells were harvested and quantified. For migration, 2 × 105 cells were added to the upper chamber of a transwell plate with a 5-μm pore size filter (Costar, Cambrige, MA), and 600 μL of chemotaxis buffer with 100 ng/mL SDF-1α (PeproTech, Rocky Hill, NJ) was added to the lower chamber. The cells that migrated against an SDF-1α gradient through the filter into the lower chamber were quantified 5 hours later.
Analysis of murine BM for human progenitor cell homing
A total of 6 to 10 × 106 BM mononuclear cells from healthy donors and patients with FA-A were intravenously injected into nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice. After 16 hours, the recipient mice were killed and BM cells were set up in progenitor CFC assays in methylcellulose medium (MethoCult GF H4434; StemCell Technologies, Vancouver, BC). These conditions were selective for human colonies because no colony growth was detected using BM from phosphate-buffered saline (PBS)–injected mice. BM homing of human GM-CFCs was expressed as a percentage of the number of GM-CFCs infused. In another set of homing assays, human BM cells were labeled with allophycocyanin (APC)–conjugated anti–human CD45 (clone HI30), PE-conjugated anti–human CD34 (clone 563), and 7-AAD (all from BD Pharmingen, San Jose, CA), and analyzed by flow cytometry. BM homing of human CD34+ progenitors was expressed as a percentage of the number of CD34+ BM cells infused. The studies involving the use of mice were conducted in accordance to the guidelines of and approved by the Institutional Animal Care and Use Committee (IACUC) of CCHMC (IACUC protocol no. 6C06041; principal investigator, Q.P.).
Assay for Cdc42 and Rac1 GTPase activities
Levels of active Cdc42 and Rac1 were determined by the GST-PAK1 pull-down assay described previously.20
Results and discussion
While clinical studies in BM transplantation settings4–6 suggest that normal HSC/progenitor cells could effectively engraft to FA BM under certain immunosuppressive conditioning, mechanistic studies on whether FA HSC/progenitor cells have cell-autonomous defects in hematopoietic engraftment remain lacking. Previously, we and others have demonstrated that FA hematopoietic progenitor cells show poor engraftment in mouse knockout models.10–17 In an effort to understand the mechanisms contributing to these engraftment defects, we have examined the homing activity, one of the early steps of FA BM cell engraftment, in vivo. We have found that relatively very low numbers of CD34+ cells in BM and mobilized PB were available in children with FA even before severe marrow failure.7 Given the inability to collect quantities of primitive HSCs sufficient to conduct the experiments described in this report, we have instead used progenitor cells harvested from FA BM. We transplanted BM mononuclear cells from healthy donors or patients with FA-A into sublethally irradiated NOD/SCID mice. The percentages of transplanted BM progenitors homing (expressed as a percentage of the total GM-CFCs input) to the BM of recipient mice were reduced by 65% in FA-A compared with healthy donor BM (Figure 1A). Additional experiments with BM cells from 8 healthy donors and 8 patients with FA-A confirmed defective BM homing (expressed as the percentage of the total CD34+ cells input) of FA-A progenitors (Figure 1B). These results suggest that FA-A BM progenitors are impaired in homing ability in the mouse model.
Figure 1.

Impaired homing and adhesion of FA-A hematopoietic cells. (A) A total of 6 to 10 × 106 low-density BM mononuclear cells from healthy donors and patients with FA-A were injected into each sublethally irradiated NOD/SCID recipient. At 16 hours after the graft, donor-derived cells were isolated from each recipient mouse and colony (CFC) assays were performed to evaluate homing capacity of human hematopoietic progenitor cells. BM homing of human GM-CFCs was expressed as a percentage of the number of GM-CFCs infused. Results represent means plus or minus SD of 2 experiments (3 pairs of samples for each experiment and 4 recipient mice for each sample). *Difference between the FA-A and normal donors is significant at P < .05. (B) A total of 6 to 10 × 106 low-density BM mononuclear cells from each healthy donor (n = 8) and patient with FA-A (n = 8) were injected into each sublethally irradiated NOD/SCID mouse. At 16 hours after the graft, BM nucleated cells were isolated from each recipient mouse and labeled with anti human CD45-APC and CD34-PE as well as 7-AAD, followed by flow cytometry to evaluate the proportion of donor-derived human progenitor cells in the bone marrow. BM homing of human CD34+ progenitors was expressed as a percentage of the number of CD34+ BM cells infused. The horizontal bar represents the median. *Difference between the patients with FA-A and the healthy donors is significant at P < .05. (C) Cell-matrix adhesion: 105 low-density BM mononuclear cells isolated from each healthy donor and patient with FA-A were seeded into a 96-well plate coated with 20 μg/mL fibronectin CH-296. After a 2-hour incubation at 37°C, nonadherent cells were removed and adherent cells were detached and quantified. Data represent the mean plus or minus SD percentage of adhesion of 3 independent experiments in triplicates with samples from 3 healthy donors and 3 patients with FA-A. * Significance between the FA-A and normal cells at *P < .05. (D) Cell-cell adhesion: 5 × 105 BM mononuclear cells from each healthy donor and patient with FA-A were added onto confluent normal BM-derived stromal monolayers and incubated for 4 hours at 37°C; the adherent cells were then subjected to CFC assay (2 pairs of healthy donors and patients with FA-A). Numbers are given as the percentage of input CFCs (mean ± SD) with 2 independent experiments in duplicates in each experiment. **P < .01. (E) Western analysis of FANCA expression in transduced normal and FA-A lymphoblasts using an anti-FANCA antibody. (F) FA-A lymphoblasts were transduced with retrovirus expression eGFP-FANCA or eGFP alone. After fluorescence-activated cell sorting (FACS) sorting, 5 × 105 GFP+ cells together with untransduced normal and FA-A lymphoblasts were added into the 24-well plates coated with 20 μg/mL fibronectin and cultured for 2 hours. The nonadherent cells were removed, and adherent cells were detached and quantified. Shown is the mean plus or minus SD percentage of adhesion in 3 independent experiments. Fibronectin-mediated adhesion was significantly reduced in FA-A or eGFP (vector)–transduced FA-A cells compared with FANCA-complemented cells (**P < .01).
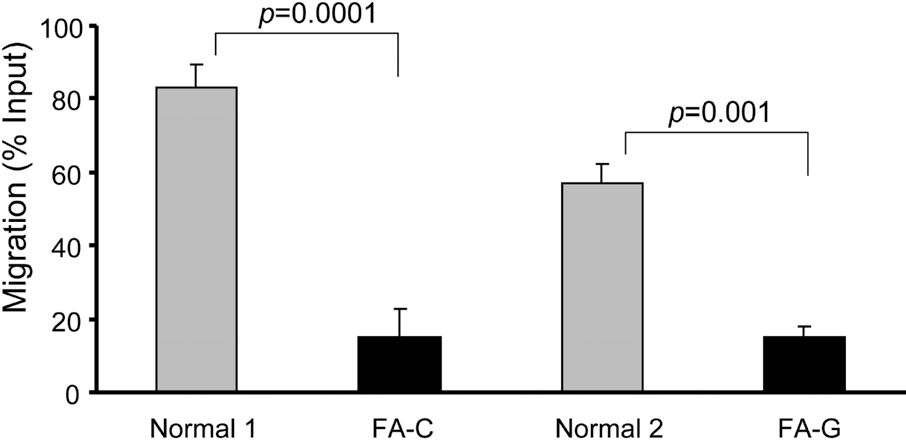
To associate the homing defect with the mechanistic steps involved in the progenitor-BM microenvironment interaction, we have examined whether there was a defect in adhesion of FA-A BM cells using both cell-cell and cell-matrix adhesion assays. Adhesion of BM cells from patients with FA-A to fibronectin was reduced by approximately 50% (mean adhesion values for BM cells from healthy donors was 18.6% (± 3.7%) in comparison with 9.1% (± 2.1%) for those from the patients with FA-A; Figure 1C). To examine whether the adhesion function was altered in FA progenitor cells, we carried out CFC assays using cells that had adhered to a healthy donor–derived stromal layer (Figure 1D). We found severe adhesion defects in cocultures of FA-A progenitor cells and normal BM-derived stroma (5.5% ± 0.6% CFC adhesion in normal cells compared with 1.7% ± 0.3% for FA-A samples). The observed defective adhesion is likely resulted from FA-A deficiency, as reexpression of FANCA in FA-A lymphoblasts (Figure 1E), but not eGFP alone, restored adhesion function to the wild-type level (31.8% ± 3.9% in FANCA-complemented cells compared with 13.4% ± 2.6% in FA-A cells transduced with eGFP-alone vector; Figure 1F). A similar defect in migration was also observed in FA-A cells (data not shown). Together, these results suggest that the impaired homing observed in FA-A BM cells could be related to a defect of the ability of FA-A cells to adhere to the appropriate BM niche.
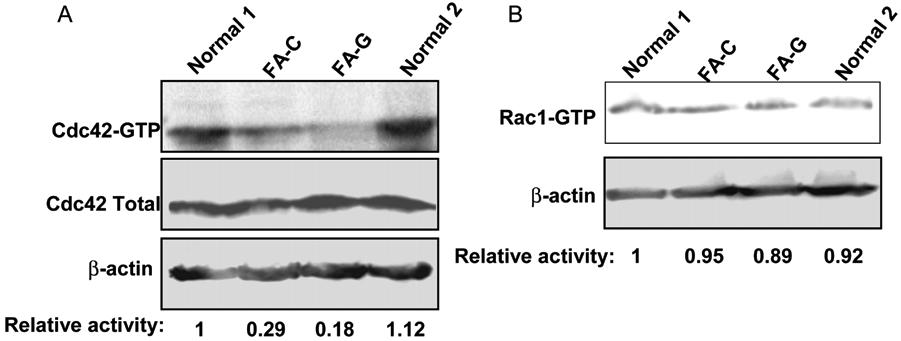
Because the Rho GTPase Cdc42 and Rac1 are critical regulators for cell adhesion and migration of many cell lineages, including hematopoietic stem/progenitors,21,22 we examined if their activities might be defective in FA-A cells. The active form of Cdc42-GTP was decreased by more than 50% in FA-A Epstein-Barr virus (EBV)–transformed lymphoblastoid cell lines compared with similar cell lines derived from healthy donor lymphocytes (Figure 2A). Complementation of FA-A lymphoblasts by a FANCA-encoding retrovirus completely restored Cdc42 activity. Interestingly, Rac1-GTP content of the FA-A cells showed a normal level similar to that of wild-type or FANCA-complemented cells (Figure 2B). Interestingly, FA-C and FA-G lymphoblasts behaved similarly in displaying significantly decreased Cdc42-GTP but not Rac1-GTP activity and the associated migration defects (Figures S1 and S2, available on the Blood website; see the Supplemental Materials link at the top of the online article). These results indicate that FA-A or other FA protein deficiencies in the FA patient samples is associated with a selective alteration in Cdc42 activity.
Figure 2.

Decreased Cdc42 activity in FA-A cells. (A) Levels of the active, GTP-bound Cdc42 (top panels) in lymphoblasts were determined by the effector-domain (GST-PAK1) pull-down assay followed by Western blotting using an antibody against Cdc42. The relative levels of active Cdc42 are indicated below each pull-down blot. The levels of total Cdc42 (middle panels) and β-actin (bottom panels) are shown as loading controls. (B) Levels of the active, GTP-bound Rac1 in lymphoblasts as determined by the effector-domain pull-down assay followed by Western blotting using an antibody against Rac1. The levels of total Rac1 and β-actin are shown as loading controls. (C) Expression of the constitutively active mutant of Cdc42 (Cdc42-F28L) containing an N-terminal HA tag in normal and FA-A lymphoblasts was analyzed by anti-HA Western blotting. (D) Forced expression of an active Cdc42 enhances adhesion of FA-A lymphoblasts. Untransduced, eGFP (vector)–transduced, or Cdc42-F28L–transduced normal or FA-A lymphoblast cells were subjected to adhesion assays. Data represent the mean plus or minus SD of 3 independent experiments in duplicates. *Difference between the Cdc42-F28L–transduced and untransduced (FA-A) or eGFP-transduced FA-A lymphoblasts is significant at P < .05.
We next determined if an increase of Cdc42 activity in FA-A cells could rescue the adhesion defect. We introduced a constitutively active form of Cdc42, Cdc42F28L,17 into FA-A lymphoblast cells (Figure 2C). Overexpression of the active Cdc42 mutant protein almost completely corrected the adhesion defect in FA-A lymphoblast cells (Figure 2D). Thus, Cdc42 down-regulation appears to account for most of the adhesion aberration in FA-A cells, although future validations in FA BM progenitors are needed to make these observations from lymphoblast cell lines therapeutically relevant.
This report shows for the first time that FA-A patient–derived hematopoietic progenitor cells possess defective adhesion and homing activities, and that these defects are associated with an aberrant regulation of Cdc42 activity. Our results implicate FA proteins in influencing hematopoietic cell homing, characterized by the regulation of cell adhesion, in addition to maintaining genomic stability. The mechanisms linking FA proteins with Cdc42 activity remain to be investigated; one possibility is that an indirect intracellular/extracellular signaling cascade involving tumor necrosis factor α (TNF-α), which is known to be up-regulated in patients with FA and is capable of down-regulating Cdc42 activity,23,24 contributes to the mechanistic modulation of FA cell behaviors.
Supplementary Material
Acknowledgments
We thank the patients with FA and their families for the donation of the samples, the Fanconi Anemia Comprehensive Care Center (FACCC), and Carrie Stevens at CCHMC for sample processing; Dr Jose Cancelas (Experimental Hematology, CCHMC) for advice on BM cells–BM stroma adhesion experiments and comments on the manuscript; Dr Lei Wang (Experimental Hematology, CCHMC) for technical assistance; the Translational Trials Development and Support Laboratory (Cincinnati Children's Research Foundation [CCRF], CCHMC) for bone marrow cell transduction; Jeff Bailey and Victoria Summey for bone marrow transplantation; and the Viral Vector Core of CCRF for the preparation of retroviruses.
This work was supported in part by National Institutes of Health grants R01 HL076712 (Q.P.), R01 HL085362 (Y.Z), R01 HL081499 (D.A.W), and by US Public Health Service grant MO1 RR 08084 (General Clinical Research Centers Program, National Center for Research Resources, National Institutes of Health). Q.P. is supported by a Leukemia & Lymphoma Society Scholar Award.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.Z. performed research, analyzed data, and wrote the paper; X.S., F.G., and K.M. performed research and analyzed data; M.K. performed research; P.K., L.R., and D.A.W. designed research; F.O.S. analyzed data and wrote the paper; and Y.Z. and Q.P. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qishen Pang or Yi Zheng, Division of Experimental Hematology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229; e-mail: qishen.pang@cchmc.org or yi.zheng@cchmc.org.
References
- 1.Bagby GC., Jr Genetic basis of Fanconi anemia. Curr Opin Hematol. 2003;10:68–76. doi: 10.1097/00062752-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy RD, D'Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 3.Rossi G, Giorgiani G, Comoli P, et al. Successful T-cell-depleted, related haploidentical peripheral blood stem cell transplantation in a patient with Fanconi anaemia using a fludarabine-based preparative regimen without radiation. Bone Marrow Transplant. 2003;31(6):437–40. doi: 10.1038/sj.bmt.1703903. [DOI] [PubMed] [Google Scholar]
- 4.Farzin A, Davies SM, Smith FO, et al. Matched sibling donor haematopoietic stem cell transplantation in Fanconi anaemia: an update of the Cincinnati Children's experience. Br J Haematol. 2007;136:633–640. doi: 10.1111/j.1365-2141.2006.06460.x. [DOI] [PubMed] [Google Scholar]
- 5.Gluckman E, Rocha V, Ionescu I, et al. Results of unrelated cord blood transplant in fanconi anemia patients: risk factor analysis for engraftment and survival. Biol Blood Marrow Transplant. 2007;13:1073–1082. doi: 10.1016/j.bbmt.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 6.Wagner JE, Eapen M, MacMillan ML, et al. Unrelated donor bone marrow transplantation for the treatment of Fanconi anemia. Blood. 2007;109:2256–2262. doi: 10.1182/blood-2006-07-036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly PF, Radtke S, Kalle C, et al. Stem cell collection and gene transfer in fanconi anemia. Mol Ther. 2007;15:211–219. doi: 10.1038/sj.mt.6300033. [DOI] [PubMed] [Google Scholar]
- 8.Liu JM, Kim S, Read EJ, et al. Engraftment of hematopoietic progenitor cells transduced with the Fanconi anemia group C gene (FANCC). Hum Gene Ther. 1999;10:2337–2346. doi: 10.1089/10430349950016988. [DOI] [PubMed] [Google Scholar]
- 9.Croop JM, Cooper R, Fernandez C, et al. Mobilization and collection of peripheral blood CD34+ cells from patients with Fanconi anemia. Blood. 2001;98:2917–2921. doi: 10.1182/blood.v98.10.2917. [DOI] [PubMed] [Google Scholar]
- 10.Haneline LS, Broxmeyer HE, Cooper S, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac−/− mice. Blood. 1998;91:4092–4098. [PubMed] [Google Scholar]
- 11.Haneline LS, Gobbett TA, Ramani R, et al. Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood. 1999;94:1–8. [PubMed] [Google Scholar]
- 12.Rathbun RK, Christianson TA, Faulkner GR, et al. Interferon-γ–induced apoptotic responses of Fanconi anemia group C hematopoietic progenitor cells involve caspase 8-dependent activation of caspase 3 family members. Blood. 2000;96:4204–4211. [PubMed] [Google Scholar]
- 13.Haneline LS, Li X, Ciccone SL, et al. Retroviral-mediated expression of recombinant Fancc enhances the repopulating ability of Fancc−/− hematopoietic stem cells and decreases the risk of clonal evolution. Blood. 2003;101:1299–1307. doi: 10.1182/blood-2002-08-2404. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Yang Y, Yuan J, et al. Continuous in vivo infusion of interferon-gamma (IFN-gamma) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in Fancc−/− mice. Blood. 2004;104:1204–1209. doi: 10.1182/blood-2004-03-1094. [DOI] [PubMed] [Google Scholar]
- 15.Si Y, Ciccone S, Yang FC, et al. Continuous in vivo infusion of interferon-gamma (IFN-γ) enhances engraftment of syngeneic wild-type cells in Fanca−/− and Fancg−/− mice. Blood. 2006;108:4283–4287. doi: 10.1182/blood-2006-03-007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Sejas DP, Qiu Y, Williams DA, Pang Q. Inflammatory reactive oxygen species promote and cooperate with Fanconi anemia mutation for hematopoietic senescence. J Cell Sci. 2007;120:1572–1583. doi: 10.1242/jcs.003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sejas DP, Rani R, Qiu Y, et al. Inflammatory reactive oxygen species-mediated hematopoietic suppression in Fancc-deficient mice. J Immunol. 2007;178:5277–5287. doi: 10.4049/jimmunol.178.8.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, et al. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet. 1996;14:320–323. doi: 10.1038/ng1196-320. [DOI] [PubMed] [Google Scholar]
- 19.Guo F, Zheng Y. Involvement of Rho family GTPases in p19Arf or p53 mediated proliferation of primary mouse embryonic fibroblasts. Mol Cell Biol. 2004;24:1426–1428. doi: 10.1128/MCB.24.3.1426-1438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Yang L, Burns K, Kuan C-Y, Zheng Y. Cdc42GAP regulates JNK-mediated apoptosis and cell number during mammalian perinatal growth. Proc Natl Acad Sci U S A. 2005;102:13484–13489. doi: 10.1073/pnas.0504420102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Yang L, Fillipi M, Williams DA, Zheng Y. Deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, migration, and engraftment. Blood. 2006;107:98–105. doi: 10.1182/blood-2005-05-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Wang L, Cancelas J, Geiger H, Mo J, Zheng Y. The Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. 2007;104:5091–5096. doi: 10.1073/pnas.0610819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosselli F, Sanceau J, Gluckman E, Wietzerbin J, Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia, II: in vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood. 1994;83:1216–25. [PubMed] [Google Scholar]
- 24.Peppelenbosch M, Boone E, Jones GE, et al. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: inhibition of Cdc42-mediated filopodium formation by TNF. J Immunol. 1999;162:837–845. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}