

Abstract
Indenoisoquinolines with lactam substituents such as ethylamino, propylamino, and butylamino have previously demonstrated potent biological activity, but optimal length has never been established. In the present study, a series of simplified indenoisoquinoline analogues possessing a linker spacing of 0–12 carbon atoms between the lactam nitrogen and the terminal amino group have been prepared, determining that 2–4 atom lengths are optimal for topoisomerase I inhibition and cytotoxicity. Using these lengths, analogues were prepared with the amino group and portions of the linker replaced by a pyridine ring. A three-carbon spacer within the pyridine series still demonstrated potent topoisomerase I inhibition.
Introduction
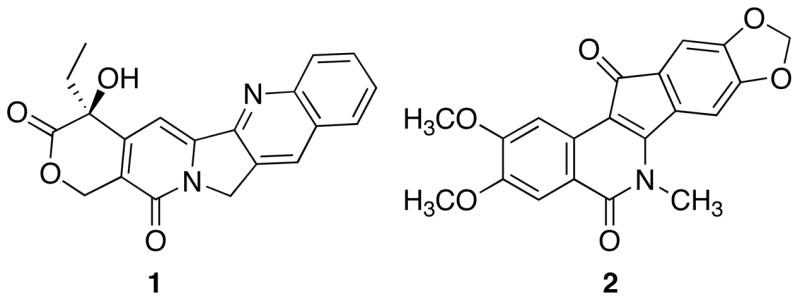
Topoisomerases are nature’s solution to the topological problems associated with manipulating double-stranded, helical DNA during essential cellular processes.1–4 The main classification of topoisomerases is dependent upon the enzyme’s ability to create a single-stranded (type I) or double-stranded (type II) nick in DNA to alleviate torsional strain.1–4 Interest in topoisomerase I (top1) as a potential therapeutic target originated from the characterization of camptothecin (1) as a selective mammalian topoisomerase I inhibitor.5–7 Validation of drug development for this target occurred with the approval of two camptothecin derivatives, topotecan and irinotecan, for the treatment of various forms of cancer.7,8 However, these drugs are less than optimal, each possessing a hydrolytically unstable lactone ring that when hydrolyzed affords a product with high affinity for serum albumin.9–12 Therefore, novel, hydrolytically stable top1 inhibitors would represent an improvement in top1-targeted anticancer chemotherapy.8
The top1 inhibitory activity of indenoisoquinoline 2 was determined after a COMPARE analysis of its cytotoxicity profile in the National Cancer Institute’s 60 cell screen indicated a high degree of correlation with camptothecin.13 Subsequent experimentation confirmed the activity of 2 as a top1 inhibitor with modest potency. The mechanism of action of the indenoisoquinolines involves the stabilization of the ternary cleavage complex by intercalation between the DNA base pairs upon single-strand cleavage by top1 and formation of a network of hydrogen bonds with top1, thus preventing re-ligation of the broken phosphodiester backbone, a mechanism identical to the camptothecins. Recent crystallographic analyses elegantly demonstrate the inhibition of top1 by these compounds and illustrate the general features of interfacial top1 inhibition by intercalation and specific contacts with top1 amino acid residues.14–17 In addition, ab initio calculations have underscored the primary importance of π-stacking interactions in the stabilization of the ternary complex.18
Previously, it was demonstrated that ethylamino-, propylamino-, and butylamino-substituted lactam side chains conferred potent biological activity for unsubstituted indenoisoquinolines.19 Indeed, the influence of these three substituents on biological activity has made them a standard functionality for incorporation into variously substituted indenoisoquinoline analogues. Furthermore, the 2–4 carbon spacing has been, and is currently, utilized for the development of highly potent indenoisoquinolines with various functional groups at the end of this linker.19–30 However, the determination of optimal spacing between the terminal amino substituent and the lactam nitrogen has not yet been confirmed experimentally and its establishment could lead to the improvement of indenoisoquinoline top1 inhibitors. Therefore, a complete series of simplified indenoisoquinoline homologues possessing a linker spacing of 0–12 carbon atoms between the lactam nitrogen and the terminal amino group have been prepared and evaluated for both cytotoxicity and top1 inhibition in an effort to determine the optimal chain length for biological activity.
Chemistry
Preparation of analogues for this study began with the monoprotection of diaminoalkanes 3–11 with Boc2O according to standard methods (Scheme 1). With compounds 12–20 suitably protected, analogues 25–36 were prepared by the treatment of 12–24 with benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (Scheme 2). Through experimentation, it was determined that the monoprotection of diaminoalkanes with lengths of less than seven carbons was not entirely necessary. Employment of excess diaminoalkanes provided a suitable method to prevent large quantities of dimerized product from forming and the diaminoalkanes were sufficiently water soluble to allow their removal during aqueous workup. Hence, compounds 25, 29, and 30 were prepared without utilization of mono-Boc-protected diaminoalkanes. Beyond a six-carbon length, however, aqueous solubility of the unprotected diaminoalkanes was negligible and purification of 31–36 was greatly facilitated by the use of a tert-butyl carbamate to protect the terminal amine on the lactam side chain. Deprotection of carbamates 26–28 and 31–36 readily occurred upon exposure to hydrochloric acid, providing the remainder of the analogues (37–47) for this study.
Scheme 1.
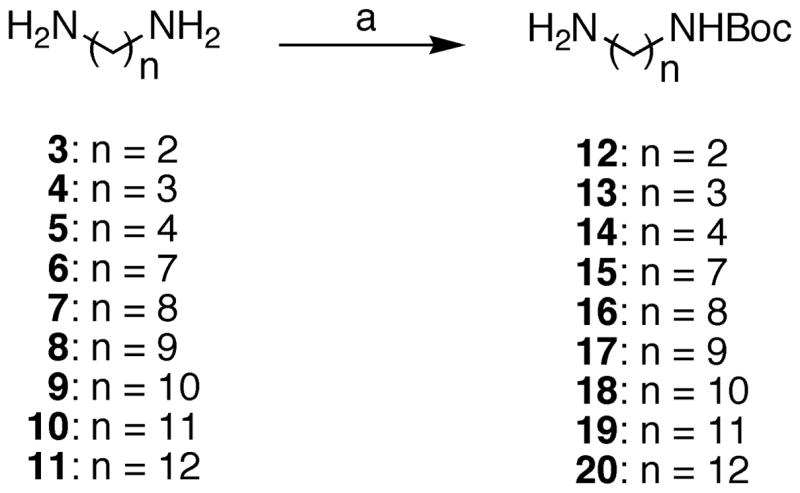
Preparation of Mono-Boc-Protected Diaminoalkanesa
aReagents and conditions: (a) Boc2O (1 equiv), CHCl3, rt.
Scheme 2.

Preparation of Indenoisoquinoline Analoguesa
aReagents and conditions: (a) CHCl3; reflux; (b) 3 M HCl in MeOH, rt.
Upon evaluation of the analogues prepared in Scheme 2, the optimal chain length for lactam substituents was determined and utilized in the preparation of pyridine-substituted indenoisoquinolines 48–51 (Scheme 3). Unlike analogues 37–47, compounds 48–51 should not be protonated at physiological pH and their biological activities, when compared to analogues 37–39, should be different due to the different roles in hydrogen-bonding (donor versus acceptor) of the analogue side chains. Treatment of 2129 with the appropriate aminopyridine derivatives readily provided compounds 48–51, with the isolation of 49 and 50 greatly facilitated by the formation of their corresponding hydrochloride salts.
Scheme 3.
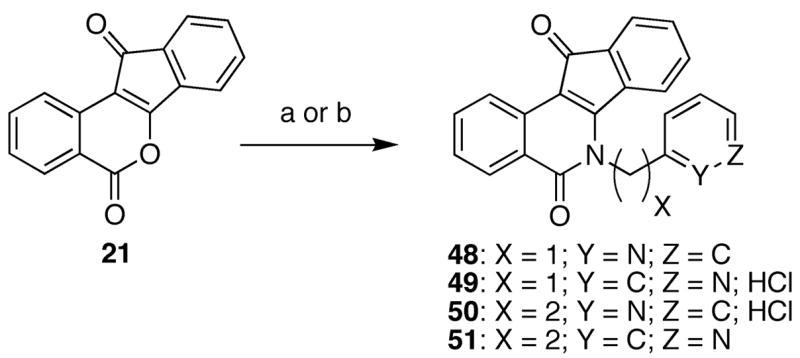
Pyridine-Substituted Indenoisoquinolinesa
aReagents and conditions: (a) aminopyridine, Et3N, CHCl3; reflux; (b) i. aminopyridine, Et3N, CHCl3; ii. 3 M HCl in MeOH.
Biological Results and Discussion
The indenoisoquinolines were examined for antiproliferative activity against the human cancer cell lines in the National Cancer Institute screen, in which the activity of each compound was evaluated with approximately 55 different cancer cell lines of diverse tumor origins.31,32 The GI50 values obtained with selected cell lines, along with the mean graph midpoint (MGM) values, are summarized in Table 1. The MGM is based on a calculation of the average GI50 for all of the cell lines tested (approximately 55) in which GI50 values below and above the test range (10−8 to 10−4 molar) are taken as the minimum (10−8 molar) and maximum (10−4 molar) drug concentrations used in the screening test. For comparison purposes, the activities of camptothecin (1), parent indenoisoquinoline 213, and previously reported compounds 37–3919 are included in the table. The relative potencies of the compounds in the production of top1-mediated DNA cleavage are listed in the table and summarized as follows: +: weak activity; ++: similar activity as the parent compound 2; +++ & ++++: greater activity than the parent compound 2; ++++: similar activity as 1 μM camptothecin (1).
Table 1.
Cytotoxicities and Topoisomerase I Inhibitory Activities of Indenoisoquinoline Analogs.
| compd | cytotoxicity (GI50 in μM)a |
MGMb | Top 1 Cleavagec | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lung HOP-62 | colon HCT-116 | CNS SF-268 | melanoma UACC-62 | ovarian OVCAR-3 | renal SN12C | prostate DU-145 | breast MDA-MB-435 | |||
| 1 | 0.010 | 0.030 | 0.010 | 0.010 | 0.220 | 0.020 | 0.010 | 0.040 | 0.0405 ± 0.0187 | ++++ |
| 2 | 1.30 | 35.0 | NT | 4.20 | 73.0 | 68.0 | 37.0 | 96.0 | 20.0 | ++ |
| 25 | 24.5 | >50.1 | 28.2 | >50.1 | >50.1 | >50.1 | >50.1 | >50.1 | 39.8 | 0 |
| 31 | NT | 17.4 | 20.9 | 20.0 | 33.9 | 74.1 | 31.6 | 81.3 | 30.9 | 0 |
| 32 | 10.7 | 25.7 | 8.71 | 15.5 | - | >50.1 | >50.1 | >50.1 | 19.9 | 0 |
| 33 | 24.5 | NT | 17.8 | 17.4 | 28.8 | 77.6 | >100 | >100 | 50.1 | - |
| 34 | 28.8 | 60.3 | 42.7 | 43.6 | >100 | >100 | 97.7 | >100 | 52.5 | 0 |
| 35 | 20.9 | 35.5 | 29.5 | 24.0 | 70.8 | >100 | 91.2 | >100 | 42.6 ± 5.35 | - |
| 36 | 32.4 | >100 | 20.4 | 27.5 | >100 | >100 | 91.2 | >100 | 51.3 | - |
| 37 | 0.620 | 0.270 | 0.210 | 0.920 | 0.710 | 0.490 | 0.760 | 0.920 | 0.530 ± 0.320 | +++ |
| 38 | 0.200 | 0.180 | 0.25 | 0.26 | 1.38 | 0.160 | 0.22 | 0.78 | 0.32 ± 0.23 | +++ |
| 39 | 0.08 | 0.10 | 0.10 | 0.05 | 0.52 | 0.04 | 0.01 | 0.84 | 0.16 ± 0.01 | +++ |
| 40 | 0.288 | 0.200 | 0.871 | 1.35 | 0.708 | 0.398 | 0.347 | 1.35 | 0.471 ± 0.054 | 0 |
| 41 | 1.29 | 0.912 | 1.23 | 1.62 | 2.00 | 1.32 | 0.603 | 2.04 | 1.32 | ++ |
| 42 | 1.20 | 1.26 | 1.78 | 2.00 | 1.70 | 1.66 | 0.832 | 2.24 | 1.66 ± 0.155 | 0 |
| 43 | 2.14 | 1.66 | 2.82 | 3.80 | 3.47 | 3.47 | 3.39 | 5.62 | 3.71 | + |
| 44 | 6.76 | 6.46 | 9.77 | 9.33 | 9.55 | 8.51 | 6.03 | 9.55 | 8.13 | 0 |
| 45 | 4.79 | 2.75 | 1.82 | 13.8 | 10.7 | 3.31 | 3.47 | 11.7 | 5.50 | 0 |
| 46 | 2.19 | 1.91 | 2.04 | 1.70 | 2.09 | 2.00 | 4.57 | 11.0 | 5.13 | + |
| 47 | 17.0 | NT | 18.2 | 14.8 | 17.8 | 13.5 | 19.1 | 19.5 | 18.2 | 0/+ |
| 48 | 11.2 | NT | 12.3 | 10.7 | 13.8 | 28.2 | 13.5 | 3.09 | 15.1 | 0 |
| 49 | 7.59 | NT | 6.76 | 8.71 | 13.8 | 13.8 | 13.8 | 42.7 | 11.5 | +++ |
| 50 | 21.4 | NT | 17.4 | 27.5 | >100 | 77.6 | 74.1 | 5.13 | 53.7 | 0 |
| 51 | 28.8 | NT | 27.5 | 89.1 | >100 | >100 | 81.3 | 4.57 | 44.7 | 0 |
| 52 | 2.8 | 0.9 | - | 2.6 | 1.7 | 2.5 | 1.2 | 2.7 | 2.4 | ++++ |
| 53 | 2.9 | 1.3 | - | 1.9 | 8.1 | 1.7 | 4.6 | 3.4 | 3.0 | ++ |
| 54 | – | 23 | - | 37 | 20 | 20 | 52 | 38 | 41 | ++++ |
| 55 | – | 43 | - | 44 | 0.88 | 33 | >100 | 68 | 59 | ++ |
The cytotoxicity GI50 values are the concentrations corresponding to 50% growth inhibition.
Mean graph midpoint for growth inhibition of all human cancer cell lines successfully tested.
The compounds were tested at concentrations ranging up to 10 μM. The activity of the compounds to produce top1-mediated DNA cleavage was expressed semi-quantitatively as follows: +: weak activity; ++: similar activity as the parent compound 2; +++ & ++++: greater activity than the parent compound 2; ++++: similar activity as 1 μM camptothecin (1).
From the biological data presented in Table 1, several general features are evident from the investigation of optimal lactam chain length. First, carbamate-protected indenoisoquinolines such as compounds 31–36 demonstrated poor cytotoxicity and top1 inhibition relative to camptothecin (1) and poor top1 inhibition relative to the parent indenoisoquinoline 2. This structure-activity relationship has been previously identified with carbamate-protected bisindenoisoquinolines33 and is believed to arise from inefficient targeting to DNA (relative to the deprotected, positively charged derivatives), detrimental steric interactions in the ternary complex, and general hydrophobicity of the molecules relative to the deprotected derivatives.
In general, potent biological activity was observed for the deprotected, alkylamino indenoisoquinolines 37–47 relative to their carbamate-protected precursors (Table 1). From the data reported in Table 1, it is clear that biological activity is greatly improved, both for cytotoxicity and top1 inhibition, with the utilization of shorter lactam side chains. Indeed, maximum biological activity was observed for the previously reported19 compounds 37–39, with 2–4 carbon chain lengths. Thus, the historical use of two and three carbon lactam substituent chain lengths has been validated and experimentally confirmed to be optimal for developing potent indenoisoquinoline top1 inhibitors. Interestingly, there are two anomalies in the generalization that “shorter chain length is better” for biological activity. First, compound 25, with a hydrazine-derived lactam side chain was poorly cytotoxic and an inactive top1 inhibitor (MGM: 39.8 μM; top1: 0). This result is believed to be due to inefficient targeting to DNA (since the side chain of 25 should not have a fixed positive charge at physiological pH). Furthermore, hypothetical binding models of 25 in ternary complex with DNA and top1 did not show any stabilizing hydrogen bonding interaction between the amino group and either the DNA base pairs at the site of intercalation or with top1. This finding was in stark contrast to the terminal amines of compounds 37–39, which displayed stabilizing hydrogen bonding interactions in hypothetical models.19 The second anomaly worth mentioning is the discrepancy between the cytotoxicity of 40 and its top1 inhibition (MGM: 0.471 μM; top1: 0). This result is not entirely without precedence and it is obvious that the cytotoxicity of this molecule is not related to its ability to inhibit top1, but must be dependent upon another, unknown biological target.19,33
The DNA cleavage patterns produced by camptothecin (1, lane 3 of each gel), the indenoisoquinoline NSC 314622, and compounds 39, 41, 43, and 45 are displayed in Figure 2. The following points are apparent from inspection of the gels: 1) The potencies of the indenoisoquinolines as top1 inhibitors are reflected in the intensities of the DNA cleavage bands. The bands produced by compound 39 (top1: +++) are slightly weaker in comparison with camptothecin, but stronger in intensity than the bands produced by 41 (top1: ++), 43 (top1: +), and 45 (top1: 0). Thus, a comparison of the gels in Figure 2 indicates that as the lactam side chains of the indenoisoquinolines increase, a corresponding decrease in the ability to inhibit top1 is observed. 2) Top1 inhibitors can be classified as top1 suppressors, which inhibit DNA cleavage, and top1 poisons, which inhibit the re-ligation reaction after DNA cleavage. As compund 39 illustrates (lanes 7 and 8), the predominant top1 cleavage site (marker 44) is trapped at lower compound concentrations and suppressed at higher concentrations, and therefore indenoisoquinoline 39 acts as a top1 poison at lower concentrations and top1 suppressor at higher concentrations. The suppression could result from binding of the drug to the DNA rendering it a poorer enzyme substrate at high drug concentration, or from a direct effect on the enzyme to suppress its ability to cleave DNA. 3) There are differences in the cleavage pattern of camptothecin vs. the indenoisoquinolines. For example, the cleavage at base pair 44 seen with the indenoisoquinolines is absent with camptothecin. This difference may indicate that the indenoisoquinolines might display antitumor spectra different from camptothecin or its clinically useful derivatives irinotecan and topotecan.
Figure 2.

Lane 1: DNA alone; lane 2: top1 alone; lane 3: + CPT (1 μM); in the first panel, lane 4: top1 + NSC 314622 (100 μM); lanes 5–8 (for compound 39) and lanes 4–7 (for compounds 41, 43, and 45): top1 + indicated compound at 0.1, 1, 10, and 100 μM, respectively. Number on left and arrows indicate cleavage site positions.
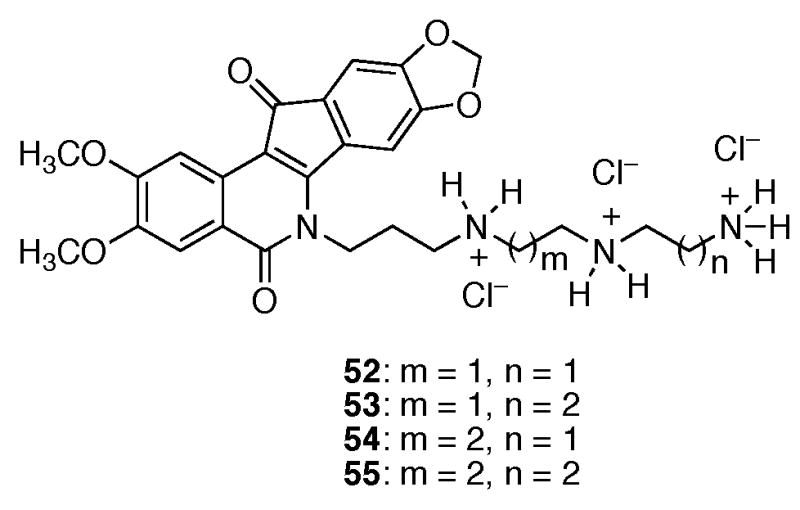
Although the knowledge that a shorter alkyl side chain is important for furthering drug development efforts for indenoisoquinolines, the cause of this result is equally important. Molecular modeling of compounds 37–47 in ternary complex with DNA and top1 (PDB code 1SC7)15 indicated that the majority of these molecules should exert similar top1 inhibitory activities, since the side chains project out into the major groove and can be readily accommodated sterically.15 However, the results from Table 1 show that as the length of the lactam side chain increases, there is a general decrease in top1 inhibition after a 4-carbon atom length. Rationalization of the biological data was therefore initially sought without the use of molecular models of the ternary complexes. Upon further inspection of Table 1 regarding only the compounds assumed to be protonated at physiological pH (37–47), it was realized that as the lactam chain length increased, there was an increase in the MGM values of the compounds (as well as the previously observed decrease in top1 inhibition). Since these compounds differed only by the incremental increase of methylene units, the effect could possibly be hydrophobic in nature. Indeed, calculation of the ClogP values of compounds 37–47 (performed in ChemDraw Ultra Version 9.0.1) and plotting against log(MGM) resulted in a linear dependence with an r2 value of 0.90, indicating a high degree of correlation (Figure 3). Thus, for this series of compounds general hydrophobicity, as measured by ClogP values, appears to be predictive of cytotoxicity in the NCI’s 60 cell screen. Inspection of the ternary complex of topotecan bound to DNA and top1 (PDB code 1K4T)14 reveals the presence of a hydrated binding pocket and appears to provide an essential clue supporting both the data in Table 1 and the relationship between ClogP and cytotoxicity (Figure 3). A hypothetical model was subsequently developed utilizing the topotecan crystal structure and the experimentally determined structural overlay between topotecan and the indenoisoquinolines (Figure 4).14,15,17 Overlaying the hypothetical models for compounds 37, 39, 41, 43, 45, and 47 (colored by atom type) in ternary complex with DNA (red) and top1 (red) allows the visualization of the extensive hydrophilic binding pocket (water molecules are indicated in green) for the indenoisoquinoline side chain. Increasing the number of methylene units beyond four in the lactam side chain of the indenoisoquinolines results in an increasingly hydrophobic linker with increasingly negative interactions with the hydrated binding pocket. In agreement with this hypothesis, polyamine-substituted indenoisoquinolines 52–55 retain significant top1 inhibitory activity in the ++ to ++++ range.24
Figure 3.

ClogP vs. Log(MGM).
Figure 4.
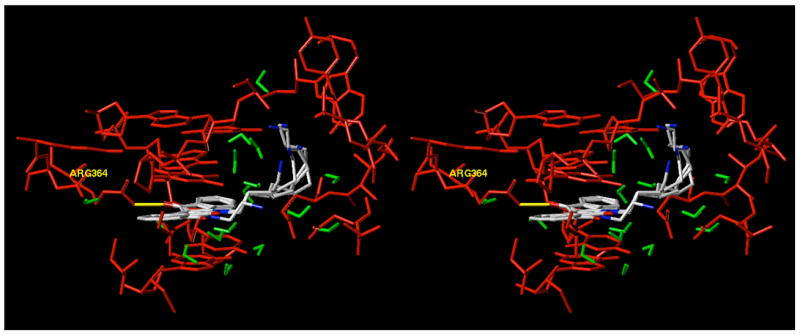
Overlay of the hypothetical models derived from PDB code 1K4T for compounds 37, 39, 41, 43, 45, and 47 (colored by atom type) in ternary complex with DNA (red) and top1 (red). Water molecules are indicated in green and the hydrogen bond between the analogues and Arg364 is shown in yellow. The figure is programmed for wall-eyed (relaxed) viewing.
Having experimentally confirmed that the optimal lactam chain length for cytotoxicity and top1 inhibition for the indenoisoquinolines was 2–4 carbon atoms between the lactam nitrogen and the terminal amino group, this relationship was further scrutinized by preparing analogues 48–51 that utilize a pyridine ring in place of a portion of the alkyl linker and the terminal amino group. However, the newly designed analogues still maintained a 2–4 carbon atom linker between the lactam nitrogen and the nitrogen atom in the pyridine ring. Although all of the analogues were less cytotoxic than their corresponding alkyl chain analogues, compound 49 displayed potent top1 inhibition equal in magnitude to compounds 37–39. With the discrepancy between cytotoxicity and top1 inhibition, it seems reasonable to infer that protonation of the terminal amino group of unsubstituted indenoisoquinolines is important for cellular cytotoxicity (perhaps by improved DNA targeting) but is not necessarily a requirement for potent top1 inhibition. Indeed, this result has been observed in another indenoisoquinoline study involving heterocycles appended to the lactam side chain and the present study of pyridine heterocycles re-affirms previously derived structure-activity relationships.34 However, the absence of protonation of the pyridine nitrogen at physiological pH should not be construed as the only reason for the discrepancies in biological activity between compounds 37–39 and 48–51. Furthermore, the pyridine-substituted analogs don’t provide strong support of the currently established optimal length of the linker chain. This could be a result of several factors including, but not limited to, reduced flexibility of the side chain, increased steric bulk, disrupted hydrogen-bonding networks with the aqueous environment, as well as the aforementioned protonation state of the pyridine nitrogen at physiological pH and DNA targeting.
Compounds 37–39 are more cytotoxic than compounds 52–55 and 48–51. Thus, incorporating polar motifs (such as polyamino groups) on the lactam side chain of the indenoisoquinolines does not necessarily translate into increased cytotoxicity but does improve Top1 inhibition. Additionally, structural changes to the lactam side chain that reduce its flexibility, such as the present case for the pyridine containing analogs, result in discrepancies between enzyme inhibition and cytotoxicity. The presently observed relationship between ClogP and cytotoxicity indicates that drug design and molecular modeling of the indenoisoquinolines should consider the environment of the lactam side chain in the ternary complex. Multiple variables in addition to lipophilicity are expected to be involved in cellular uptake, distribution, and ultimately cytotoxicity of the indenoisoquinolines (as well as other DNA targeting drug molecules). However, lipophilicity has previously been demonstrated to affect the nonspecific binding of anthracycline antitumor drugs to serum albumin and it seems reasonable to assume that nonspecific binding (or unintended localization) could occur intracellularly with the indenoisoquinolines and result in discrepant biological activities.35,36 Differences in structure, such as the replacement of the indenoisoquinoline side chain with a pyridine ring or polyamino groups, are also expected to result in different rates of uptake, distribution, metabolism, and excretion within the cancer cell. Hence, discrepancies will ultimately arise between enzyme-based assays and the NCI 60 cell line screen for cytotoxicity.
In conclusion, a series of indenoisoquinolines have been prepared to determine the optimal carbon chain length between the lactam nitrogen and the terminal amine of the side chain. The results of this study confirmed that the optimal carbon linker for biological activity (both top1 inhibition and cytotoxicity) is 2–4 atoms, thereby validating the use of this length in past and future efforts to develop indenoisoquinolines as top1 inhibitors. Interestingly, the study indicated a strong correlation between ClogP values and MGM values for indenoisoquinolines with protonated terminal amines on the lactam side chain. Furthermore, the substitution of a pyridine ring in indenoisoquinoline 49 in place of a portion of the linker and the terminal amino group resulted in a discrepancy between top1 inhibition and cytotoxicity, indicating that a protonated terminal amino group on the indenoisoquinolines is more important for maintaining cytotoxicity than top1 inhibition.
Experimental Section
General
Melting points were determined using capillary tubes with a Mel-Temp apparatus and are uncorrected. Infrared spectra were obtained using CHCl3 as the solvent unless otherwise specified. The proton nuclear magnetic resonance (1H NMR) spectra were recorded using an ARX300 300 MHz Bruker NMR spectrometer. IR spectra were recorded using a Perkin-Elmer 1600 series FTIR spectrometer. Combustion microanalyses were performed at the Purdue University Microanalysis Laboratory and the reported values were within 0.4% of the calculated values. Analytical thin-layer chromatography was carried out on Baker-flex silica gel IB2-F plates and compounds were visualized with short wavelength UV light. Silica gel flash chromatography was performed using 230–400 mesh silica gel. Compounds 12–14, 26–28, and 37–39 were prepared as previously described.19
General Procedure for the Preparation of Mono-Boc-Protected Diamines 12–20
Boc2O (0.500 g, 2.291 mmol) was dissolved in CHCl3 (10 mL) and the solution was added dropwise to a solution of diamine (11.45 mmol) in CHCl3 (50 mL). The reaction mixture was allowed to stir at room temperature for 24 h, concentrated, and purified by flash column chromatography (SiO2), eluting with a solution of 1% Et3N/10% MeOH in CHCl3, to provide the mono-Boc-protected diamine.
Mono-Boc-1,7-diaminoheptane (15).37
The general procedure provided the desired compound as a colorless semisolid (0.473 g, 90%). 1H NMR (CDCl3) δ 4.52 (bs, 1 H), 3.12 (q, J = 6.2 Hz, 2 H), 2.70 (t, J = 6.8 Hz, 2 H), 1.43-1.23 (m, 19 H).
Mono-Boc-1,8-diaminooctane (16).38
The general procedure provided the desired compound as a colorless semisolid (0.492 g, 88%). 1H NMR (CDCl3) δ 4.51 (bs, 1 H), 3.12 (q, J = 6.5 Hz, 2 H), 2.69 (t, J = 6.8 Hz, 2 H), 1.43-1.23 (m, 21 H).
Mono-Boc-1,9-diaminononane (17).39
The general procedure provided the desired compound as a colorless semisolid (0.125 g, 21%). 1H NMR (CDCl3) δ 4.50 (bs, 1 H), 3.12 (q, J = 6.5 Hz, 2 H), 2.70 (t, J = 6.8 Hz, 2 H), 1.44-1.22 (m, 23 H).
Mono-Boc-1,10-diaminodecane (18).39
The general procedure provided the desired compound as a colorless semisolid (0.192 g, 31%). 1H NMR (CDCl3) δ 1H NMR (CDCl3) δ 4.50 (bs, 1 H), 3.13 (q, J = 6.3 Hz, 2 H), 2.71 (t, J = 6.9 Hz, 2 H), 1.44-1.18 (m, 27 H).
Mono-Boc-1,11-diaminoundecane (19)
The general procedure provided the desired compound as a colorless solid (0.555 g, 85%): mp 30–34 °C. IR (film) 3370, 2919, 2851, 1687, and 1522 cm−1; 1H NMR (CDCl3) δ 4.49 (bs, 1 H), 3.11 (q, J = 6.5 Hz, 2 H), 2.71 (t, J = 6.8 Hz, 2 H), 1.44-1.27 (m, 29 H); ESIMS m/z (rel intensity) 287 (MH+, 100). Anal. (C16H34N2O2) C, H, N.
Mono-Boc-1,12-diaminododecane (20).39,40
The general procedure provided the desired compound as a colorless semisolid (0.191 g, 28%). 1H NMR (CDCl3) δ 4.48 (bs, 1 H), 3.11 (q, J = 6.2 Hz, 2 H), 2.76 (t, J = 6.9 Hz, 2 H), 1.44-1.26 (m, 31 H).
6-Amino-5,6-dihydro-5,11-diketo-11H-indeno[1,2-c]isoquinoline (25)
Benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.150 g, 0.604 mmol) was treated with hydrazine (22) (0.255 g, 7.964 mmol) in CHCl3 (50 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, diluted with CHCl3 (150 mL), and washed with sat NaHCO3 (2 × 50 mL). The solution was dried over sodium sulfate and concentrated to provide a red-orange solid (0.120 g, 76%): mp 272–274 °C. IR (film) 3448, 3305, 1686, 1663, 1610, 1507, 1312, 762 cm−1; 1H NMR (CDCl3) δ 8.54 (d, J = 7.8 Hz, 1 H), 8.51 (d, J = 7.2 Hz, 1 H), 8.24 (d, J = 7.4 Hz, 1 H), 7.85 (m, 1 H), 7.60-7.45 (m, 4 H), 6.19 (s, 2 H); EIMS m/z (rel intensity) 262 (M+, 100). Anal. (C16H10N2O2·0.25 H2O) C, H, N.
General Procedure for the Preparation of Mono-Boc-Protected Indenoisoquinolines 31–36
Mono-Boc-protected diamine (2.054 mmol) was added to a solution of benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.255 g, 1.027 mmol) in CHCl3 (100 mL). The reaction mixture was heated at reflux for 24 h, concentrated, and purified by flash column chromatography (SiO2), eluting with CHCl3, to provide the mono-Boc-protected indenoisoquinoline.
6-(7′-tert-BOC-Aminoheptyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (31)
The general procedure provided the desired compound as a yellow-orange solid (0.451 g, 95%): mp 112–116 °C. IR (film) 3369, 1697, 1664, 1503, and 1172 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 8.1 Hz, 1 H), 8.23 (d, J = 8.2 Hz, 1 H), 7.84-7.79 (m, 1 H), 7.71 (d, J = 7.7 Hz, 1 H), 7.63-7.50 (m, 4 H), 6.76 (m, 1 H), 4.50 (t, J = 7.4 Hz, 2 H), 2.90 (q, J = 6.2 Hz, 2 H), 1.77 (m, 2 H), 1.46-1.28 (m, 17 H); ESIMS m/z (rel intensity) 483 (MNa+, 100). Anal. (C28H32N2O4) C, H, N.
6-(8′-tert-BOC-Aminooctyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (32)
The general procedure provided the desired compound as a yellow-orange solid (0.466 g, 97%): mp 140–143 °C. IR (film) 3368, 2929, 1698, 1665, 1504, and 1172 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 7.9 Hz, 1 H), 8.23 (d, J = 8.1 Hz, 1 H), 7.84-7.78 (m, 1 H), 7.71 (d, J = 7.5 Hz, 1 H), 7.63-7.47 (m, 4 H), 6.75 (m, 1 H), 4.50 (t, J = 7.4 Hz, 2 H), 2.91 (q, J = 6.6 Hz, 2 H), 1.78 (m, 2 H), 1.46-1.26 (m, 19 H); ESIMS m/z (rel intensity) 497 (MNa+, 100). Anal. (C29H34N2O4) C, H, N.
6-(9′-tert-BOC-Aminononyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (33)
The general procedure provided the desired compound as an orange solid (0.145 g, 77%): mp 91–95 °C. IR (film) 3371, 2928, 1698, 1666, 1504, and 1172 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 8.0 Hz, 1 H), 8.23 (d, J = 7.4 Hz, 1 H), 7.84-7.79 (m, 1 H), 7.71 (d, J = 7.4 Hz, 1 H), 7.63-7.50 (m, 4 H), 6.74 (m, 1 H), 4.50 (t, J = 7.3 Hz, 2 H), 2.91 (q, J = 6.6 Hz, 2 H), 1.78 (m, 2 H), 1.47-1.24 (m, 21 H); ESIMS m/z (rel intensity) 511 (MNa+, 100). Anal. (C30H36N2O4) C, H, N.
6-(10′-tert-BOC-Aminodecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (34)
The general procedure provided the desired compound as a yellow-orange solid (0.220 g, 78%): mp 135–137 °C. IR (film) 3368, 2927, 1698, 1666, 1504, and 1172 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 7.9 Hz, 1 H), 8.23 (dd, J = 8.1 Hz and 0.7 Hz, 1 H), 7.84 (dt, J = 7.2 Hz and 1.4 Hz, 1 H), 7.71 (d, J = 7.5 Hz, 1 H), 7.63-7.47 (m, 4 H), 6.76 (m, 1 H), 4.50 (t, J = 7.4 Hz, 2 H), 2.90 (q, J = 6.5 Hz, 2 H), 1.77 (m, 2 H), 1.46-1.23 (m, 23 H); ESIMS m/z (rel intensity) 525 (MNa+, 100). Anal. (C31H38N2O4) C, H, N.
6-(11′-tert-BOC-Aminoundecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (35)
The general procedure provided the desired compound as a yellow-orange solid (0.445 g, 86%): mp 111–114 °C. IR (KBr) 3364, 2918, 2850, 1678, 1660, 1534, 1505, and 758 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 8.1 Hz, 1 H), 8.23 (d, J = 7.4 Hz, 1 H), 7.84-7.79 (m, 1 H), 7.71 (d, J = 7.5 Hz, 1 H), 7.62-7.50 (m, 4 H), 6.74 (m, 1 H), 4.50 (t, J = 7.4 Hz, 2 H), 2.90 (q, J = 6.5 Hz, 2 H), 1.78 (m, 2 H), 1.46-1.22 (m, 25 H); ESIMS m/z (rel intensity) 539 (MNa+, 100). Anal. (C32H40N2O4) C, H, N.
6-(12′-tert-BOC-Aminododecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (36)
The general procedure provided the desired compound as a yellow-orange solid (0.177 g, 66%): mp 129–134 °C. IR (film) 3369, 2926, 1698, 1666, 1504, and 1172 cm−1; 1H NMR (DMSO-d6) δ 8.58 (d, J = 8.0 Hz, 1 H), 8.22 (d, J = 7.4 Hz, 1 H), 7.84-7.78 (m, 1 H), 7.71 (d, J = 7.5 Hz, 1 H), 7.62-7.50 (m, 4 H), 6.74 (m, 1 H), 4.50 (t, J = 7.3 Hz, 2 H), 2.90 (q, J = 6.6 Hz, 2 H), 1.77 (m, 2 H), 1.46-1.22 (m, 27 H); ESIMS m/z (rel intensity) 553 (MNa+, 100). Anal. (C33H42N2O4) C, H, N.
6-(5-Aminopentyl)-5,6-dihydro-5,11-diketo-11H-indeno[1,2-c]isoquinoline Hydrochloride (40)
Benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) was treated with diamine 23 (0.206 g, 2.014 mmol) in CHCl3 (40 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature and washed with water (3 × 15 mL). The solution was dried over sodium sulfate, filtered, and treated with 2 M HCl in Et2O (5 mL). After 30 min, the reaction mixture was filtered and the filter pad was washed with CHCl3 (50 mL) and hexanes (50 mL) to provide an orange solid (0.122 g, 82%): mp 265–268 °C. IR (film) 3432, 3077, 2856, 1707, 1635, 1611, 1549, and 1504 cm−1; 1H NMR (CDCl3) δ 8.59 (d, J = 8.1 Hz, 1 H), 8.23 (d, J = 8.1 Hz, 1 H), 7.85-7.80 (m, 3 H), 7.74 (d, J = 7.4 Hz, 1 H), 7.63-7.51 (m, 4 H), 4.52 (t, J = 7.3 Hz, 2 H), 2.81 (m, 2 H), 1.83 (m, 2 H), 1.65-1.52 (m, 4 H); ESIMS m/z (rel intensity) 333 (MH+, 100). Anal. (C21H21ClN2O2·0.75 H2O) C, H, N.
6-(6-Aminohexyl)-5,6-dihydro-5,11-diketo-11H-indeno[1,2-c]isoquinoline Hydrochloride (41)
Benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) was treated with diamine 24 (0.234 g, 2.014 mmol) in CHCl3 (40 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature and washed with water (3 × 25 mL). The solution was dried over sodium sulfate, filtered, and treated with 2 M HCl in Et2O (5 mL). After 30 min, the reaction mixture was filtered and the filter pad was washed with CHCl3 (50 mL) and hexanes (50 mL) to provide an orange solid (0.125 g, 81%): mp 195 °C (dec). IR (film) 3435, 1660, 1630, 1610, and 1504 cm−1; 1H NMR (CDCl3) δ 8.59 (d, J = 7.8 Hz, 1 H), 8.23 (d, J = 8.1 Hz, 1 H), 7.85-7.71 (m, 4 H), 7.61-7.51 (m, 4 H), 4.52 (t, J = 7.3 Hz, 2 H), 2.78 (m, 2 H), 1.79 (m, 2 H), 1.59-1.39 (m, 6 H); ESIMS m/z (rel intensity) 347 (MH+, 100). Anal. (C22H23ClN2O2·0.5 H2O) C, H, N.
General Procedure for the Preparation of Indenoisoquinoline Hydrochloride Salts 42–47
3 M HCl in MeOH (10 mL) was slowly added to a solution of mono-Boc-protected indenoisoquinoline (0.100 g, 0.188–0.217 mmol) in CHCl3 (50 mL) at room temperature. After 2 h, the reaction mixture was concentrated and the residue was triturated with Et2O. Filtration of the obtained solid provided the indenoisoquinoline as a hydrochloride salt.
6-(7-Aminoheptyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (42)
The general procedure provided the desired compound as a yellow-orange solid (0.085 g, 99%): mp 228–231 °C. IR (KBr) 3436, 2931, 1702, 1650, 1611, 1549, 1504, and 759 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 8.1 Hz, 1 H), 8.24 (d, J = 8.2 Hz, 1 H), 7.85-7.80 (m, 1 H), 7.73 (d, J = 7.4 Hz, 1 H), 7.63-7.49 (m, 6 H), 4.53 (t, J = 7.0 Hz, 2 H), 2.78 (t, J = 7.1 Hz, 2 H), 1.80 (m, 2 H), 1.55-1.35 (m, 8 H); ESIMS m/z (rel intensity) 361 (MH+, 100). Anal. (C23H25ClN2O2·0.5 H2O) C, H, N.
6-(8-Aminooctyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (43)
The general procedure provided the desired compound as an orange solid (0.083 g, 95%): mp 182–185 °C. IR (KBr) 3436, 2930, 1661, 1505, and 761 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 8.5 Hz, 1 H), 8.24 (d, J = 7.0 Hz, 1 H), 7.86-7.81 (m, 1 H), 7.73 (d, J = 7.6 Hz, 1 H), 7.63-7.52 (m, 6 H), 4.52 (t, J = 7.9 Hz, 2 H), 2.78 (t, J = 7.3 Hz, 2 H), 1.79 (m, 2 H), 1.50 (m, 4 H), 1.31 (m, 6 H); ESIMS m/z (rel intensity) 375 (MH+, 100). Anal. (C24H27ClN2O2·0.75 H2O) C, H, N.
6-(9-Aminononyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (44)
The general procedure provided the desired compound as an orange solid (0.082 g, 94%): mp 204–207 °C. IR (KBr) 3435, 2927, 1702, 1662, 1610, 1549, 1504, 1427, and 759 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 8.3 Hz, 1 H), 8.24 (d, J = 9.3 Hz, 1 H), 7.83-7.81 (m, 1 H), 7.73 (d, J = 7.8 Hz, 1 H), 7.63-7.51 (m, 6 H), 4.52 (t, J = 8.3 Hz, 2 H), 2.78 (t, J = 7.3 Hz, 2 H), 1.79 (m, 2 H), 1.51 (m, 4 H), 1.28 (m, 8 H); ESIMS m/z (rel intensity) 389 (MH+, 100). Anal. (C25H29ClN2O2·0.75 H2O) C, H, N.
6-(10-Aminodecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (45)
The general procedure provided the desired compound as an orange solid (0.087 g, 91%): mp 189–192 °C. IR (KBr) 3443, 2925, 2851, 1705, 1646, 1611, 1550, 1504, 1467, and 759 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 7.9 Hz, 1 H), 8.23 (d, J = 7.5 Hz, 1 H), 7.83 (m, 1 H), 7.73 (d, J = 8.0 Hz, 1 H), 7.63-7.51 (m, 6 H), 4.52 (t, J = 7.4 Hz, 2 H), 2.76 (m, 2 H), 1.79 (m, 2 H), 1.49 (m, 4 H), 1.27 (m, 10 H); ESIMS m/z (rel intensity) 403 (MH+, 100). Anal. (C26H31ClN2O2·0.5 H2O) C, H, N.
6-(11-Aminoundecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (46)
The general procedure provided the desired compound as an orange solid (0.085 g, 88%): mp 125–129 °C. IR (KBr) 3436, 2922, 2851, 1662, 1610, 1549, 1504, 1426, and 758 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 8.0 Hz, 1 H), 8.23 (d, J = 7.5 Hz, 1 H), 7.85 (m, 1 H), 7.72 (d, J = 7.5 Hz, 1 H), 7.63-7.51 (m, 6 H), 4.51 (t, J = 8.0 Hz, 2 H), 2.77 (t, J = 7.6 Hz, 2 H), 1.78 (m, 2 H), 1.48 (m, 4 H), 1.25 (m, 12 H); ESIMS m/z (rel intensity) 417 (MH+, 100). Anal. (C26H31ClN2O2·1 H2O) C, H, N.
6-(12-Aminododecyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (47)
The general procedure provided the desired compound as a yellow solid (0.087 g, 91%): mp 175–178 °C. IR (KBr) 3435, 2927, 2850, 1704, 1644, 1506, 1466, and 762 cm−1; 1H NMR (DMSO-d6) δ 8.59 (d, J = 7.8 Hz, 1 H), 8.23 (d, J = 7.5 Hz, 1 H), 7.83 (m, 1 H), 7.72 (d, J = 7.8 Hz, 1 H), 7.63-7.51 (m, 6 H), 4.51 (t, J = 7.5 Hz, 2 H), 2.76 (m, 2 H), 1.78 (m, 2 H), 1.51 (m, 4 H), 1.24 (m, 14 H); ESIMS m/z (rel intensity) 431 (MH+, 100). Anal. (C28H35ClN2O2·1.25 H2O) C, H, N.
5,6-Dihydro-5,11-dioxo-6-(2-pyridylmethyl)-11H-indeno[1,2-c]isoquinoline (48)
2-(Aminomethyl)pyridine (0.054 g, 0.504 mmol) was added to a solution of benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) in CHCl3 (50 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, washed with H2O (3 × 25 mL), sat NaCl (25 mL), dried over sodium sulfate, and concentrated. The residue was washed with EtOAc, hexanes, and dried to provide a yellow solid (0.110 g, 81%): mp 240–242 °C. IR (KBr) 1698, 1655, 1618, 1501, 1427, and 755 cm−1; 1H NMR (DMSO-d6) δ 8.62 (d, J = 8.0 Hz, 1 H), 8.56 (d, J = 4.9 Hz, 1 H), 8.22 (d, J = 8.1 Hz, 1 H), 7.97 (dt, J = 7.8 Hz and 1.7 Hz, 1 H), 7.89 (m, 1 H), 7.61-7.37 (m, 7 H), 5.91 (s, 2 H); ESIMS m/z (rel intensity) 339 (MH+, 100). Anal. (C22H14N2O2) C, H, N.
5,6-Dihydro-5,11-dioxo-6-(3-pyridylmethyl)-11H-indeno[1,2-c]isoquinoline Hydrochloride (49)
3-(Aminomethyl)pyridine (0.054 g, 0.504 mmol) was added to a solution of benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) in CHCl3 (50 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, washed with H2O (3 × 25 mL), sat NaCl (25 mL), dried over sodium sulfate, and concentrated. The residue was diluted with CHCl3 (40 mL), 3 M HCl in MeOH (10 mL) was added, and the reaction mixture was allowed to stir at room temperature for 2 h. The reaction mixture was concentrated, and the residue was washed with CHCl3 to provide a pink solid (0.146 g, 97%): mp 274 °C (dec). IR (KBr) 2343, 2106, 1695, 1655, 1610, 1551, 1501, and 754 cm−1; 1H NMR (DMSO-d6) δ 8.98 (s, 1 H), 8.78 (d, J = 5.2 Hz, 1 H), 8.64 (d, J = 8.1 Hz, 1 H), 8.36 (d, J = 9.1 Hz, 1 H), 8.23 (d, J = 7.5 Hz, 1 H), 7.90 (m, 2 H), 7.59-7.39 (m, 5 H), 5.89 (s, 2 H); ESIMS m/z (rel intensity) 339 (MH+, 100). Anal. (C22H15ClN2O2) C, H, N.
5,6-Dihydro-5,11-dioxo-6-(2-pyridylethyl)-11H-indeno[1,2-c]isoquinoline Hydrochloride (50)
2-(2-Aminoethyl)pyridine (0.098 g, 0.806 mmol) was added to a solution of benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) in CHCl3 (50 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, washed with H2O (3 × 25 mL), sat NaCl (25 mL), dried over sodium sulfate, and concentrated. The residue was diluted with CHCl3 (40 mL), 3 M HCl in MeOH (10 mL) was added, and the reaction mixture was allowed to stir at room temperature for 2 h. The reaction mixture was concentrated, and the residue was washed with CHCl3 to provide a yellow solid (0.146 g, 93%): mp 240 °C (dec). IR (KBr) 2307, 1698, 1659, 1610, 1548, 1504, 1429, and 760 cm−1; 1H NMR (DMSO-d6) δ 8.78 (d, J = 5.5 Hz, 1 H), 8.57 (d, J = 8.0 Hz, 1 H), 8.33 (m, 1 H), 8.02 (d, J = 8.0 Hz, 1 H), 7.97 (d, J = 8.0 Hz, 1 H), 7.90 (d, J = 8.0 Hz, 1 H), 7.80 (m, 2 H), 7.61-7.44 (m, 4 H), 4.92 (t, J = 6.5 Hz, 2 H), 3.59 (t, J = 6.3 Hz, 2 H); ESIMS m/z (rel intensity) 353 (MH+, 100). Anal. (C23H17ClN2O2) C, H, N.
5,6-Dihydro-5,11-dioxo-6-(3-pyridylethyl)-11H-indeno[1,2-c]isoquinoline (51)
3-(2-Aminoethyl)pyridine (0.172 g, 0.604 mmol) was added to a solution of benz[d]indeno[1,2-b]pyran-5,11-dione (21)29 (0.100 g, 0.403 mmol) in CHCl3 (50 mL). Triethylamine (0.224 mL, 1.612 mmol) was added and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, washed with H2O (3 × 25 mL), sat NaCl (25 mL), dried over sodium sulfate, and concentrated. The obtained precipitate was washed with EtOAc, hexanes, and dried to provide an orange solid (0.140 g, 99%): mp 220 °C (dec). IR (KBr) 1691, 1660, 1609, 1549, 1504, 1424, and 765 cm−1; 1H NMR (DMSO-d6) δ 8.90 (s, 1 H), 8.73 (d, J = 5.6 Hz, 1 H), 8.59 (d, J = 8.1 Hz, 1 H), 8.44 (d, J = 7.9 Hz, 1 H), 8.10 (d, J = 8.1 Hz, 1 H), 7.88 (m, 3 H), 7.61-7.48 (m, 4 H), 4.87 (t, J = 6.7 Hz, 2 H), 3.37 (t, J = 6.3 Hz, 2 H); ESIMS m/z (rel intensity) 353 (MH+, 100). Anal. (C23H16N2O2·0.55 H2O) C, H, N.
Top1-Mediated DNA Cleavage Reactions
Human recombinant Top1 was purified from Baculovirus as described previously.41 The 161 bp fragment from pBluescript SK(−) phagemid DNA (Stratagene, La Jolla, CA) was cleaved with the restriction endonuclease Pvu II and Hind III (New England Biolabs, Beverly, MA) in supplied NE buffer 2 (50 μL reactions) for 1 h at 37 °C, and separated by electrophoresis in a 1% agarose gel made in 1X TBE buffer. The 161 bp fragment was eluted from the gel slice using the QIAEX II kit (QIAGEN Inc., Valencia, CA). Approximately 200 ng of the fragment was 3′-end labeled at the Hind III site by fill-in reaction with [alpha-32P]-dGTP and 0.5 mM dATP, dCTP, and dTTP, in React 2 buffer (50 mM Tris-HCl, pH 8.0, 100 mM MgCl2, 50 mM NaCl) with 0.5 units of DNA polymerase I (Klenow fragment). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3′-end-labeled 161 bp fragment was collected. Aliquots (approximately 50,000 dpm/reaction) were incubated with Top1 at 22 °C for 30 min in the presence of the tested drug. Reactions were terminated by adding SDS (0.5% final concentration). The samples (10 μL) were mixed with 30 μL of loading buffer (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue, pH 8.0). Aliquots were separated in denaturing gels (16% polyacrylamide, 7 M urea). Gels were dried and visualized by using a Phosphoimager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Molecular Modeling
The structure of the ternary complex, containing topoisomerase I, DNA, and topotecan, was downloaded from the Protein Data Bank (PDB code 1K4T).14 One molecule of PEG and the topotecan carboxylate form were deleted. Several of the atoms were then fixed according to Sybyl® atom types. Hydrogens were added and minimized using the MMFF94s force field and MMFF94 charges. The structure of indenoisoquinolines 37–47, constructed in Sybyl® and energy minimized with the MMFF94s force field and MMFF94 charges, was overlapped with the structure of topotecan according to the proposed structural similarity16,17 in the ternary complex, and the structure of topotecan was then deleted. The new whole complex was subsequently subjected to energy minimization using the MMFF94s force field with MMFF94 charges. During energy minimization, the structure of the indenoisoquinoline was allowed to move while the structures of the protein, nucleic acid, and water molecules were frozen. The energy minimization was performed using the Powell method with a 0.05 kcal/mol·Å energy gradient convergence criterion and a distance-dependent dielectric constant.
Supplementary Material
Elemental analyses for compounds 19, 25, 31–36, and 40–51. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.
Representative topoisomerase I inhibitors.
Acknowledgments
This work was supported by Research Grant UO1 CA89566, Training Grant ST32 CA09634-12, by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and the American Chemical Society Division of Medicinal Chemistry and Pfizer Global Research and Development for predoctoral support of A.M. This research was conducted in a facility constructed with support from Research Facilities Improvement Program Grant C06-14400 from the National Institutes of Health.
References
- 1.Wang JC. DNA Topoisomerases. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Nitiss JL. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim Biophys Acta. 1998;1400:63–81. doi: 10.1016/s0167-4781(98)00128-6. [DOI] [PubMed] [Google Scholar]
- 3.Champoux JJ. DNA Topoisomerases: Structure, Function, and Mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 4.Wang JC. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 5.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin Induces Protein-Linked DNA Breaks via Mammalian DNA Topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 6.Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of Action of Eukaryotic DNA Topoisomerase I and Drugs Targeted to the Enzyme. Biochim Biophys Acta. 1998;1400:83–106. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 7.Takimoto CH, Wright J, Arbuck SG. Clinical Applications of the Camptothecins. Biochim Biophys Acta. 1998;1400:107–119. doi: 10.1016/s0167-4781(98)00130-4. [DOI] [PubMed] [Google Scholar]
- 8.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 9.Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-Activity Study of the Actions of Camptothecin Derivatives on Mammalian Topoisomerase I: Evidence for a Specific Receptor Site and a Relation to Antitumor Activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 10.Minami H, Beijnen JH, Verweij J, Ratain MJ. Limited Sampling Model for Area Under the Concentration Time Curve of Total Topotecan. Clin Cancer Res. 1996;2(1):43–46. [PubMed] [Google Scholar]
- 11.Danks MK, Pawlik CA, Whipple DO, Wolverton JS. Intermittent Exposure of Medulloblastoma Cells to Topotecan Produces Growth Inhibition Equivalent to Continuous Exposure. Curr Topics Med Chem. 1997;3(10):1731–1738. [PubMed] [Google Scholar]
- 12.Haas NB, LaCreta FP, Walczak J, Hudes GR, Brennan JM, Ozols RF, O’Dwyer PJ. Phase-I Pharmacokinetic Study of Topotecan by 24-Hour Continuous-Infusion Weekly. Cancer Res. 1994;54(5):1220–1226. [PubMed] [Google Scholar]
- 13.Kohlhagen G, Paull KD, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 14.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analog. Proc Natl Acad Sci US A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower DLS, Burgin AB. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 16.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. Synthesis and Mechanism of Action Studies of a Series of Norindenoisoquinoline Topoisomerase I Poisons Reveal an Inhibitor with a Flipped Orientation in the Ternary DNA-Enzyme-Inhibitor Complex As Determined by X-ray Crystallographic Analysis. J Med Chem. 2005;48:4803–4814. doi: 10.1021/jm050076b. [DOI] [PubMed] [Google Scholar]
- 17.Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y. A Novel Norindenoisoquinoline Structure Reveals a Common Interfacial Inhibitor Paradigm for Ternary Trapping of the Topoisomerase I-DNA Covalent Complex. Mol Cancer Ther. 2006;5:287–295. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao X, Cushman M. An Ab Initio Quantum Mechanics Calculation that Correlates with Ligand Orientation and DNA Cleavage Site Selectivity in Camptothecin-DNA-Topoisomerase I Ternary Cleavage Complexes. J Am Chem Soc. 2005;127:9960–9961. doi: 10.1021/ja042485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 20.Strumberg D, Pommier Y, Paull K, Jayaraman M, Nagafuji P, Cushman M. Synthesis of Cytotoxic Indenoisoquinoline Topoisomerase I Poisons. J Med Chem. 1999;42:446–457. doi: 10.1021/jm9803323. [DOI] [PubMed] [Google Scholar]
- 21.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J Med Chem. 2000;43:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 22.Jayaraman M, Fox BM, Hollingshead M, Kohlhagen G, Pommier Y, Cushman M. Synthesis of New Dihydroindeno[1,2-c]isoquinoline and Indenoisoquinolinium Chloride Topoisomerase I Inhibitors Having High In Vivo Anticancer Acitivity in the Hollow Fiber Animal Model. J Med Chem. 2002;45:242–249. doi: 10.1021/jm000498f. [DOI] [PubMed] [Google Scholar]
- 23.Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker BL, Stewart L, Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Alkenylindenoisoquinoline Topoisomerase I Inhibitors and Indenoisoquinoline-Camptothecin Hybrids. J Med Chem. 2003;46:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 24.Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Featuring Polyamine Side Chains on the Lactam Nitrogen. J Med Chem. 2003;46:5712–5724. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- 25.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 26.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Aminoalkenylindenoisoquinoline and 11-Diaminoalkenylindenoisoquinoline Topoisomerase I Inhibitors. Bioorg Med Chem. 2004;12:5147–5160. doi: 10.1016/j.bmc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 28.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 29.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2006;16:1846–1849. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Morrell A, Jayaraman M, Nagarajan M, Fox BM, Meckley MR, Ioanoviciu A, Pommier Y, Antony S, Hollingshead M, Cushman M. Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Using a Hollow Fiber Assay. Bioorg Med Chem Lett. 2006;16:4395–4399. doi: 10.1016/j.bmcl.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 31.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 32.Boyd MR, Paull KD. Some Practical Considerations and Applications of the National Cancer Institute In-Vitro Anticancer Drug Discovery Screen. Drug Development Res. 1995;34:91–109. [Google Scholar]
- 33.Nagarajan M, Morrell A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. Synthesis and Biological Evaluation of Bisindenoisoquinolines as Topoisomerase I Inhibitors. J Med Chem. 2006;49:5129–5140. doi: 10.1021/jm060046o. [DOI] [PubMed] [Google Scholar]
- 34.Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Hollingshead M, Pommier Y, Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J Med Chem. 2006;49:6283–6289. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demant EJF, Friche E. Equilibrium Binding of Anthracycline Cytostatics to Serum Albumin and Small Unilamellar Phospholipid Vesicles as Measured by Gel Filtration. Biochemical Pharmacology. 1998;55:27–32. doi: 10.1016/s0006-2952(97)00437-1. [DOI] [PubMed] [Google Scholar]
- 36.Demant EJF, Friche E. Kinetics of Anthracycline Accumulation in Multidrug-resistant Tumor Cells: Relationship to Drug Lipophilicity and Serum Albumin Binding. Biochemical Pharmacology. 1998;56:1209–1217. doi: 10.1016/s0006-2952(98)00255-x. [DOI] [PubMed] [Google Scholar]
- 37.Stermitz FR, Gillespie JP, Amoros LG, Romero R, Stermitz TA. Synthesis and Biological Activity of Some Antitumor Benzophenanthridinium Salts. J Med Chem. 1975;18:708–713. doi: 10.1021/jm00241a014. [DOI] [PubMed] [Google Scholar]
- 38.Cushman M, Mohan P, Smith ECR. Synthesis and Biological Activity of Structural Analogues of the Anticancer Benzophenanthridine Alkaloid Nitidine Chloride. J Med Chem. 1984;27:544–547. doi: 10.1021/jm00370a021. [DOI] [PubMed] [Google Scholar]
- 39.Fang SD, Wang LK, Hecht SM. Inhibitors of DNA Topoisomerase I Isolated from the Roots of Zanthoxylum nitidum. J Org Chem. 1993;58:5025–5027. [Google Scholar]
- 40.Sethi ML. Inhibition of Reverse Transcriptase Activity by Benzophenanthridine Alkaloids. J Nat Prod. 1979;42:187–196. [PubMed] [Google Scholar]
- 41.Pourquier P, Ueng LM, Fertala J, Wang D, Park HK, Essigmann JM, Bjornsti MA, Pommier Y. Induction of Reversible Complexes Between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-Oxoguanine and 5-Hydroxycytosine. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analyses for compounds 19, 25, 31–36, and 40–51. This material is available free of charge via the Internet at http://pubs.acs.org.