

Abstract
In order to acquire accurate structural and dynamical information on complex biomolecular machines using single-molecule fluorescence resonance energy transfer (sm-FRET), a large flux of donor and acceptor photons are needed. To achieve such fluxes one may use higher laser excitation intensity, however this induces increased rates of photobleaching. Anti-oxidants additives have been extensively used for reducing acceptor’s photobleaching. Here we focus on deciphering the initial step along the photobleaching pathway. Utilizing an array of recently-developed single molecule and ensemble spectroscopies, and doubly-labeled Acyl-CoA binding protein and double-stranded DNA as model systems, we study these photobleaching pathways which place fundamental limitations on sm-FRET experiments. We find that: (i) acceptor photobleaching scales with FRET efficiency; (ii) acceptor photobleaching is enhanced under picosecond pulsed (vs. continuous wave) excitation; and (iii) acceptor photobleaching scales with the intensity of only the short wavelength (donor) excitation laser. We infer from these findings that the main pathway for acceptor’s photobleaching is through absorption of a short wavelength photon from the acceptor’s first excited singlet state and that donor’s photobleaching is usually not a concern. We conclude by suggesting using short pulses for donor excitation, among other possible remedies, for reducing acceptor’s photobleaching in sm-FRET measurements.
Introduction
The application of single molecule fluorescence methods to life sciences has taken a stronghold in recent years1–6. In particular, Single-Molecule Fluorescence Resonance Energy Transfer (sm-FRET) has become a sensitive and powerful tool for determining bioconformational dynamics and biomolecular interactions via accurate measurements of inter- and intra-molecular distances5–8. Beyond measuring energy transfer efficiency, multiparameter fluorescence detection (MFD) can monitor and globally analyze additional fluorescence parameters such as emission intensities, lifetimes and anisotropies, providing further dynamic molecular information9–11. In order to achieve high temporal resolution and to minimize shot-noise in single molecule measurements, however, a relatively high excitation power is needed, which often leads to enhanced photobleaching12–14. As a result, considerable effort has been recently dedicated to optimizing conditions for sm-FRET experiments, mostly through the addition of anti-oxidents additives to the buffer solution for preventing/reducing acceptors’ photobleaching15–17. These works focused mostly on carbocyanine dyes (Cy5), which display excitation intensity dependent cis/trans isomerization using CW excitation. The timescales for the resulting blinking/switching behavior span microseconds to seconds, introducing significant noise to sm-FRET measurements18–20.
More insight to the problem of acceptor’s bleaching in sm-FRET experiments could be gained by delineating the actual photobleaching pathway and investigating where anti-oxidents additives act along it. It has been recently shown that short laser pulses (femtoseconds to picoseconds, and in some cases even continuous wave) could lead to the excitation of higher electronic states and accelerate photobleaching, limiting the number of emitted photons and thus, reducing the structural and temporal resolution and accuracy in single molecule FRET measurements21.
Here we implement novel photophysical tools to study the very initial step along the photobleaching pathway. We have recently introduced a variant of the MFD methodology, dubbed Alternating Laser EXcitation (ALEX), which is capable of revealing both structural and stoichiometric information5, 6, 22, 23. Of particular significance to this work, ALEX can efficiently separate single molecule events (photon bursts) with photo-active fluorophores from those with bleached fluorophores or incomplete labeling. Furthermore, varying the pulse durations and/or pulse separations in ALEX can also provide information on the acceptor’s photobleaching pathway.
We previously implemented ALEX with continuous wave (CW) lasers that were modulated on the microsecond timescale (tens to hundreds of kHz, μs-ALEX) using electro-optical modulators (EOM) combined with polarizers5, 22 or acousto-optic modulators (AOM)24. In these embodiments of μs-ALEX, modulation depths, or extinction ratios (ratio between the laser ‘on’ intensity and the laser ‘off’ intensity) of several hundreds were achieved. Because of the finite modulation rise and fall times (~3μs), however, it was necessary to add a delay of several microseconds between successive modulated excitation periods (with both lasers being in the off state during this delay)22.
A simplified implementation of μs-ALEX can be realized by direct modulation of novel solid-state lasers, dubbed hereafter direct modulation ALEX, or dm-ALEX. Here we modulated iPulse and iBeam lasers (Toptica Photonics, Westfield, MA) using a Digital to Analog PC board (PCI-6602, National Instruments, Austin, TX). This implementation achieved extinction ratios > 300 with rise and fall times of several tens of nanoseconds, eliminating the need for inserting a delay between modulation periods. With this direct modulation scheme, alternation frequencies of up to 100MHz can be attained, ~2 orders of magnitude higher than achievable with AOMs and EOMs.
For the photobleaching study described below, we combined dm-ALEX with another technique dubbed nanosecond-ALEX (ns-ALEX), in which picoseconds pulsed excitations are alternated on nanosecond timescales10 (similar to recently reported PIE technique25). Both dm-ALEX and ns-ALEX excitations were implemented on the same microscope with a common detection path, enabling a direct comparison between the two excitation schemes on the same samples and thus more reliable determination of the initial step along the acceptor bleaching process..
In order to probe the various potential pathways leading to acceptor photobleaching, single-molecule and ensemble measurements were performed using dm-ALEX, ns-ALEX and single laser excitations. Single molecule burst analysis and ensemble photon emission rate analysis were then applied to evaluate the contributions from different mechanisms along the bleaching process. Based on the major contributions we give out experimental suggestions for minimizing the acceptor photobleaching.
The Acceptor’s Bleaching Problem
In single molecule experiments, a trade-off is usually made between the photon emission rate (signal strength), photobleaching rate, and triplet blinking/saturation, all of which increase with laser excitation power. Photobleaching is the result of an increased reactivity of the fluorophore in its excited states, leading to irreversible loss of emission (fluorophore destruction), usually due to a permanent change in the chemical structure of the fluorophore (photochemical reaction). Photobleaching has long been one of the major impediments in single molecule spectroscopy. Although it has been demonstrated that anti-oxidant additives can reduce photobleaching16, 26, 27 by preventing oxidation when in triplet state, this is only a partial solution. Better understanding of the pathways leading to the fluorophore’s destruction should allow the optimization of experimental conditions without chemically altering the molecular environment and possibly future synthesis and high throughput screening of more resilient dyes.
In a single-molecule ALEX-FRET experiment, a macromolecule is doubly-labeled with a donor fluorophore (D) and an acceptor fluorophore (A), which differ in their emission wavelengths (Fig. 1). The donor, upon absorption of a blue photon, is excited from its ground to first excited singlet state ( ), and can then relax to its ground state either by emitting a photon or by non-radiative energy transfer to the nearby acceptor (FRET; Fig. 1A, green dashed arrows). The acceptor can be excited ( ) either directly by a red photon or via FRET, after which it has the choice of three different pathways: (1) relaxation to by emission of a red photon (downward red arrow); (2) absorption of another (blue or red) photon to reach a higher excited states (upward blue or orange arrows in the center); (3) inter-system crossing (ISC) to its triplet state followed by relaxation to its ground state by phosphorescence (black curved arrow) or absorption of another (blue or red) photon to reach a higher triplet excited states (upward blue and orange arrows on the right). Higher-order excited states are more sensitive to photochemical reactions, and can lead to isomerization that might result in blinking or irreversible bond breakage and hence cessation of emission (photobleaching). Several pathways can lead to such higher-energy excited states: (i) energy transfer to a high vibronic state of ; (ii) inter-system crossing ; (iii) excited-state absorption of a second (blue or red) photon from or . In the studies reported here, acceptors are subjected to direct blue and red excitations and to excitation via energy transfer and could therefore be bleached by a combination of different pathways. Donor bleaching was not observed in this study and therefore will be discussed only briefly.
Figure 1.

(A) Jablonski diagram of a donor-acceptor pair, adapted for FRET, and showing multiple possible pathways for acceptor photobleaching. The donor, upon excitation by a blue photon during the blue excitation period (upward blue arrow) reaches its first excited state and relaxes either by emitting a photon (downward solid green arrow) or by transferring its energy to the acceptor (dashed green curved arrow). The acceptor is excited from its ground state to its first excited state either by energy transfer from the donor, by a blue photon during the blue excitation period (not shown), or by a red photon during the red excitation period (upward orange arrow). The acceptor relaxes to its ground state by emitting a red photon (downward red arrow) or to its triplet state by intersystem crossing (horizontal black wavy arrow) followed by phosphorescence (downward black wavy arrow), or absorbs another blue or red photon to reach a higher excited state . The acceptor can also absorb another blue or red photon while in its triplet state, to reach a higher excited triplet state . (B) Excitation and emission spectra of Alexa Fluor 488 (blue, excitation; green, emission) and Alexa Fluor 647 (orange, excitation; red, emission). Excitation wavelengths for dm-ALEX and ns-ALEX were 467–470nm for blue (dashed blue line) and 635–640nm for red (dashed red line) respectively. The overlap between the donor’s emission and the acceptor’s absorption is highlighted by the hatched green area. (C) Structure diagram of ACBP, with the donor (Alexa Fluor 488) and acceptor (Alexa Fluor 647) labeled at position 17 and 86. (D) Schematic diagram of double-stranded DNA.
By implementing dm-ALEX and ns-ALEX on the same setup and exciting single-molecule and ensemble samples, we are able to separate subpopulation of molecules with photo-active acceptors from those with bleached acceptors. This constitutes a quantitative measure of the level of photobleaching, which we monitor as a function of excitation intensity, excitation duration (CW or pulsed), and time delay between blue and red excitations. We can therefore distinguish the contributions to acceptor bleaching from the blue laser versus the red laser, from pulsed excitation versus CW excitation, and from high FRET efficiency versus low FRET efficiency. We are then able to optimize the excitation parameters for a specific molecular construct, specific fluorophores and specific experimental conditions.
Materials and Methods
Photon Burst analysis
Individual molecules (bursts) were separated from the background signal using a previously described burst-search algorithm28. For each of these bursts, FRET efficiency (E) and stochiometry ratio (S) were calculated according to Eq.1 and Eq.2, respectively 23:
| (1) |
| (2) |
FFRET is the acceptor’s emission due to FRET, corrected for background, donor cross-talk and acceptor’s direct excitation (by the D-excitation wavelength) contributions; is the donor’s emission during the D-excitation period, corrected for background contribution; is the acceptor’s emission during the A-excitation period, corrected for background; and γ is the ratio of the donor and acceptor product of the detection efficiency and quantum yield. Fig. 2 summarizes in 2D E-S histograms simulations of freely diffusing doubly labeled single molecules24. Molecules with active D and A are identified as a subpopulation with corresponding E and S values determined by the distance between the labeling positions and the brightness of each fluorophore (Fig. 2A). Molecules with a bleached acceptor (identified as donor-only, or D-only subpopulation) have FFRET ≈ 0 and , and therefore appear on the 2D E-S histogram as a subpopulation with E value close to 0 and S value close to 1 (solid black square, Fig. 2B). When the acceptor’s photobleaching time constant is comparable to or shorter than the molecule’s transit (diffusion) time through the confocal excitation volume (0.2–2 ms, depending on molecular size and viscosity of the solution), the acceptor’s emission could cease as traversing the excitation volume (while leaving the donor in its active emitting state). Such events result in a burst with a reduced apparent acceptor brightness and are visualized as a smear-like “bridge” populating the area between the D-only (solid black square) and the FRET (dashed black square) subpopulations on the 2D histogram (Fig. 2B). In extracting bursts corresponding to a specific subpopulation we define the vertical (E) and horizontal (S) boundaries to minimize contamination from these “bridge” events24, so that D-only subpopulation corresponds only to molecules with an inactive acceptor during the whole bursts and the D-A subpopulation corresponds only to molecules with both the donor and the acceptor active during the whole bursts.
Figure 2.

Simulated single molecule 2D E-S ALEX histograms of (A) pure doubly-labeled FRET species (dashed black square) and (B) a scenario where the acceptor is partially bleached, showing donor only (solid black square), FRET species (dashed black square) and ‘bridge’ events.
In our study each identified burst represents the crossing of the confocal volume by a single molecule. If the donor and acceptor have the same emission rates (brightness) and the donor and the acceptor detection channel have the same detection efficiency (γ=1), the number of accumulated bursts per subpopulation will be proportional to that subpopulation’s concentration. Thus, a larger D-only subpopulation indicates a higher level of acceptor bleaching. In our analyses we infer the degree of acceptor photobleaching by counting the number of D-only bursts and calculating their fraction in the whole sample.
dm-ALEX
μs-ALEX was previously implemented using two CW lasers with external EOMs22 or AOMs24. In the first case, light amplitude modulation is achieved by providing an on/off voltage (square wave function) to an electrooptical crystal which in turn rotates the polarization of a linearly polarized laser. This polarization rotation is then converted into an amplitude modulation by a polarizer. For a two-color ALEX scheme, two such EOMs are needed, driven by two complementary square waves.
Despite its great merits, this configuration entails a few problems: (i) the amplitude modulation relies on polarization modulation and therefore is susceptible to polarization noise (laser polarization fluctuations and other fluctuations in the optical path); (ii) the intrinsic response time constant associated with charging/discharging the electrooptic crystal limits its temporal response to several microsecounds. Previously, a short interval of zero voltage was inserted into the two complementary square waves during the ‘on’ and ‘off’ transitions to avoid overlapping excitation periods; (iii) the ALEX excitation path contains quite a few optical components and residual polarization ellipticity is accumulated as the light traverses these components, reducing extinction ratios to about 100:1 (1% leakage is noticeable and needs to be taken into account in the data analysis).
In the second case, the laser beams pass through an AOM and the directions of their first diffraction orders are alternated according to wavelength and applied RF power, directing them into or out of an optical fiber used to deliver the excitation light into the microscope. Despite the greater stability and extinction ratios achieved using this technique, it still has a response time of ~3 μs thereby necessitating the use of a non-excitation delay and restricting faster alternation frequencies.
Recently, Toptica Photonics AG has introduced the iPulse and the iBeam solid-state laser sources that can be directly modulated up to 200MHz while maintaining good beam characteristics and full modulation depth (Fig. S1), outperforming external EOM/AOM modulators. They have plausible stability and can be directly modulated by simple TTL voltage signals. Additionally the response time is as fast as tens of ns, with achievable extinction ratios of > 300:1, making them a more efficient instrumentation for ALEX over a wider dynamic range.
Ensemble bleaching experiments
Alternating laser excitation under ensemble conditions can also be used to study the mechanism of acceptor bleaching. When a fluorophore is bleached at a rate faster than the replenishment of molecules into the observation volume, the ensemble-averaged count rate will decrease significantly. We therefore used this average count rate of fluorophores as an indication of bleaching events. The ensemble measurements were conducted as a function of: (1) blue pulse or CW intensity; (2) red pulse or CW intensity; (3) nanosecond separation of alternating pulsed lasers; (4) microsecond separation of alternating CW lasers. Thus we could compare the contributions to acceptor photobleaching from blue and red photon excitation, and from singlet absorption or triplet absorption, which enabled us to further elucidate the acceptor bleaching pathway.
Optical Setup
The combined dm-ALEX/ns-ALEX setup is described in Fig. 3. We used similar wavelengths for both excitation schemes, namely, CW 470nm/640nm for dm-ALEX (iPulse470, and iBeam640, Toptica GmbH, Westfield, MA) and picosecond pulsed 467nm/635nm for ns-ALEX (LDH-P-C-470 and LDH 635-B, PicoQuant GmbH, Berlin). Both pulsed lasers were synchronized to the same 40 MHz source from the laser driver (PDL-800B, PicoQuant GmbH, Berlin). A fixed time delay of 14ns between the blue and the red pulses was accomplished by a fixed length coaxial delay cable. The iPulse and the iBeam beams (similarly with the picosecond pulsed beams) were combined with a dichroic mirror (550DRLP, OMEGA Optical, Brattleboro, VT). We switched between the two excitation schemes by using a flippable mirror to couple either the iPulse/iBeam lasers or the pulsed lasers into an optical fiber (QSMJ-A3A, 3AF-488-3.5/125-3-2, OzOptics, Ottawa). The output beams were collimated, coupled into the microscope objective (100X Apochromat, NA 1.4, Zeiss, Jiena) and then focused into the sample. Fluorescence from the samples was collected through the same objective and refocused onto a 100 μm pinhole to reject chromatic aberrations. The emitted photons were then separated by a polarized beam splitter cube (TECH SPEC Polarizing Cube Beam splitters, Edmund Optics, Barrington, NJ) and further split by wavelength using dichroic mirrors (630DRLP, OMEGA Optical, Brattleboro, VT), and finally detected by four avalanche photodiodes (SPCM-AQR-14, PerkinElmer, Fremont, CA).
Figure 3.

Combined dm-ALEX/ns-ALEX set-up. The two picosecond pulsed lasers are synchronized with the same driver. Their beams are combined with a dichroic mirror DM2 (550DRLP, Omega Optical, Brattleboro, VT) and coupled into an optical fiber (QSMJ-A3A, 3AF-488-3.5/125-3-2, OzOptics, Ottawa). Alternatively, the beams of the CW iPulse and the iBeam are directly modulated by the DAC board (National Instruments, Austin, TX, PCI-6602), and are combined and coupled into the same fiber in a similar fashion. Switching between the two excitation schemes is achieved through the flippable mirror (FM) just before coupling the light into the fiber. The light output of the fiber is collimated, polarized and reflected by DM3 (400-535-635TBDR, Omega Optical, Brattleboro, VT), and focused by the microscope objective (100X Apochromat, NA 1.4, Zeiss, Jiena) into the sample. Collected photons are split by a polarizing beam splitter (PBS) into vertical (V) and horizontal (H) polarizations, and split again by DM3 and DM4 (630DRLP, Omega Optical, Brattleboro, VT) into D and A emissions. Bandpass filters BP1-BP4 (BP1&3: 580DF30; BP2&4: 661AGLP, Omega Optical, Brattleboro, VT) are used to exclude photons of the incorrect wavelength. The signals are detected by avalanche photodiodes APD1-APD4 (SPCM-AQR-14, PerkinElmer, Fremont, CA).
We generated TTL signals using a customized LabVIEW program and output the signals through a National Instruments PXI-6602 DAQ board. The iPulse/iBeam lasers were set “off” with 0V and “on” with 5V. Since both excitation and data recording commands were sent through the same DAQ board, data acquisition could be conveniently triggered and synchronized with the excitation modulation.
The alternation frequency and duty-cycle were controlled via the LabVIEW (National Instruments, Austin, TX) program. In order to demonstrate the modulation performance of the lasers, we used a photodiode to monitor the beam intensities and recorded the signals with a 500MHz digital oscilloscope (TDS3052B, Tektronix, Richardson, TX) as a function of the modulation frequency (Fig. S1). Although the modulation at higher frequencies deviates from a square wave (generated by the DAQ board), good modulation performance could be achieved (with the current PC board) up to 10 MHz (100 ns modulation period). Thus the current modulation performance is one order of magnitude faster than the fastest performance achieved with AOM/EOM modulators (~1MHz). For subsequent FRET experiments, modulation periods of 10μs to 50μs were used.
In the ensemble bleaching experiment, we achieved series of pulse separations by using the following technique: for ns-ALEX, the blue and red picosecond pulsed lasers were triggered by a common, external oscillator operating at a repetition rate of 40MHz (25ns period); The two trains of pulses were therefore fully synchronized with one train delayed with respect to the other using an electronic delay box (425A, Ortec, TN); for dm-ALEX, the TTL waveform frequency (laser alternation frequency) was set to 200 kHz, with a 1μs ‘on’ period and a 4 μs ‘off’ period (for both blue and red pulse trains); the red excitation train was delayed with respect to the blue train with an interval continuously varying between 0 and 3.5 μs.
Sample Preparation
Acyl-CoA binding protein (ACBP, Fig. 1C) was purified and labeled with donor (Alexa Fluor 488, Invitrogen, Carlsbad, CA) at position 17 and acceptor (Alexa Fluor 647, Invitrogen, Carlsbad, CA) at the C–terminus as previously described10. dsDNA: Oligodeoxyribonucleotides (Fig. 1D) were prepared by automated synthesis22, labeled, and hybridized to form D-A double-stranded DNA (dsDNA) fragments. For the three DNA fragments set, the top-strand sequence was: 5′-CGATAACAGGTAAGTGATTGCCATTAGTCCGATAAGCAGTAAAACG-3′, with amino-C6-dT residues (Glen Research, Sterling, VA) at position 24; the two bottom strands were 3′-GCTATTGTCCATTCACTAACG-5′ and: 3′-GTAATCAGGCTATTCGTCATTTTGC-5′, modified with amino-C6-dT residues at positions 13, 17 or 20 of the second bottom strand, respectively. Oligodeoxyribonucleotides were HPLC-purified, and labeled with the same fluorophores with N-hydroxy-succinimidyl esters as linkers using manufacturer’s instructions, and HPLC-purified again. dsDNA was formed by hybridization of top and bottom strands in 40 mM Tris-HCl pH 8, 500 mM NaCl after heating for 2 min at 95°C and cooling to 25°C overnight.
Results and Discussion
Single Molecule ALEX
Bleaching as revealed from E-S histograms
ACBP was measured with dm-ALEX and ns-ALEX at two different denaturant conditions with two average excitation powers (50 μW and 100 μW, Fig. 4). By varying denaturant concentration, the protein chain expands (in a two state fashion) from a compact folded (native) state to an expanded unfolded (denatured) state, where the distance between the two fluorophores increases (and therefore the FRET efficiency decreases). The dashed black squares highlight the subpopulation with both fluorophores active (D-A subpopulation), while the solid black rectangles highlight the D-only subpopulation. As mentioned earlier, the size and position of these selections were made so that D-only subpopulation corresponds only to molecules with an inactive acceptor during the whole bursts and the D-A subpopulation corresponds only to molecules with both the donor and the acceptor active during the whole bursts. The least possible number of bridge events24 was counted in either of the selected subpopulations. In doing so we excluded bursts with more than one molecule in the focal volume and bursts with acceptor bleached during traversing the focal spot. However, since acceptor bleaching is directly reflected by the percentage of D-only subpopulation, only the solid black rectangle selection influences the calculation of bleaching percentages. Thus in this work we used the same selection criteria for all molecules in all experiments to ensure the consistency and reproducibility of our calculations. According to Equation (1), E=0.9 for 0M [GuHCl] (folded protein) and E=0.2 for 6.0M [GuHCl] (denatured protein) were calculated, while the calculated S value increases with increased blue excitation intensity, as predicted by Eq. (2) ( and FFRET are increased while stays constant).
Figure 4.

Single molecule dm-ALEX and ns-ALEX histograms of ACBP under various denaturation and average blue excitation power conditions (red excitation power is fixed). (A) dm-ALEX, 6.0M [GuHCl], 50 μW; (B) dm-ALEX, 6.0M [GuHCl], 100 μW; (C) ns-ALEX, 6.0M [GuHCl], 50 μW; (D) ns-ALEX, 6.0M [GuHCl], 100 μW; (E) dm-ALEX, 0.0M [GuHCl], 50 μW; (F) dm-ALEX, 0.0M [GuHCl], 100 μW; (G) ns-ALEX, 0.0M [GuHCl], 50 μW; (H) ns-ALEX, 0.0M [GuHCl], 100 μW. Solid black squares highlight events with bleached acceptor (donor-only), and dashed black squares highlight doubly labeled FRET events.
Increasing the donor excitation power for the low E sample (6.0M [GuHCl]) increased the donor-only subpopulation for both excitation schemes (4A vs. 4B for dm-ALEX and 4C vs. 4D for ns-ALEX). However, the fraction of the D-only subpopulation was significantly larger for the ns-ALEX than of the dm-ALEX (4D vs. 4B). A much stronger trend was observed when the high E sample (0.0M [GuHCl]) (4E vs. 4F for dm-ALEX and 4G vs. 4H for ns-ALEX) was measured. The D-only subpopulation in the ns-ALEX was 4 times larger compared to the dm-ALEX (4H vs. 4F). In both GuHCl concentrations (low and high E), Alexa Fluor 647 bleaching rate was faster using ns-ALEX.
These experiments were repeated for average excitation powers ranging from 50 μW to 100 μW for the blue excitation (while maintaining the red excitation at 20μW) and 10 μW to 20 μW for the red excitation (while maintaining the blue excitation at 60μW). The guideline for choosing the constant power of the second laser while varying the power of the first laser was to maintain the effective excitations from both colors while not bleaching the fluorophores by the second laser. The results are summarized in Fig. 5A (denatured ACBP) and 5B (native ACBP). The ranges of average powers were selected according to values normally applied in single-molecule experiment for Alexa Fluor 488 and Alexa Fluor 647, and adjusted such that the emissions rates following blue excitation are comparable to those following the red excitation. This ensured S values close to 0.5 where sensitivity to stoichiometry changes is highest22.
Figure 5.
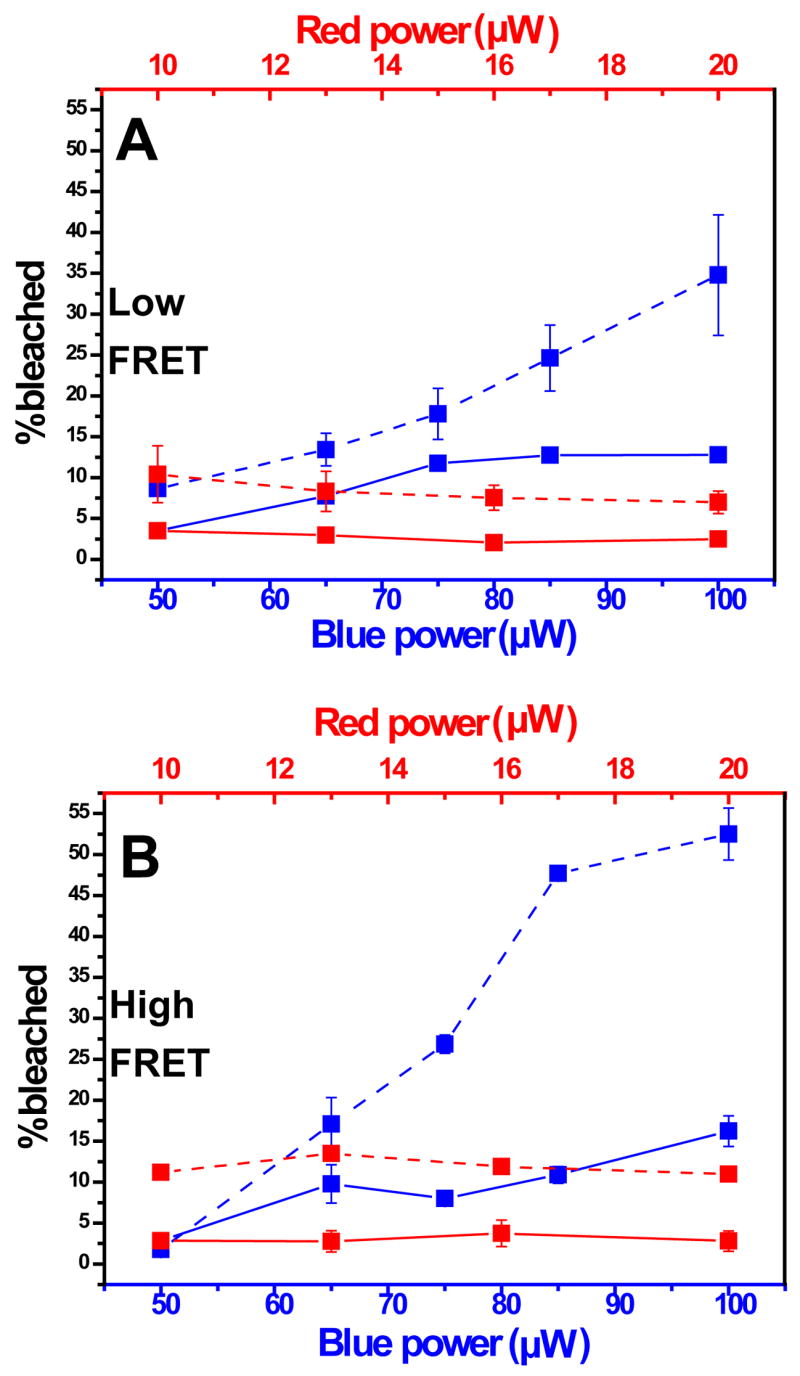
Fraction of D-only events as a function of the excitation scheme and average excitation powers. (A) denatured ACBP (6.0 M [GuHCl], low FRET); (B) folded ACBP (0.0 M [GuHCl, high FRET]). All blue lines represent the fraction of D-only events as function of average blue excitation power; all red lines represent the fraction of D-only events as function of average red excitation power. Solid: dm-ALEX scheme; dashed: ns-ALEX scheme.
As can be seen from Fig. 4 & 5, the acceptor’s photobleaching rate is increased upon raising the average blue excitation power for both dm-ALEX and ns-ALEX, but at a faster rate for ns-ALEX. This result agrees with the conclusions on other acceptor fluorophores reported by Eggeling et al.21, where a comparison between CW and pulsed green (502nm) excitation of several red dyes and a FRET dye pair (Rh110/Cy5) indicated a significant difference in the acceptor’s photobleaching (and a modest difference in acceptor’s brightness), due to nonlinear excited state absorption21.
To distinguish between the contributions from blue and red excitations, shown in Fig. 5 (solid and dashed red curves) is the acceptor’s photobleaching rate as function of the average red excitation power. Doubling the red power from 10 μW to 20 μW while maintaining the blue power (at 60μW) resulted in no increase in bleaching rate for both dm-ALEX and ns-ALEX. For the high E ACBP sample the average emission rate of red acceptor photons within detected bursts was 13kHz for 20 μW of direct red excitation and 11kHz for acceptor excitation via FRET with 100 μW of blue excitation. Therefore the FRET excitation rate of the acceptor for most of the experiments was somewhat lower than the direct excitation by the red laser, as the first excited states were more populated when 20 μW of direct red excitation was applied. The photobleaching rate, nonetheless, was higher for the blue excitation, suggesting that the main pathway for acceptor’s photobleaching involves excited state absorption of blue photons.
As in Eggeling et al.21, direct excitation of the red acceptor dye by the D-excitation laser is negligible (even more so in our case, where 467nm rather than 502nm excitation was used). Thus it is unlikely that the acceptor’s photobleaching is the result of direct excitation by a blue photon. This conclusion is further supported by the ensemble experiment (see below). Eggeling et al.21 suggested that the pathways for acceptor’s photobleaching in FRET experiment involves resonant energy transfer of donor excitation into either or followed by excited state absorption of the green photon (blue photon in our case) to or . In water (or other polar solvent), these highly excited states efficiently couple to ionic states, leading to an enhanced reactivity with free radicals12, 14. Moreover, a pulsed picosecond excitation leads to even stronger nonlinear excited state absorption and hence enhanced photobleaching. With the combination of dm-ALEX and ns-ALEX we aim to separate these processes and extract the first major step in the acceptor bleaching pathway, as shown below.
The photobleaching rate of the folded (E=0.9) ACBP molecule is higher than that of the denatured (E=0.2) ACBP molecule, when both are excited at the same excitation irradiance, confirming that the FRET process is involved in the bleaching mechanism. This is likely because higher FRET efficiencies result in increases in the (and possibly ) populations and therefore higher probability of acceptor’s excited state absorption (of a blue photon).
Based on these dm-ALEX and ns-ALEX experiments, we conclude that: (i) short pulse excitation bleached the acceptor more severely than CW excitation; (ii) blue excitation, even at a lower excitation rate, induced more bleaching to the acceptor than red excitation; and (iii) high FRET resulted in more bleaching. These conclusions were further supported by photon arrival time analysis within the bursts21 (the mean-arrival-time relative to the beginning of the burst of blue and red photons shows acceptor bleaching during the burst, data not shown).
Effect of protein denaturant in solution
To rule out the possible contributions to bleaching from GuHCl, we performed control experiments using double-stranded DNA (dsDNA) samples doubly labeled with the same fluorophore pair, in 0 and 6M [GuHCl]. Since dsDNA can be considered a rigid rod under these conditions, and the distance between the fluorophores was fixed, any effects observed would originate solely from the solution environment of the fluorophores. We used ns-ALEX excitation scheme to seek maximum bleaching effect on DNA samples, and we studied 46 bases long dsDNA with 10, 14 and 17 bases separating between the two fluorophores, resulting in E = 0.61, 0.54 and 0.31 respectively.
Addition of GuHCl increased the acceptor photobleaching rate for the three DNA samples (Fig. 6A), probably due to a solvent induced photophysical effect, while this rate decreased for the labeled proteins (Fig. 6B). The opposing photobleaching trends displayed by adding GuHCl to dsDNA vs. protein samples support the conclusion that the bleaching rate is proportional to the FRET efficiency and is not a solvent induced artifact. In fact, the increase of acceptor bleaching due to increase of FRET (on ACBP from 5M to 0M GuHCl) is much higher than shown because part of the bleaching at low FRET (5M GuHCl) is due to the denaturant. Moreover, the dsDNA with the shortest base separation between fluorophores (10 bases, highest FRET) also shows the highest bleaching rate (Fig. 6A).
Figure 6.

Effect of GuHCl on bleaching. (A) Fraction of bleached molecules as function of GuHCl measured for 3 dsDNA molecules with dye separations of 10 bases (solid); 14 bases (dashed); and 17 bases (dotted). (B) Fraction of bleached molecules as function of GuHCl measured for ACBP molecules by dm-ALEX (solid) and ns-ALEX (dotted).
Ensemble Measurements of Bleaching Rates
To further elucidate the pathway leading to acceptor’s bleaching, we studied the same protein samples at the ensemble level (10nM). We used an identical confocal geometry and laser alternation schemes to measure the acceptor’s average photon count rate emitted by an ensemble of donor-acceptor pairs occupying the confocal volume. We expect this count rate to decrease when acceptors are bleached at a faster rate than the replenishment of new pairs into the observation volume. In addition to the alternating blue and red lasers of different intensities (as in single molecule ALEX measurements), ACBP molecules were excited by individual blue and red lasers, probing the bleaching response to excitation by photons of different energy.
A pronounced plateau in the acceptor’s emission rate (at a level of 50kHz) was observed when a blue-only pulsed excitation was varied from 10μW to 160μW; the donor emission, on the other hand, stayed linear with power in this range (Fig. 7A). The plateau of the acceptor’s emission is interpreted to be due to photobleaching; the linear increase in donor emission suggests negligible donor bleaching in this range. Since only blue excitation was applied, the acceptor’s bleaching must involve blue photons. In contrast, when red-only pulsed excitation was applied (varied from 1μW to 70μW) the acceptor emission rate increased well above 200kHz before saturation (Fig. 7B). Compared to the 50 kHz saturation level using the blue-only excitation, we infer that red excitation is less detrimental to the acceptor. These measurements were highly reproducible with little statistical error (bars shown in the inset of Fig. 7A). Similar trends were found when CW blue-only and red-only excitations were used, but with higher saturation levels (Fig. 7C and 7D). We therefore conclude that excited-state absorption of a blue photon from to or from to are the first step on the acceptor’s pathway to photobleaching.
Figure 7.

Ensemble count rates measured from pump-probe experiments. (A) Donor (solid) and acceptor (dashed) emission as function of blue excitation power using ns-ALEX. (B) Acceptor emission as function of blue excitation power (solid) and red excitation power (dashed) using ns-ALEX. (C) Donor (solid) and acceptor (dashed) emission as function of blue excitation power using dm-ALEX. (D) Acceptor emission as function of blue excitation power (solid) and red excitation power (dashed) using dm-ALEX.
In order to confirm the effect of blue photon absorption and to determine if this absorption is from or from , we studied the bleaching kinetics on the nanosecond and on the microsecond timescales using the same dm-ALEX and ns-ALEX schemes (but at a 10nM concentration). The ability to electronically vary the time delay between the picosecond blue and red pulses provides a simple scheme for pump-probe spectroscopy, directly probing the singlet state ( ) absorption. The delay (Δτ) between the blue and the red pulses was incremented by steps of 0.5 ns (tuned over the 0 – 25 ns range) and the average donor and acceptor emission rates were recorded as function of this time delay. When the two pulses overlapped ( Δτ = 0 ns and Δτ = 25 ns, Fig. 8A), the acceptor’s emission rate reached a minimum, indicating maximal bleaching. The donor emission, on the other hand, reached a maximum value (bleached acceptors are less likely to quench the donors, thus donor emission was increased). As the red pulse was further delayed from the preceding blue pulse, the acceptor emission rate showed an abrupt rise to a constant value up to a delay of ~18 ns. When the delay was between 18 ns and 25ns, the acceptors’ emission rate gradually decreased towards its minimum. This gradual decay as a function of pulse separation confirms that the major contribution to bleaching is through the acceptor’s singlet excited-state absorption of a blue photon: When the acceptor is excited to its first excited state , it will dwell in this state, on average, for the acceptor lifetime. If a blue photon is applied during this time (before relaxation), the acceptor is able to absorb a blue photon and be excited to , resulting in a higher probability for photobleaching. The gradual decrease between 18 ns and 25ns is therefore an indirect measure of the acceptor’s lifetime. The sharp rise from 0 ns to 2 ns suggests, however, that a second red photon is unlikely to be the main bleaching source, since when the red excitation was applied after the blue excitation with no overlap; the acceptor count rate was constant and was independent of the pulse separation between the two lasers.
Figure 8.

(A) Ensemble donor (black) and acceptor (grey) emission rates as function of the time delay between a consecutive red and blue picosecond pulses (ns-ALEX excitation scheme). Order of pulse arrivals is shown in cartoons below the figure. (B) Acceptor emission rate as function of time delay (grey) and a fit to the model described by Eq. 3 (dashed black). (C) Donor (black) and acceptor (grey) emission rates as function of time delay between microsecond modulated CW pulse trains (dm-ALEX excitation scheme). (D) Acceptor’s emission rate normalized to the donor’s emission rate as function of excitation power in a static cell (solid) and in a continuous-flow mixing device (dashed).
If indeed the sole contribution to the acceptor bleaching was from a second blue photon, the acceptor emission rate would be determined by the population of , and exhibit a decay as a function of the pulse separation according to:
| (3) |
where τA is the acceptor fluorescence lifetime, Δτ is the pulse separation, c and A are normalization factors. By fitting the acceptor emission response to pulse separation with this model we obtained τA of 2.2 ns (Fig. 8B) compared to the directly measured acceptor lifetime of 1.5 ns. This deviation indicates that there might be other contributions to the acceptor’s photobleaching. For example, the red pulse could also bleach a small portion of the acceptors before the blue pulse arrives (resulting in a count rate that is not linearly proportional to ). A more elaborate model is needed to fully describe the observed decay in Fig. 8B. Nevertheless this does not alter the main conclusion.
Similar experiments were performed on the microsecond timescale using dm-ALEX, probing possible contributions to bleaching from triplet states. This was achieved by delaying one TTL pulse train (red) continuously with respect to the other train (blue). Here again the acceptor’s emission rate reached a minimum when both excitations overlapped, but was completely constant when they did not (between 1 and 3.5 μs, Fig. 8C). Donor emission was also constant in this non-overlapping region. Since triplet states lifetimes are in the microsecond range, and since no dependence on time delay was observed, we conclude that the contribution of excited triplet state absorption to acceptor photobleaching is insignificant. This conclusion is already confirmed by our preliminary results on the effect of an oxygen scavenger (MEA). We observed that the bleaching of Alexa 647 is not affected by addition of MEA (data not shown), which is generally applied to suppress bleaching via triplet state absorption. Thus the photobleaching of Alexa 647 does not involve triplet state and cannot be alleviated by oxygen scavenging additives.
Photobleaching vs. Saturation
In order to verify that the decrease in acceptor emission is indeed caused by photobleaching rather than by saturation of the excited state, we repeated the ensemble measurements described in Fig. 7 in a continuous-flow mixing-device, where fresh, unbleached, molecules are continuously replenishing the confocal observation volume (with a transit time of ~50 μs). In this device, bleached molecules are flushed out in a unidirectional way, minimizing the probability of bleached molecules re-entering the observation volume. Hence we expect the average count rate to be independent of bleaching and only dependent on saturation effects. Fig 8D shows very little relative decrease in the acceptors’ count rate (normalized to the donors’ emission) when the measurement is performed in the mixer (dashed) but a significant decrease when performed in a static cell (solid). This observation indicates that bleaching and not excited-state saturation is the source of the plateau in acceptor emission vs. blue excitation power (Fig. 7) and again supports the proposed mechanism for bleaching (excited state absorption of a blue photon from ).
Conclusions
We introduced four technical innovations: (i) direct modulation μs-ALEX (dm-ALEX), (ii) merging of dm-ALEX with ns-ALEX in a single experimental setup, (iii) application of the merged dm-ALEX/ns-ALEX to photophysical pump-probe ensemble measurements of donor and acceptor emission rates, and (iv) implementation of fast flowing device to replenish bleached molecules. These advances were used to study the pathway for photobleaching in single molecule FRET experiments. We found that the acceptor photobleaching probability is proportional to the FRET efficiency, that it is higher for short (picosecond) pulsed excitation (compared to CW excitation) and that the main pathway for acceptor’s bleaching is through absorption of a blue (donor excitation) photon from the first acceptor’s excited singlet (and not triplet) state. Thus, we suggest that applying shorter pulses for the blue laser (donor excitation) can effectively reduce acceptor bleaching, since the probability of absorbing another blue photon, after being excited to the first excited state by FRET, is decreased. This certainly results in higher peak irradiance on the donor if the same average power is maintained, which usually might cause bleaching to the donor. However, we have shown that under typical sm-FRET experimental conditions there is no significant donor photobleaching. And since the donor → acceptor energy transfer rate is in the range of nanoseconds to tens of picoseconds (proportional to the inverse of the quenched donor’s lifetime) which is comparable to the pulse durations, decreasing the blue pulse duration can efficiently reduce the probability of exciting the acceptor in S1 and prevent significant bleaching to the acceptor. In addition, in both ns- and dm-ALEX, lowering the blue laser power will reduce acceptor bleaching, though it will also result in lower detected count rates and increased shot-noise broadening.
The choice and optimization of a FRET pair for a particular system under study requires a careful consideration of the system, the distance and distance changes to be measured, the Förster radius, the spectral overlap, photostability, quantum yields, emission colors, assay format, use of anti-oxidents and more. This work and previously reported results21 suggest that the photobleaching of the acceptor via its excited state absorption of a donor excitation photon is an important parameter in choosing the pair.
It is yet to be determined if the results reported by Eggeling et al. and here are general for all FRET pairs, or particular to the pairs used in these two studies (Rh110/Cy5 in Eggeling et al. and Alexa Fluor 488/Alexa Fluor 647 here), i.e. if the requirement for resonance between the donor emission and acceptor absorption also dictates a ‘resonance’ in promoting the transition from to via the absorption of a donor’s excitation photon (Fig. 9A), or other pairs could be found that satisfy the FRET resonance condition without the acceptor excited state absorption, i.e. if donor/acceptor states alignment could be found such that the FRET resonance condition is obeyed, but the absorption of a blue photon from is banned (Fig. 9B). Such a solution, if could be found (or engineered), will eliminate the need for anti-oxidants additives.
Figure 9.

Simplified Jablonski diagrams for a donor-acceptor pair where the absorption of a donor excitation photon by the acceptor in is (A) allowed; (B) prohibited.
Future investigations will focus on downstream steps in the pathway, other dye pairs, the effects of anti-oxidants additives on the different parts in the bleaching pathway, and further optimization for sm-FRET experiments.
Supplementary Material
The laser beam modulation (in dm-ALEX) as a function of alternation frequency. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We would like to thank Dr. Marcus Jäger for providing protein samples and Dr. Xavier Michalet for helpful discussions. We thank Toptica Photonics, Inc. for the help with the excitation sources for dm-ALEX. This work was funded by the NIH grant #GM069709-01, the FIBR program at the NSF grant #0623664 and the NSF Center for Biophotonics Science and Technology, University of California, Davis, under Cooperative Agreement No. PHY0120999 (to S. W.). E. Nir is supported by the Human Frontier Science Program (HFSP).
References
- 1.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci U S A. 1996;93(13):6264–8. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nie S, Zare RN. Optical detection of single molecules. Annu Rev Biophys Biomol Struct. 1997;26:567–96. doi: 10.1146/annurev.biophys.26.1.567. [DOI] [PubMed] [Google Scholar]
- 3.Xie XS, Trautman JK. Optical studies of single molecules at room temperature. Annu Rev Phys Chem. 1998;49:441–80. doi: 10.1146/annurev.physchem.49.1.441. [DOI] [PubMed] [Google Scholar]
- 4.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S. A single-molecule study of RNA catalysis and folding. Science. 2000;288(5473):2048–51. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 5.Kapanidis AN, Margeat E, Laurence TA, Doose S, Ho SO, Mukhopadhyay J, Kortkhonjia E, Mekler V, Ebright RH, Weiss S. Retention of transcription initiation factor sigma70 in transcription elongation: single-molecule analysis. Mol Cell. 2005;20(3):347–56. doi: 10.1016/j.molcel.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Margeat E, Kapanidis AN, Tinnefeld P, Wang Y, Mukhopadhyay J, Ebright RH, Weiss S. Direct observation of abortive initiation and promoter escape within single immobilized transcription complexes. Biophys J. 2006;90(4):1419–31. doi: 10.1529/biophysj.105.069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Ren X, Ying L, Balasubramanian S, Klenerman D. Measuring single-molecule nucleic acid dynamics in solution by two-color filtered ratiometric fluorescence correlation spectroscopy. Proc Natl Acad Sci U S A. 2004;101(40):14425–30. doi: 10.1073/pnas.0404295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camacho A, Korn K, Damond M, Cajot JF, Litborn E, Liao B, Thyberg P, Winter H, Honegger A, Gardellin P, Rigler R. Direct quantification of mRNA expression levels using single molecule detection. J Biotechnol. 2004;107(2):107–14. doi: 10.1016/j.jbiotec.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Rothwell PJ, Berger S, Kensch O, Felekyan S, Antonik M, Wohrl BM, Restle T, Goody RS, Seidel CA. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc Natl Acad Sci U S A. 2003;100(4):1655–60. doi: 10.1073/pnas.0434003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurence TA, Kong X, Jager M, Weiss S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc Natl Acad Sci U S A. 2005;102(48):17348–53. doi: 10.1073/pnas.0508584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navon A, Ittah V, Landsman P, Scheraga HA, Haas E. Distributions of intramolecular distances in the reduced and denatured states of bovine pancreatic ribonuclease A. Folding initiation structures in the C-terminal portions of the reduced protein. Biochemistry. 2001;40(1):105–18. doi: 10.1021/bi001946o. [DOI] [PubMed] [Google Scholar]
- 12.Eggeling C, Volkmer A, Seidel CA. Molecular photobleaching kinetics of Rhodamine 6G by one- and two-photon induced confocal fluorescence microscopy. Chemphyschem. 2005;6(5):791–804. doi: 10.1002/cphc.200400509. [DOI] [PubMed] [Google Scholar]
- 13.Eggeling C, Fries JR, Brand L, Gunther R, Seidel CA. Monitoring conformational dynamics of a single molecule by selective fluorescence spectroscopy. Proc Natl Acad Sci U S A. 1998;95(4):1556–61. doi: 10.1073/pnas.95.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eggeling C, Widengren J, Rigler R, Seidel C. Photobleaching of fluorescent dyes under conditions used for single-molecule detection: Evidence of two-step photolysis. Anal Chem. 1998;70(13):2651–2659. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- 15.Buschmann V, Weston K, Sauer M. Spectroscopic study and evaluation of red-absorbing fluorescent dyes. Bioconjugate Chem. 2003;14(1):195–204. doi: 10.1021/bc025600x. [DOI] [PubMed] [Google Scholar]
- 16.Rasnik I, McKinney SA, Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods. 2006;3(11):891–3. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 17.Tinnefeld P, Buschmann V, Weston K, Sauer M. Direct observation of collective blinking and energy transfer in a bichromophoric system. J Phys Chem A. 2003;107(3):323–327. [Google Scholar]
- 18.Heilemann M, Margeat E, Kasper R, Sauer M, Tinnefeld P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J Am Chem Soc. 2005;127(11):3801–3806. doi: 10.1021/ja044686x. [DOI] [PubMed] [Google Scholar]
- 19.Sabanayagam C, Eid J, Meller A. Using fluorescence resonance energy transfer to measure distances along individual DNA molecules: Corrections due to nonideal transfer. J Chem Phys. 2005;122:6. doi: 10.1063/1.1854120. [DOI] [PubMed] [Google Scholar]
- 20.White S, Li H, Marsh R, Piper J, Leonczek N, Nicolaou N, Bain A, Ying L, Klenerman D. Characterization of a single molecule DNA switch in free solution. J Am Chem Soc. 2006;128(35):11423–11432. doi: 10.1021/ja0614870. [DOI] [PubMed] [Google Scholar]
- 21.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CA. Analysis of photobleaching in single-molecule multicolor excitation and Forster resonance energy transfer measurements. J Phys Chem A Mol Spectrosc Kinet Environ Gen Theory. 2006;110(9):2979–95. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 22.Kapanidis AN, Lee NK, Laurence TA, Doose S, Margeat E, Weiss S. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc Natl Acad Sci U S A. 2004;101(24):8936–41. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys J. 2005;88(4):2939–53. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nir EMX, Hamadani K, Laurence TA, Neuhauser D, Kovchegov Y, Weiss S. Shot-noise limited single-molecule FRET histogram: comparison between theory and experiments. J Phys Chem. 2006;110(44):22103–22124. doi: 10.1021/jp063483n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller BK, Zaychikov E, Brauchle C, Lamb DC. Pulsed interleaved excitation. Biophys J. 2005;89(5):3508–22. doi: 10.1529/biophysj.105.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha T, Xu J. Photodestruction intermediates probed by an adjacent reporter molecule. Phys Rev Lett. 2003;90(22):223002. doi: 10.1103/PhysRevLett.90.223002. [DOI] [PubMed] [Google Scholar]
- 27.Penttila A, Boyle CR, Salin ML. Active oxygen intermediates and chlorophyllin bleaching. Biochem Biophys Res Commun. 1996;226(1):135–9. doi: 10.1006/bbrc.1996.1322. [DOI] [PubMed] [Google Scholar]
- 28.Eggeling C, Berger S, Brand L, Fries JR, Schaffer J, Volkmer A, Seidel CA. Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J Biotechnol. 2001;86(3):163–80. doi: 10.1016/s0168-1656(00)00412-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The laser beam modulation (in dm-ALEX) as a function of alternation frequency. This material is available free of charge via the Internet at http://pubs.acs.org.