

Abstract
Adenoviruses (Ad) with the early region E4 deleted (E4-deleted virus) are defective for DNA replication and late protein synthesis. Infection with E4-deleted viruses results in activation of a DNA damage response, accumulation of cellular repair factors in foci at viral replication centers, and joining together of viral genomes into concatemers. The cellular DNA repair complex composed of Mre11, Rad50, and Nbs1 (MRN) is required for concatemer formation and full activation of damage signaling through the protein kinases Ataxia-telangiectasia mutated (ATM) and ATM-Rad3-related (ATR). The E4orf3 and E4orf6 proteins expressed from the E4 region of Ad type 5 (Ad5) inactivate the MRN complex by degradation and mislocalization, and prevent the DNA damage response. Here we investigated individual contributions of the MRN complex, concatemer formation, and damage signaling to viral DNA replication during infection with E4-deleted virus. Using virus mutants, short hairpin RNA knockdown and hypomorphic cell lines, we show that inactivation of MRN results in increased viral replication. We demonstrate that defective replication in the absence of E4 is not due to concatemer formation or DNA damage signaling. The C terminus of Nbs1 is required for the inhibition of Ad DNA replication and recruitment of MRN to viral replication centers. We identified regions of Nbs1 that are differentially required for concatemer formation and inhibition of Ad DNA replication. These results demonstrate that targeting of the MRN complex explains the redundant functions of E4orf3 and E4orf6 in promoting Ad DNA replication. Understanding how MRN impacts the adenoviral life cycle will provide insights into the functions of this DNA damage sensor.
Virus infection activates host cell responses that serve to limit virus replication and progeny production. We previously demonstrated that the cellular DNA repair machinery presents an obstacle to productive infection for human adenoviruses (55). The DNA repair apparatus consists of an elaborate network of cellular factors that monitor the integrity of genomic DNA and activate signaling pathways in response to DNA damage (reviewed in references 28 and 43). The Mre11, Rad50, and Nbs1 proteins form the MRN complex, which is involved in the cellular DNA damage response and is required for damage signaling (15, 32, 58, 69). The phosphatidylinositol 3-kinase-like kinases (PIKKs) ataxia-telangiectasia mutated (ATM) and ATM-Rad3-related (ATR) activate a central part of the cellular response to DNA damage. These protein kinases phosphorylate multiple substrates involved in cell cycle checkpoints (29, 42, 53). The MRN complex is required for full activation of ATM in response to double-stranded breaks in genomic DNA (9, 34-36, 61, 62) and plays a role in mediating signaling by ATR in response to some types of damage (17, 27, 54). The Nbs1 protein is required for nuclear localization of Mre11 and Rad50 (16) and for DNA binding and unwinding by the complex (33, 47). The C terminus of Nbs1 contains separate motifs required for direct interaction with Mre11 (16, 72) and ATM (11, 21, 72). In addition to its roles as a sensor of DNA damage (48) and as a tethering scaffold for breaks in double-stranded DNA (69), the MRN complex is also required for removal of the covalently attached Spo11 protein during meiotic recombination (5).
The adenovirus (Ad) genome is a double-stranded DNA molecule of 36 kb that is maintained as linear monomers during wild-type infection. Infection with Ads lacking the early region E4 (E4-deleted virus) results in activation of the cellular DNA damage response and signaling through the ATM and ATR pathways (9, 55). Infection with E4-deleted Ads also results in the formation of virus genome concatemers in a process that requires the MRN complex, as well as proteins involved in the cellular nonhomologous-end-joining DNA repair pathway (6, 55, 65). DNA damage signaling responses and genome concatemerization are both prevented during infection with wild-type Ad5 (9, 55). The E4orf3 and E4orf6 proteins are products from the E4 region that play redundant functions in promoting viral DNA replication and late protein synthesis (reviewed in references 60 and 67). E4orf3 and E4orf6 from Ad5 can also prevent genome concatemerization and modulate ATM and ATR pathways (55, 65). Induction of proteasome-mediated degradation of the MRN complex by E4orf6, in conjunction with the viral E1b55K protein, blocks concatemer formation and damage signaling (9, 55). The E1b55K/E4orf6 complex also induces degradation of DNA ligase IV protein (2), and this may contribute to the inhibition of DNA repair during infection (6). Expression of the viral E4orf3 protein results in mislocalization of the MRN complex (1, 19, 20, 55, 56), which also inhibits concatemer formation (19, 55, 56) and ATR signaling (C. T. Carson and M. D. Weitzman, unpublished observations).
The contribution of concatemerization, the MRN complex, and DNA damage signaling to the replication and late protein defects of E4-deleted virus remains unclear. The formation of genome concatemers may be detrimental to virus growth for multiple reasons. It has been suggested that concatemerization interferes with viral DNA replication (2) and late gene expression (26) and that concatemers are too large to be packaged efficiently into virus particles (2, 55, 65). The MRN complex is required for concatemer formation, but it might also have additional inhibitory effects on virus growth. Degradation of the Mre11 protein during infection with wild-type Ad5 correlates with the time at which viral replication initiates (41), and it has been suggested that MRN has a detrimental effect on the initiation of viral DNA replication (20, 41). The MRN complex is required for full activation of the cellular DNA damage response to Ad infection (9), and these signaling cascades could also interfere with efficient viral infection.
Here we examine the impact of DNA damage signaling and viral genome concatemerization on DNA replication. We developed a quantitative PCR (qPCR) assay to measure accumulation of viral DNA in infected cells, and we compared infections in matched cell lines. Using mutant E1b55K proteins that differentially degrade cellular substrates, we demonstrate that viral replication and late protein production are enhanced in the absence of MRN, independently of concatemer formation. The E4orf3 protein from Ad4 cannot mislocalize the MRN complex (56), and we show that replication of Ad4 virus is more sensitive to the deletion of E1b55K than Ad5. These results establish that inactivation of MRN through degradation by E4orf6/E1b55K and mislocalization by E4orf3 promotes viral DNA replication, and this can explain the redundancy in phenotypes observed for mutations in these virus genes (8, 24, 25). Our results also show that Ad DNA replication is not impacted by concatemerization of Ad genomes and DNA damage signaling. The defective DNA replication of E4-deleted virus is rescued in Nijmegan breakage syndrome (NBS) cells that lack functional MRN, while inhibition can be restored by complementing with wild-type full-length Nbs1. We demonstrate that the Mre11-binding region in the C terminus of Nbs1 is required for complete nuclear localization of Rad50, recruitment to viral replication centers, and the inhibition of Ad DNA replication. In addition, we show that the final 47 residues in Nbs1 are required to suppress Ad DNA replication but are dispensable for concatemer formation. We conclude that there are multiple steps at which the cellular DNA repair machinery acts as a barrier to productive Ad infection.
MATERIALS AND METHODS
Cell lines.
HeLa, U2OS, and 293 cells were purchased from the American Tissue Culture Collection. W162 cells for growth of E4-deleted viruses were a gift from G. Ketner. Stable cell lines derived from HeLa and U2OS that express wild-type and mutant E1b55K from retrovirus vectors have been described previously (9). A-T cells (AT221JET and complemented version) were gifts from Y. Shiloh. HeLa cells expressing short hairpin RNA (shRNA) to Mre11 were a gift from C. Her (63). The U2OS-derived cells that express inducible wild-type (GW33) and kinase-dead (GK41) ATR proteins were gifts from S. Schreiber and were induced with doxycycline as previously described (46). The ligase IV hypomorphic cell line, 180 BRM, was a gift from G. Iliakis. Immortalized NBS (NBS-ILB1) cells transduced with retrovirus expressing the Nbs1 cDNA or the empty vector were previously described (9, 13). NBS cells expressing the FR5 and 652 fragments of Nbs1, and Mre11-NLS were gifts from P. Concannon and have been previously described (10, 11, 16). All cells were maintained as monolayers in Dulbecco modified Eagle medium supplemented with 10 or 20% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2.
Plasmids and transfections.
Retroviral plasmids based on pLXIN and those expressing full-length Nbs1 and the Myc-tagged FR5 fragments were gifts from K. Cerosaletti and have been previously described (13, 16). Plasmids used as standards for qPCR replication assays contained cloned cDNAs for the Ad5-DBP and Ad4-E4orf3 genes (56). To generate retroviruses expressing mutant FR5 fragments of Nbs1, a retrovirus vector plasmid containing FR5 with a Myc-epitope tag was mutated by site-directed mutagenesis kit (Stratagene). Primers used to generate the FR5-2K and FR5-Δ47 mutants are listed in Table 1. These plasmids were used to generate retrovirus vectors that were used to transduce NBS-ILB1 cells, which were subsequently selected with G418 (1 mg/ml) as described previously (13, 16).
TABLE 1.
Primers used for PCR and mutagenesis
| Gene | Direction | Sequence (5′-3′) | Purpose |
|---|---|---|---|
| Ad4orf3 | F | GGAATTCCATATGAGGGTGTGC | qPCR/RT-PCR |
| Ad4orf3 | R | CCCAAGCTTATCCAAACGGTCTCG | qPCR/RT-PCR |
| Ad5 DBP | F | GCCATTGCGCCCAAGAAGAA | qPCR |
| Ad5 DBP | R | CTGTCCACGATTACCTCTGGTGAT | qPCR |
| Ad4 E1b55K | F | CGTCCAGAAACAGTGTGGTGG | RT-PCR |
| Ad4 E1b55K | R | GCAGGTCAGCATCTGGTAG | RT-PCR |
| Ad5 E4orf3 | F | ATGATTCGCTGCTTGAGGCTG | RT-PCR |
| Ad5 E4orf3 | R | CTATTAAGTGAACGCGCTCC | RT-PCR |
| Ad5 E1b55K | F | AGAAGTATTCCATAGAGCAGC | RT-PCR |
| Ad5 E1b55K | R | GGTATTGTTAAACCCATAGAA | RT-PCR |
| FR5-2K | F | GGTCAACTAAAAAATTTCGAGGAATTCAAAAAGGTCACATAT | Mutagenesis |
| FR5-2K | R | GGATATGTGACCTTTTTGAATTCCTCGAAATTTTTTAGTTGACC | Mutagenesis |
| FR5-Δ47 | F | GGAGGATCAGATTAATAGCTCATCATGC | Mutagenesis |
| FR5-Δ47 | F | GCATGATGAGCTATTAATCTGATCCTCC | Mutagenesis |
| Ad4 E1b del | F | GGTGCTGACTAGGTCTTCGAGTGGTCGGGAGAGGGGTATTAAGCGGGAGAGGCATGATGA CCTGTGACGGAAGATCACTTCG | Mutagenesis |
| Ad4 E1b del | R | CTCGCAGGCTCGACACCTGGTCTTGTATTCATCATATCTTAGAATCTTCCACACTTCCACCTGAGGTTCTTATGGCTCTTG | Mutagenesis |
| Ad4 E1b del | Selection | AGGTCTTCGAGTGGTCGGGAGAGGGGTATTAAGCGGGAGAGGCATGATGAGTGGAAGTGTGGAAGATTCTAAGATATGATGAATACAAGACCAGGTGTCG | Mutagenesis |
Viruses and infections.
Wild-type Ad5 was obtained from the American Tissue Culture Collection. The mutant viruses dl1004 (ΔE4), dl110 (ΔE1b55K), dl1016 (ΔE1b55K/ΔE4orf3), and dl3112 (ΔE1b55K/ΔE4orf3) have previously been described (8, 23, 52) and were gifts from G. Ketner and D. Ornelles. Wild-type Ad5, dl110, dl1016, and dl3112 were propagated in 293 cells. The dl1004 virus was propagated on W162 cells (66). The Ad4ΔE1b55K mutant was generated in a bacterial artificial chromosome (BAC) by recombination in E. coli and is deleted from 2201 to 3170 bp in the Ad4 genome. The Ad4 genome was captured and modified as previously described (44). Briefly, purified human Ad4 (strain RI-6) viral DNA was cotransformed into induced SW102 bacteria with a modified BAC-based vector based on pBeloBAC11 (New England Biolabs) in which the left-hand (bp 1 to 452) and right-hand (bp 35135 to 35994) ends of the Ad4 genome had been cloned. Chloramphenicol-resistant colonies were grown, and BAC DNA was extracted and digested with the appropriate enzyme to confirm the Ad4 genome had been successfully cloned. E1b55K (bp 2201 to 3170) was deleted by using “recombineering” technology (64) with a selectable marker cassette that allows both positive and negative selection (sacB/lacZ/Ampr). The selectable marker was amplified by using PCR primers that contain homology to the targeted regions of the Ad4 genome (see Table 1). After successful recombination the selectable marker was then removed using a single-stranded DNA oligonucleotide leaving the appropriate deletion (Table 1). The Ad4 genome in the BAC vector is flanked by PacI recognition sites, which were used to recover viral DNA for transfection into 293 cells in order to generate virus. Viral DNA was isolated, digested, and sequenced to confirm the predicted genomic structure. All viruses were purified by two sequential rounds of ultracentrifugation in cesium chloride gradients and stored in 40% glycerol at −20°C. Infections were performed on monolayers of cells in Dulbecco modified Eagle medium supplemented with 2% FBS using a multiplicity of infection (MOI) of 10, unless otherwise indicated. After 2 to 4 h at 37°C the cells were washed with phosphate-buffered saline (PBS) and replaced with fresh medium containing 10% FBS. Infections with Ad4 and Ad4ΔE1b55K were performed at an MOI of 10, and equal input DNA was recovered from infected cells at 4 h postinfection (hpi), although the amount was less than from Ad5-infected cells.
Antibodies.
Primary antibodies to the following proteins were purchased from commercial sources and used at the indicated dilutions: Nbs1 (1:1,000; Novus Biologicals), ATR (1:500; Santa Cruz), Rad50 (1:500; GeneTex), Mre11 (1:500; GeneTex), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 1:50,000; Research Diagnostics, Inc.), ATM (1:500; Epitomics), FLAG (1:5,000; Sigma), DNA ligase IV (1:500; Serotype), HA (hemagglutinin; 1:2,000; Covance), Ku86 (1:1,000; Santa Cruz), adenoviral Hexon protein (1:5,000; Abcam), and adenoviral fiber protein (1:10,000; Neomarkers). Antibodies to adenoviral proteins E1B55K (used at 1:500) and DBP (used at 1:1,000) were gifts from A. Levine. Secondary antibodies were from Jackson Laboratories or Eurogentec.
Immunoblotting.
Cells were lysed in PBS containing 1% NP-40, 0.25% Triton, 0.1% sodium dodecyl sulfate, and proteasome inhibitors (sodium vanadate, beta-glycerol phosphate, sodium fluoride, and phenylmethylsulfonyl fluoride). A Lowry assay (Bio-Rad) was used to determine the protein concentrations of each lysate. Equal amounts of each lysate were separated by electrophoresis on polyacrylamide gels (NuPAGE; Invitrogen). Proteins were transferred to Hybond ECL membrane and blocked with 5% dry milk in PBS-Tween overnight at 4°C. Primary antibodies were incubated with the membranes for 1 h at room temperature in PBS with 3% bovine serum albumin and 1× sodium azide. Membranes were probed with secondary goat anti-mouse or anti-rabbit horseradish peroxidase-conjugated antibodies for 1 h at room temperature in PBS with 3% bovine serum albumin. Visualization of the immunoblots was performed by using enhanced chemiluminescence reagent (Perkin-Elmer).
Real-time qPCR analysis of DNA replication.
Cells were harvested over a time course of infection, and total DNA was extracted from cells by using a DNeasy kit (Qiagen). qPCR analysis was performed on all samples in triplicate using primers to the Ad5-DBP or Ad4-E4orf3 genes (see Table 1 for primers). Amplified products were detected on an ABI Prism 7900 sequence detection system with Sybr green. Plasmid standards for Ad5-DBP and Ad4-E4orf3 were used to quantitate the number of viral genomes. The 4-h time point was used for normalization to input internalized viral DNA. The amount of input DNA recovered at the 4-h time point for each virus was the same within each cell line comparison. The results are presented as the mean and standard error of the mean from triplicate experiments. We further analyzed data by Student t test, comparing fold changes in replication to appropriate controls.
PFGE.
Cells were infected with wild-type Ad5 or the E4-deleted mutant dl1004. At 48 hpi viral DNA was analyzed for concatemer formation, as previously described (55). Cells were incorporated into agarose plugs that were loaded onto a 1.2% high gelling temperature agarose gel and subjected to pulsed-field gel electrophoresis (PFGE) for 16 h. DNA was visualized by staining the gel in Sybr green or ethidium bromide.
Reverse transcription-PCR (RT-PCR).
For analysis of E1b55K and E4orf3 expression in cells infected with Ad4 and Ad5 viruses with the E1b55K gene deleted, RNA was isolated from infected cells by using an RNAqueous kit (Ambion). Total RNA was DNase treated using the Turbo DNA free kit (Ambion), and 1 μg of RNA was then used to make cDNA with random hexamers and a High-Fidelity cDNA synthesis kit (ABI). PCR was performed on 4 μl of cDNA reaction for Ad4 E1b55K, Ad4 E4orf3, Ad5 E1b55K, or Ad5 E4orf3 using the oligonucleotide primers listed in Table 1. PCR products were separated on a 1% agarose gel and visualized on an Alpha Imager.
RESULTS
Late protein production is enhanced by MRN degradation, independently of concatemer formation.
We previously identified mutants of E1b55K that can distinguish between degradation targets (9). In the presence of E4orf6, the R240A mutant induces degradation of the MRN complex but not p53, whereas the H354 mutant degrades p53 but not MRN. We used stable cell lines expressing these E1b55K proteins to determine what effect degradation of the MRN complex has on late protein production (Fig. 1). HeLa-derived cell lines (9) were infected with ΔE1b55K/ΔE4orf3 mutant Ads (dl1016 or dl3112), and lysates were analyzed by immunoblotting at 30 hpi. The defective virus was complemented in the cell line expressing wild-type E1b55K and, together with the E4orf6 expressed from the virus, E1b55K expression resulted in degradation of the MRN complex (Fig. 1A). In these cells there was robust expression of late Ad gene products, as detected by immunoblotting with antibodies to the hexon and fiber proteins. Similar results were obtained in cells expressing the R240A mutant, which retains the ability to degrade MRN. In contrast, in the green fluorescent protein (GFP) control cell line or in the presence of the H354 mutant, MRN was not degraded and the production of late proteins was significantly diminished. Both the R240A and the H354 mutants retained the ability to degrade DNA ligase IV (Fig. 1A), which was recently identified as an E1b55K/E4orf6 substrate (2). The same pattern of protein production was observed in stable E1b55K-expressing cell lines derived from U2OS (data not shown). These data demonstrate a correlation between MRN degradation and production of late viral proteins.
FIG. 1.
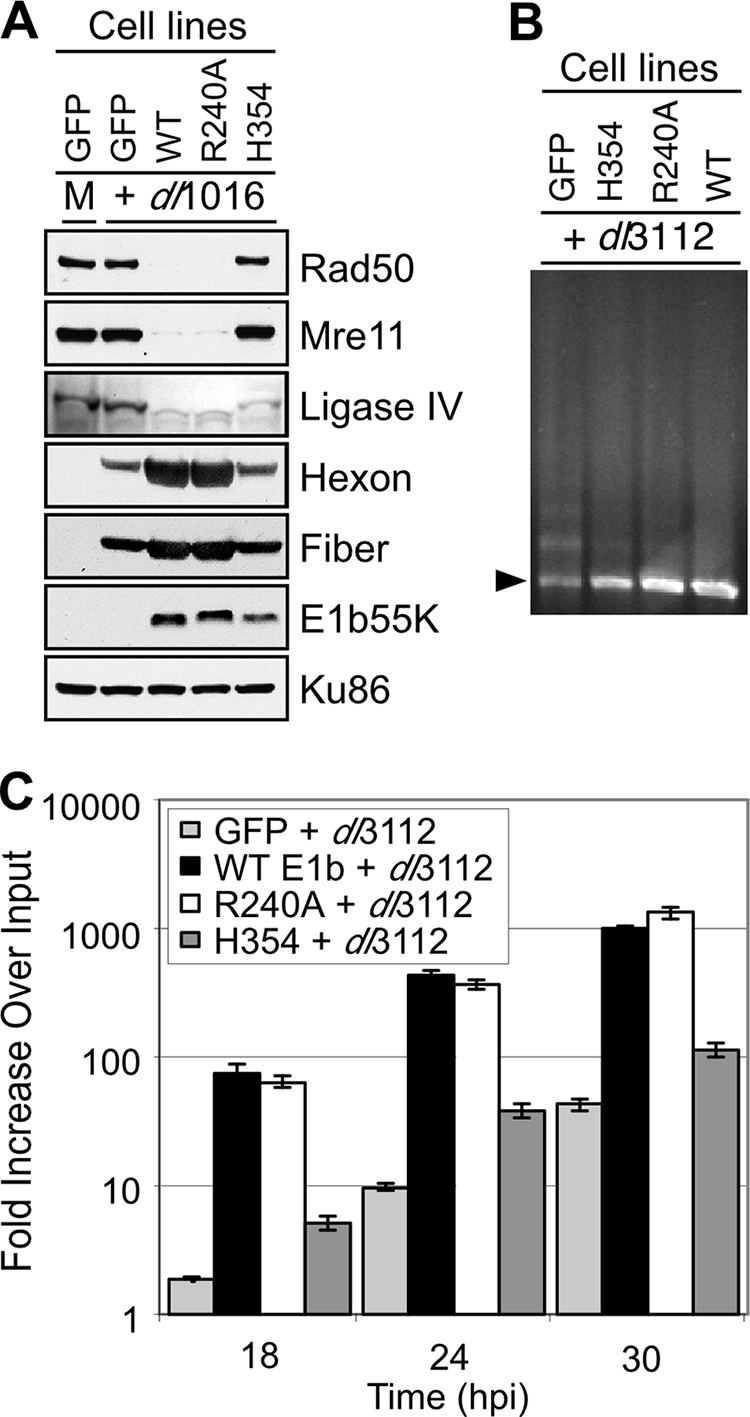
Degradation of the MRN complex by the E1b55K/E4orf6 proteins correlates with enhanced Ad late protein production and DNA replication. Stable cell lines expressing either E1b55K proteins or GFP were infected with ΔE1b55K/ΔE4orf3 mutant Ads (either dl1016 or dl3112). (A) Immunoblot analysis of viral and cellular proteins in lysates from infected cells harvested at 30 hpi. Ku86 served as a loading control. (B) Concatemer formation was examined at 30 hpi by PFGE analysis of DNA. Viral DNA was visualized by staining the gel in Sybr green, and the position of linear viral genome is indicated by an arrow. (C) Viral DNA was extracted from cells harvested at 4, 18, 24, and 30 hpi and analyzed by qPCR. Accumulation of viral DNA is represented as the fold increase over input viral DNA, as determined at the 4-h time point. WT, wild type.
The decreased late protein production observed in the presence of the MRN complex could reflect defects in viral DNA replication, RNA processing, protein translation, or a combination of factors. It has previously been suggested that concatemers affect late protein production during infection with E4-deleted mutants (26). We therefore first assessed concatemer formation during infection of the E1b55K mutant cell lines (Fig. 1B). Cells were infected with ΔE1b55K/ΔE4orf3 mutant Ad and concatemers were examined by PFGE. Concatemers were detected during infection of the GFP control cell line, but in all three E1b55K cell lines the virus DNA was predominantly present as linear monomers. This assay is not quantitative and is therefore not used to measure differences in the total amount of accumulated viral DNA. The lack of concatemers reflects the fact that the R240A and H354 mutants both retain the ability to degrade DNA ligase IV (Fig. 1A) and shows that this is sufficient to prevent concatemers. This result indicates that the defect of late protein production is not due to concatemer formation.
Inactivation of the MRN complex by E4 proteins of Ad5 correlates with increased virus DNA replication.
To assess whether MRN degradation could promote viral DNA replication, we used qPCR to measure the accumulation of total Ad genomic DNA over a time course of infection (Fig. 1C). The GFP and E1b55K cell lines were infected with the ΔE1b55K/ΔE4orf3 mutant virus and extracted viral DNA was analyzed using primers to the Ad5-DBP gene. The results demonstrated robust DNA replication of the mutant virus in cells that expressed E1b55K proteins that degrade MRN (wild type and R240A) and defective replication in cells that could not degrade MRN (GFP and H354). Therefore, MRN degradation correlates with increased virus DNA replication. Together, these data demonstrate that Ad5 late protein production and virus DNA replication are defective when the MRN complex is not degraded, but these defects are both independent of concatemer formation.
The E4orf3 protein of Ad5 serves functions redundant to E1b55K/E4orf6 in its requirement for efficient virus production (8, 24, 25) and inhibition of concatemer formation (55, 65). The Ad5-E4orf3 protein also inactivates the MRN complex through redistribution of components into intranuclear track-like structures and cytoplasmic aggregates (1, 19, 20, 55, 56). We therefore assessed whether E4orf3 was sufficient to promote DNA replication in an Ad5 virus that lacked E1b55K and was unable to induce MRN degradation (Fig. 2A). Replication of the E1b55K-deleted virus dl110 (23) was very similar to wild-type Ad5. Similar results were obtained with the Ad5-derived virus dl1017 (7) in which both E1b55K and E4orf6 are deleted (data not shown). In contrast, replication of an E4-deleted virus that lacks both E4orf3 and E4orf6 was defective in HeLa cells. To confirm that the MRN complex is the target of the E4 proteins, we compared virus DNA accumulation in a HeLa-derived cell line that stably expresses an shRNA to knock down Mre11 protein levels (63). Knockdown of Mre11 was confirmed by immunoblotting (data not shown). The DNA replication defect of the E4-deleted virus was rescued to wild-type levels in this cell line (Fig. 2A), in agreement with previous reports (20, 41). These results suggest that viruses unable to inactivate MRN are defective for viral DNA replication.
FIG. 2.
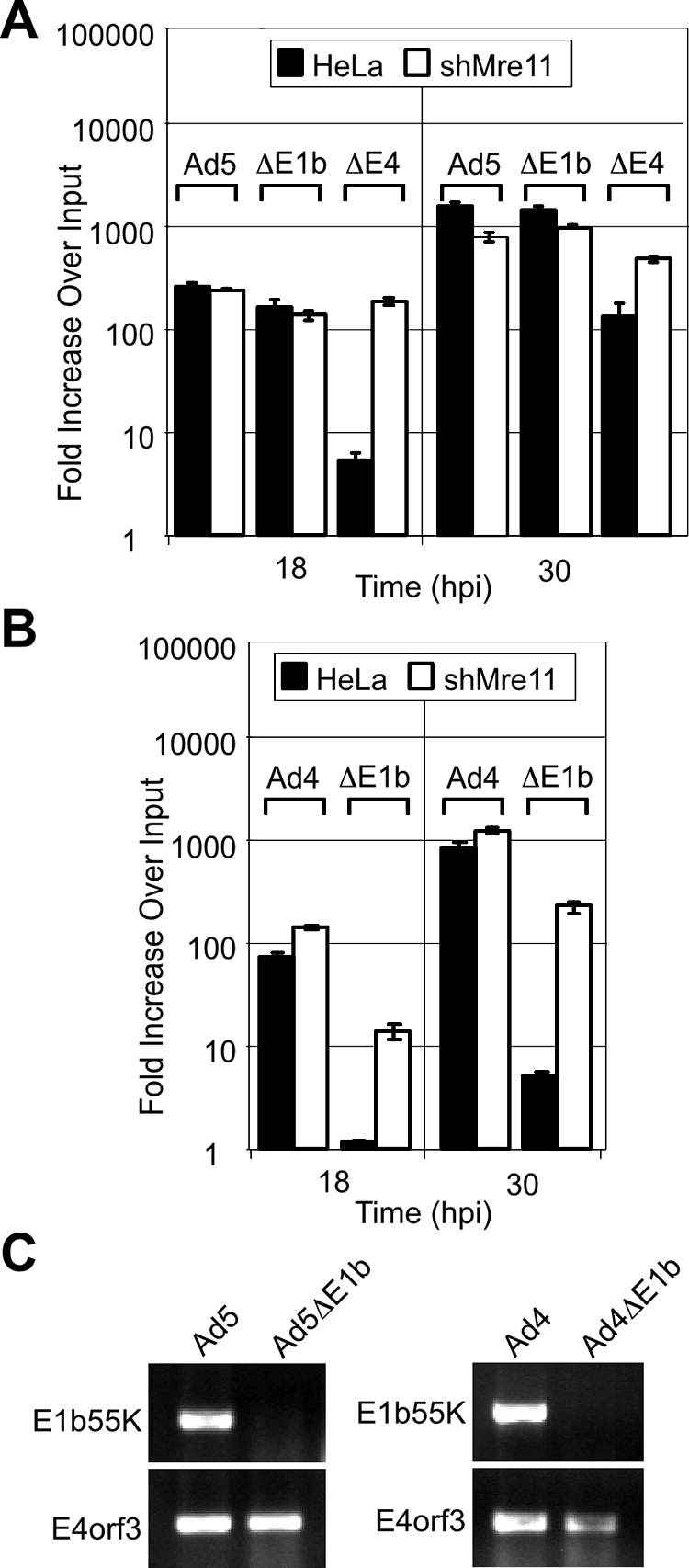
Mislocalization of the MRN complex by the viral Ad5-E4orf3 protein correlates with enhanced Ad DNA replication. Viral DNA replication was assessed by qPCR analysis of DNA extracted from cells harvested at 4, 18, and 30 h after infection with either wild-type or mutant forms of Ad5 (A) and Ad4 (B). Accumulation of viral DNA is represented as the fold increase over the input viral DNA, which was determined at the 4-h time point. Infections were compared between HeLa cells and HeLa-derived cells stably expressing shRNA directed to reduce the level of Mre11 protein. (C) Transcription of viral genes was demonstrated by RT-PCR analysis of RNA extracted from infected lysates using primer sets specific to gene and serotype.
We have previously reported that the E4orf3 protein from serotype Ad4 is unable to target the MRN complex, although the Ad4-E1b55K/E4orf6 complex retains the ability to induce MRN degradation (56). We therefore examined replication of wild-type Ad4 and a mutant that does not express E1b55K (Fig. 2B). Compared to wild-type Ad4 virus, the E1b55K mutant was severely defective for virus DNA replication in the HeLa cells, but this defect was rescued to a significant extent in the shMre11 knockdown cells. Transcription of viral proteins was confirmed by RT-PCR analysis of mRNA extracted from infected cells using serotype-specific primers (Fig. 2C). Together, these results suggest that mislocalization and inactivation of the MRN complex by the Ad5-E4orf3 protein promotes viral DNA replication.
The MRN complex is responsible for the replication defect of E4-deleted viruses.
Based on the results from E1b55K and E4orf3 mutants, we also examined viral DNA replication in NBS cell lines that lack functional MRN complex (Fig. 3). We infected the NBS-ILB1 cell line that is derived from a patient with NBS and harbors a hypomorphic Nbs1 mutation and compared it to a matched cell line that has been complemented with the wild-type Nbs1 cDNA (13). Immunoblotting of cell lysates from cells infected with the E4-deleted virus (dl1004) and harvested over a time course of infection demonstrated appropriate Nbs1 expression and comparable levels of Ad5-DBP (Fig. 3A) and other viral proteins (data not shown). Replication of the E4-deleted mutant virus dl1004 was defective in NBS cells complemented with Nbs1 but was rescued to wild-type levels in the NBS cells transduced with an empty vector (Fig. 3B). Wild-type Ad5 DNA replication was not significantly affected by the presence of Nbs1, presumably because the E4 proteins inactivate MRN. These data in matched cell lines demonstrate that the MRN complex has a detrimental effect on DNA replication of E4-deleted Ad, a finding consistent with results in Mre11 knockdown cells (Fig. 2A).
FIG. 3.
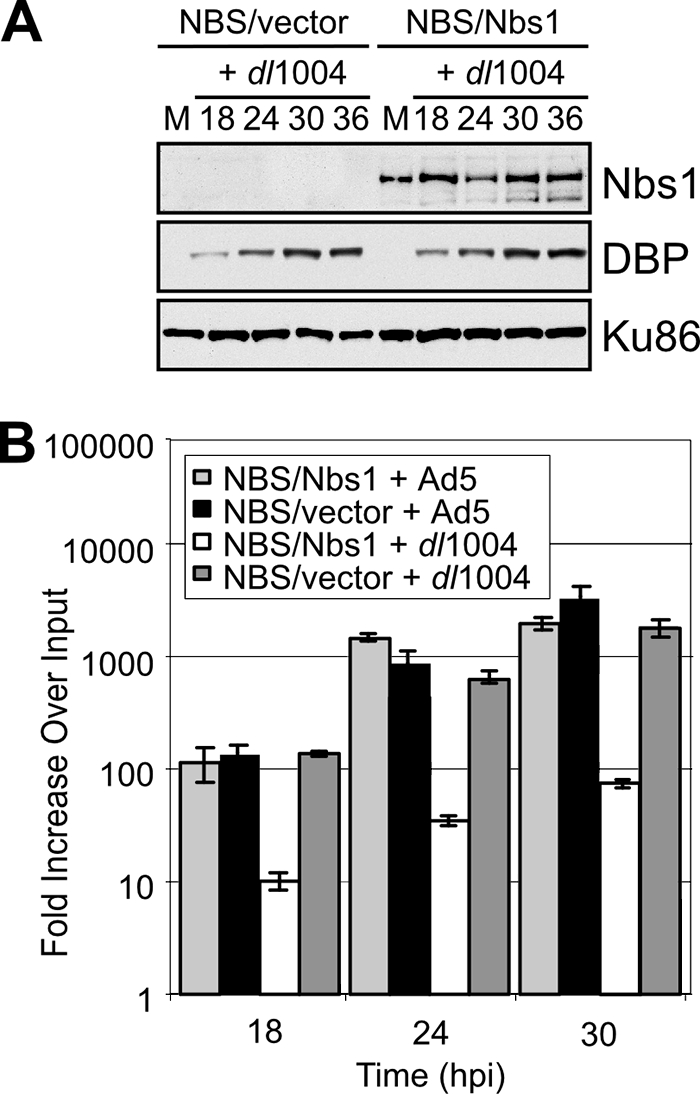
The replication defect of E4-deleted mutant Ad5 is rescued in the absence of functional MRN complex. (A) Immunoblot of lysates from NBS cells infected with the E4-deleted virus dl1004. Infections were compared in NBS cells transduced with an empty retrovirus vector or with a vector that expresses full-length Nbs1 protein. DBP demonstrated that early virus protein synthesis was comparable in the two cell lines. M represents lysates from mock infection, and Ku86 served as a loading control. (B) Replication of the E4-deleted virus is similar to wild-type Ad5 in NBS cells transduced with empty vector but is significantly compromised in cells expressing Nbs1. Cells were infected with either wild-type Ad5 or dl1004, and viral DNA extracted over a time course was analyzed by qPCR. Accumulation of viral DNA is represented as the fold increase over the input viral DNA, determined at the 4-h time point.
Concatemerization and DNA damage signaling are not responsible for the inhibition of Ad DNA replication.
Since we have previously demonstrated that the MRN complex is required for virus genome concatemerization and also for activation of ATM and ATR signaling in response to E4-deleted Ad (9, 55), we addressed whether DNA damage signaling mediated by MRN and concatemers have any impact on DNA replication. Viral DNA replication was analyzed by infections of cells with inactivated PIKKs (Fig. 4). We first examined infections in cells derived from an ataxia telangiectasia A-T patient with a mutation in ATM that renders them defective for ATM-dependent damage signaling. Replication of dl1004 mutant virus was defective in A-T cells, demonstrating that the absence of ATM signaling does not rescue the E4-deleted virus phenotype. We also examined infection in A-T cells treated with caffeine at a concentration (5 mM) that inhibits ATR activity (50) and found that replication of mutant virus was not rescued by caffeine treatment (Fig. 4A). Replication of wild-type Ad5 was normal in these cells, in the presence or absence of caffeine (data not shown). To test further the effect of ATR signaling, we performed infections in cells expressing an inducible kinase-dead trans-dominant-negative version of ATR (Fig. 4B) and in cells expressing shRNA to ATR (data not shown). In both cases, replication of the E4-deleted virus was defective and was not rescued by ATR inactivation. Inactivation of ATR was confirmed by reduced phosphorylation of ATR substrates (Chk1-S345 and Nbs1-S343), as assessed by immunoblotting with phospho-specific antibodies (data not shown). Together, these data show that damage signaling through ATM and ATR does not affect viral DNA replication. We also examined concatemer formation during these infections. We found that blocking ATM or ATR signaling alone had no effect on concatemerization, but concatemers were prevented in A-T cells by caffeine treatment (Fig. 4C; data not shown). This result suggests that damage signaling may be involved in concatemerization but that the defective replication of E4-deleted virus is independent of concatemer formation. In addition, we examined viral replication in cells with a hypomorphic mutation in DNA ligase IV that prevents concatemer formation (55). In these cells the E4-deleted virus was still defective for DNA replication (Fig. 4D), supporting the conclusion that concatemer formation is not responsible.
FIG. 4.

DNA damage signaling does not affect Ad replication. Cells were infected with either wild-type Ad5 or dl1004 mutant. Viral DNA was extracted at the indicated time postinfection for analysis by qPCR and PFGE. Accumulation of viral DNA is represented as the fold increase over input viral DNA, determined at the 4-h time point. (A) Replication of E4-deleted virus was defective in A-T cells with or without 5 mM caffeine. (B) E4-deleted virus was not rescued when ATR signaling was compromised by overexpression of inducible kinase dead ATR. Cell lines derived from U2OS that express inducible wild type (ATR-WT) or kinase dead (ATR-KD) ATR transgenes were induced with doxycycline and infected with Ad5 and dl1004 for 30 h. (C) Caffeine treatment (5 mM) blocks concatemer formation in A-T cells. DNA was prepared at 30 hpi and analyzed by PFGE. Viral DNA was visualized by staining the gel in Sybr green, and the position of linear viral genome is indicated by an arrow. (D) Viral replication in cells with mutant DNA ligase IV. The 180 BRM cells were infected with Ad5 and dl1004 (MOI = 100) and harvested for qPCR analysis at 48 hpi. Replication of the E4-deleted virus was not rescued in these cells, where concatemers were not formed.
The C terminus of Nbs1 is sufficient to inhibit Ad DNA replication.
There are a number of distinct functional domains within the Nbs1 protein (Fig. 5A). At the N terminus are forkhead-associated (FHA) and BRCA C-terminal (BRCT) domains, protein-protein interaction motifs that are required for the accumulation of MRN at ionizing radiation-induced foci (12, 16, 73) and association with chromatin (30, 31, 39, 59). Two ATM/ATR-dependent phosphorylation sites are located at S278 and S343 (22, 37, 71). At the C terminus is a region that binds Mre11 (16, 72) and a conserved motif that interacts with ATM (11, 21, 72). To gain further insights into the functions of the MRN complex that impact the viral life cycle, we first determined which domains of Nbs1 are required for inhibition of Ad DNA replication. We infected NBS cells complemented with truncated forms of Nbs1 (16) with wild-type Ad5 and dl1004. Expression of wild-type and truncated Nbs1 proteins in these cells was confirmed by immunoblotting (Fig. 5B). DNA extracted from cells over a time course of infection with the E4-deleted virus dl1004 was analyzed for replication by qPCR (Fig. 5C) and for concatemers by PFGE (data not shown). The 652 N-terminal fragment of Nbs1 contains the FHA and BRCT protein interaction domains, but these were not required for the inhibition of Ad replication or concatemer formation. The FR5 fragment restored both inhibition of Ad DNA replication and concatemers, demonstrating that the C terminus of Nbs1 is necessary and sufficient for these activities.
FIG. 5.

The C terminus of Nbs1 is required for inhibition of Ad replication. (A) Schematic of the domains in wild-type Nbs1 and Nbs1 truncation mutants. (B) Immunoblot of lysates from NBS cells expressing wild-type and mutant Nbs1 proteins. GAPDH served as a loading control. (C) The C terminus of Nbs1 is required for inhibiting mutant Ad DNA replication. NBS cells expressing wild-type or mutant Nbs1 were infected with the E4-deletion mutant virus (dl1004), and DNA was extracted over a time course of infection for analysis by qPCR. Accumulation of viral DNA is represented as the fold increase over input viral DNA, determined at the 4-h time point. (D) Localization of Nbs1 and Rad50 proteins in the absence or presence of virus infection. NBS-derived cell lines either uninfected or infected with E4-deleted virus dl1004 were fixed and analyzed by immunofluorescence at 24 hpi. Virus replication centers were visualized in infected cells by staining with an antibody to DBP. Cell nuclei were marked by staining with DAPI.
We also used immunofluorescence to examine the localization of mutant proteins and recruitment to viral replication centers (Fig. 5D). The full-length Nbs1 protein, as well as the 652 and FR5 fragments, were localized in the nucleus of NBS cells, as previously reported (16). The Nbs1 and FR5 proteins retain interaction with Mre11 and relocalized Rad50 from the cytoplasm to the nucleus in NBS cells. In contrast, the 652 fragment lacks the Mre11 interaction domain, and Rad50 remained predominantly cytoplasmic. During infection with E4-deleted virus dl1004, the nuclear localized Rad50 accumulated at viral replication centers in cells expressing wild-type Nbs1 and FR5. These results demonstrate that the C terminus of Nbs1 is sufficient to localize MRN components into the nucleus and into virus replication centers.
We reasoned that if the only role of the Nbs1 protein is to recruit Mre11 into the nucleus, then expression of Mre11 independently localized to the nucleus of NBS cells via a nuclear localization signal (NLS) should inhibit Ad DNA replication. We therefore examined E4 mutant virus replication in NBS cells transduced with a retrovirus that expresses the Mre11 transgene with an NLS at the C terminus (11). Immunofluorescence demonstrated that this Mre11 protein was localized to the nucleus (Fig. 6A). Surprisingly, we observed that Rad50 remained predominantly cytoplasmic, even though this Mre11-NLS fusion protein has been shown to interact with Rad50 (11). As a control we used an NBS cell line expressing wild-type Mre11 without an NLS, which remained predominantly cytoplasmic. When the NBS cell lines were infected with E4-deleted virus, accumulation of viral DNA was only inhibited when cells were complemented with wild-type Nbs1 (Fig. 6B). This shows that nuclear localization of Mre11 is not sufficient to inhibit virus replication and suggests that nuclear localization of Rad50 and Nbs1 is also required.
FIG. 6.
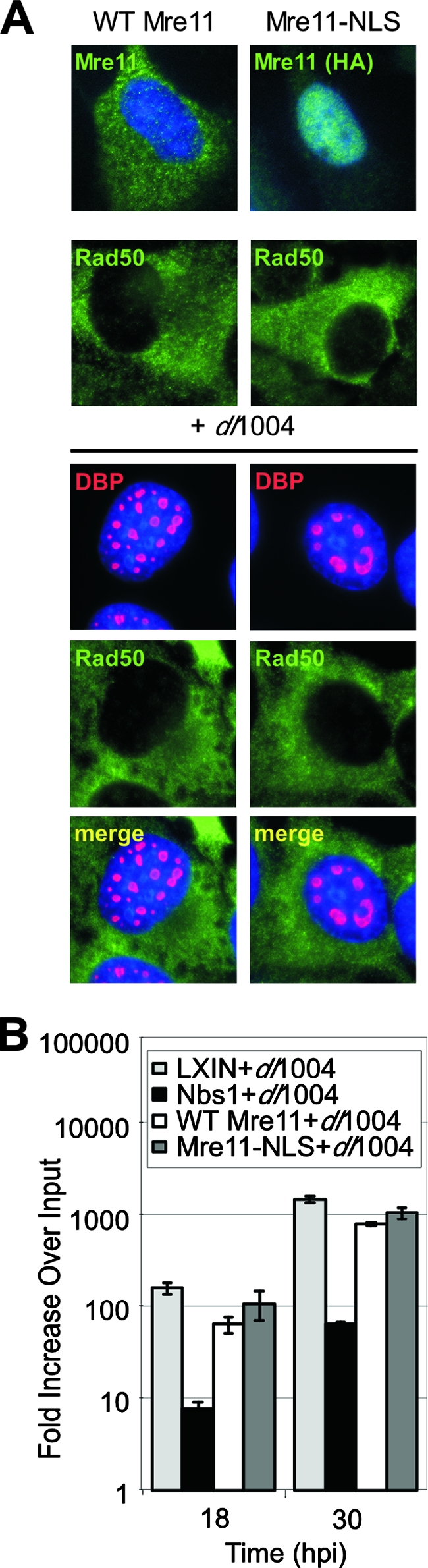
Nuclear localization of Mre11 is not sufficient to inhibit Ad replication. (A) Localization of Mre11 and Rad50 proteins in the absence and presence of virus infection. NBS cells overexpressing wild-type (WT) Mre11 and Mre11-NLS were infected with dl1004 mutant Ad and analyzed by immunofluorescence at 24 hpi. The Mre11-NLS protein was detected with an antibody to the HA epitope tag. Virus replication centers were visualized by staining with antibody to DBP, and cell nuclei were marked by staining with DAPI. (B) Replication of E4-deleted virus is inhibited by Nbs1 but not by Mre11-NLS. NBS cells transduced with empty vector (LXIN) or vectors expressing Nbs1, wild-type Mre11, and Mre11-NLS were infected with the E4-deleted mutant virus dl1004. Viral DNA was extracted over a time course of infection for analysis by qPCR. Accumulation of viral DNA is represented as fold increase over input viral DNA, determined at the 4-h time point.
Inhibition of Ad DNA replication requires Mre11 binding by the C terminus of Nbs1.
The C terminus of Nbs1 encompassed by the FR5 fragment contains sequences known to interact with Mre11 and ATM (11, 16, 21, 72). In order to ascertain whether these domains are involved in inhibition of Ad DNA replication, we generated FR5 fragments with specific mutations that disrupt the Mre11 and ATM interactions and introduced them into NBS cells by retrovirus transduction (Fig. 7). The FR5-2K fragment contains the KK685/686EE mutations that disrupt Mre11 binding (72). The FR5-Δ47 fragment contains a stop codon at residue 706 that results in deletion of the last 47 amino acids at the C terminus, and thus this mutant lacks the ATM binding region (11, 72). Expression of these proteins was confirmed by immunoblotting (Fig. 7B), and all of the C-terminal fragments showed nuclear localization by immunofluorescence (Fig. 7C). The FR5 and FR5-Δ47 fragments contain the Mre11-binding domain, and they both recruited Rad50 into the nucleus, where it was found colocalized with viral replication centers (Fig. 7C). In contrast, cells expressing the binding mutant FR5-2K displayed predominantly cytoplasmic localization of Rad50 both with or without infection, similar to what was observed in the LXIN and 652 cells (see Fig. 5). Cells were infected with the E4-deleted virus dl1004 in order to assess viral DNA replication by qPCR (Fig. 7D) and concatemer formation by PFGE (Fig. 7E). The FR5 fragment was sufficient to inhibit replication of mutant Ad, whereas both FR5-2K and FR5-Δ47 did not inhibit viral DNA accumulation. Concatemer formation was observed in cells expressing the FR5 fragment and the FR5-Δ47 mutant but not in cells with FR5-2K. This result shows that Nbs1 binding to Mre11 is required for concatemer formation. The loss of mutant Ad replication inhibition by both FR5-2K and FR5-Δ47 mutants suggests that binding to an additional factor at the end of the Nbs1 protein contributes to the inhibition of virus DNA replication.
FIG. 7.

Requirements for inhibition of viral DNA replication and for concatemer formation by the C terminus of Nbs1. (A) Schematic of motifs in the C terminus of Nbs1 proteins and the mutations generated. (B) Immunoblot of lysates from NBS cells transduced with retroviruses expressing wild-type and mutant Nbs1 proteins. GAPDH served as a loading control. (C) Localization of Nbs1 and Rad50 proteins in NBS cells in the absence and presence of virus infection. NBS-derived cell lines either uninfected or infected with dl1004 were fixed and analyzed by immunofluorescence at 24 hpi. Virus replication centers were visualized in infected cells with an antibody to DBP, and cell nuclei were marked by staining with DAPI. (D) Effect of mutations in the C terminus FR5 fragment of Nbs1 on mutant Ad DNA replication. NBS cells expressing wild-type or mutant Nbs1 proteins were infected with mutant virus dl1004, and DNA was extracted over a time course for quantitation by qPCR. Accumulation of viral DNA is represented as the fold increase over input viral DNA, determined at the 4-h time point. (E) Complementation of concatemer formation by C-terminal fragments of Nbs1. NBS cells expressing FR5, FR5-2K and FR5-Δ47 were infected with dl1004, and DNA was prepared at 30 hpi for analysis by PFGE. Viral DNA was visualized by staining the gel in ethidium bromide, and the position of linear viral genome is indicated by an arrow.
DISCUSSION
Deletion of the early E4 region of Ad5 is associated with defects in viral DNA replication, viral mRNA nucleocytoplasmic transport, and late protein synthesis (reviewed in references 60 and 67). Infection with E4-deleted viruses also leads to activation of DNA damage signaling and the formation of viral concatemers, both of which require the cellular MRN complex (6, 9, 55, 65). In the present study we developed a qPCR assay that specifically measures the accumulation of viral DNA in infected cells, and we used multiple cell systems to examine the effects of MRN and damage signaling on Ad DNA replication. We utilized viral mutants with distinct defects to demonstrate that MRN inactivation promotes viral replication. We also compared infections in hypomorphic cells and their matched counterparts. Finally, we used isogenic cells that differ only in expression of a single shRNA targeted to a specific repair factor. Our results demonstrate that the MRN complex has a detrimental effect on replication of E4-deleted Ad and that this is counteracted by E4 proteins that target the MRN proteins. We provide evidence that damage signaling and concatemers are not responsible for the inhibition of mutant Ad DNA replication. These observations extend studies from Evans and Hearing (20) and Mathew and Bridge (40, 41) that suggested an effect of MRN on virus replication. Furthermore, we identify an essential role for the C terminus of Nbs1 but demonstrate that there is a distinction in the requirements for inhibition of mutant virus replication and for concatemer formation.
The E4 proteins have evolved to attenuate cellular innate defenses against viral infection (68). The E4orf3 and E4orf6 proteins of Ad5 can compensate for each other to promote efficient viral DNA replication and progeny production (8, 24, 25). The MRN complex is a common target of both the E4orf6/E1b55K ubiquitin ligase activity and the E4orf3 protein (9, 19, 20, 55). Our data demonstrate that both are independently sufficient to inactivate MRN and that this target is relevant to the promotion of Ad5 DNA replication. Targeting of MRN may therefore explain the known redundancy between E4orf3 and E4orf6. It has been suggested that all of the phenotypes of viruses with E1b55K/E4orf6 deleted may be due to the lack of E3 ubiquitin ligase activity (4, 70) and that proteasome activity is required for E4orf6 stimulation of late gene expression (14). We show that degradation of the MRN complex is sufficient to explain the DNA replication phenotype. It will be interesting to see whether this substrate is also responsible for all other phenotypes. Our analysis of late protein production with E1b55K mutants (Fig. 1A) indicates that enhanced expression correlates with MRN degradation but does not separate this from increased virus DNA replication. We previously showed that degradation of MRN is a feature conserved across E1b55K/E4orf6 proteins from different Ad serotypes but that mislocalization of MRN by E4orf3 is unique to subgroup C viruses (56). Consistent with the importance of inactivating MRN to promote efficient viral DNA replication, we found that replication of the Ad4 serotype was less efficient than Ad5 in HeLa cells. Deleting E1b55K had minimal effect on Ad5 DNA replication but had a dramatic effect on Ad4 in HeLa cells (Fig. 2). This suggests that mislocalization of MRN by the E4orf3 protein of subgroup C viruses provides a growth advantage and could contribute to their high prevalence in the human population compared to other serotypes.
The MRN complex acts as a sensor of DNA damage and is required for signaling cascades that activate checkpoints to block cellular replication (3, 32, 43). We have previously demonstrated that MRN is required for activation of damage signaling in response to virus infection (9); however, our data now show that the ATM and ATR kinase pathways do not affect viral DNA replication. The defective DNA replication of E4-deleted Ad is also not due to concatemer formation, as demonstrated by the following observations: (i) the E1b55K mutants R240A and H354 both prevent concatemers through degradation of DNA ligase IV but only R240A, which degrades MRN, can promote Ad replication; (ii) concatemers were prevented by blocking both ATM and ATR signaling through treatment of A-T cells with caffeine, but this did not rescue the defective replication of E4-deleted Ad; (iii) the FR5-Δ47 mutant promotes concatemers but does not inhibit replication; and (iv) concatemers were not observed in DNA ligase IV mutant cells, but the E4-deleted virus was still defective for replication in these cells. Defective replication of ΔE4 and ΔE1b55K/ΔE4orf3 viruses is also not rescued in cells with mutant DNA-PKcs that do not form concatemers (40, 52). This conclusion is also supported by E4orf3 mutants that show genetic separation of function in Ad DNA replication and inhibition of concatemers (19). Our qPCR assay measures the accumulation of total virus DNA but does not give an indication of the impact on final progeny virus production. It was previously suggested that concatemer formation contributes to defective late protein production (26); however, we now show that preventing concatemers does not rescue the late protein production defect of ΔE1b55K/ΔE4orf3 mutant Ad (see late protein immunoblots from infections with H354 in Fig. 1A). Therefore, concatemer formation does not appear to be responsible for either the DNA replication or late protein defects of E4-deleted viruses. Concatemers, however, will be too large to be packaged into virus particles and therefore may need to be prevented in order to maximize production of infectious viral progeny. Although DNA damage signaling does not directly affect virus replication, it may lead to modification of cellular or viral proteins that could have a negative impact on late protein production (S. S. Lakdawala and M. D. Weitzman, unpublished observations).
Replication of the Ad genome is initiated by a viral terminal protein that is covalently attached to the 5′ end of each strand of the viral DNA (49). We have suggested that the MRN complex may be involved in removal of the terminal protein from the end of the viral genome (55). The yeast Mre11 protein has been implicated to function with the Sae2 protein in removal of covalently linked Spo11p from double-strand breaks that initiate meiotic recombination (45). Removal of the Ad terminal protein may be analogous to Spo11p cleavage and could lead to inhibition of replication initiation, as well as joining of viral genomes. Support for this hypothesis comes from the observation that Mre11 with a mutation in the nuclease domain cannot complement hypomorphic cells for concatemer formation (55). It has recently been suggested that Mre11 can physically associate with the Ad genome in a manner that requires Nbs1 (40). Consistent with this proposal, we show that the C terminus of Nbs1 is required for inhibition of mutant Ad DNA replication. The result of the Mre11-NLS fusion protein suggests that nuclear localization of Mre11 is not sufficient to inhibit mutant Ad DNA replication (see Fig. 5) (40). However, this experiment is difficult to interpret because Rad50 does not appear to be recruited into the nucleus by Mre11-NLS, suggesting that the protein may be misfolded or functionally impaired. The FR5-2K mutant suggests that Nbs1 interaction with Mre11 is required for complete nuclear localization of Mre11/Rad50, recruitment to viral replication centers, and inhibition of virus DNA replication. Interestingly, the FR5-Δ47 mutant is able to recruit Rad50 to viral replication centers but does not inhibit viral DNA replication, demonstrating that localization is not sufficient. Although the FHA and BRCT domains of Nbs1 are required for checkpoint activation and focus formation in response to irradiation (12, 18, 30, 73), the N terminus was not required for the accumulation of MRN at viral replication centers or for concatemer formation. These observations imply that recognition of viral genomes is different from sensing of DNA breaks in the context of cellular chromatin. The results with the FR5-Δ47 mutant suggest that binding to another factor is also required for inhibition of viral replication. The Nbs1 C terminus has been shown to regulate apoptosis in response to irradiation (18, 57), although the proteins that interact with this region have not been identified. Other factors may function cooperatively with the MRN proteins or modulate their activities (38, 51). Understanding how the MRN complex works with other cellular proteins to affect the adenoviral life cycle will provide further insights into the functions of this complex in DNA repair and replication.
Acknowledgments
We thank K. Cerosaletti, P. Concannon, C. Her, G. Iliakis, G. Ketner, A. Levine, D. Ornelles, Y. Shiloh, and S. Schreiber for generous gifts of reagents. We thank members of the Weitzman lab, Travis Stracker, Zhongsheng You, and Karen Cerosaletti for discussions and critical reading of the manuscript. We acknowledge the James B. Pendleton Charitable Trust for providing the Pendleton Microscopy Facility.
This study was supported by NIH grant CA97093 (M.D.W.) and by gifts from the H. A. and Mary K. Chapman Charitable Trust, Joe W. and Dorothy Dorsett Brown Foundation, and the Lebensfeld Foundation. B.P.M. and G.W.W. acknowledge funding from the MRC, the BBSRC, and the Wellcome Trust. R.A.S. was supported in part by a scholarship from the ARCS Foundation.
Footnotes
Published ahead of print on 18 June 2008.
REFERENCES
- 1.Araujo, F. D., T. H. Stracker, C. T. Carson, D. V. Lee, and M. D. Weitzman. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 7911382-11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, A., K. J. Rohleder, L. A. Hanakahi, and G. Ketner. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 817034-7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartek, J., and J. Lukas. 2007. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 19238-245. [DOI] [PubMed] [Google Scholar]
- 4.Blanchette, P., K. Kindsmuller, P. Groitl, F. Dallaire, T. Speiseder, P. E. Branton, and T. Dobner. 2008. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires the E4orf6 ubiquitin ligase activity. J. Virol. 822642-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borde, V. 2007. The multiple roles of the Mre11 complex for meiotic recombination. Chromosome Res. 15551-563. [DOI] [PubMed] [Google Scholar]
- 6.Boyer, J., K. Rohleder, and G. Ketner. 1999. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263307-312. [DOI] [PubMed] [Google Scholar]
- 7.Bridge, E., and G. Ketner. 1990. Interaction of adenoviral E4 and E1b products in late gene expression. Virology 174345-353. [DOI] [PubMed] [Google Scholar]
- 8.Bridge, E., and G. Ketner. 1989. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 63631-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carson, C. T., R. A. Schwartz, T. H. Stracker, C. E. Lilley, D. V. Lee, and M. D. Weitzman. 2003. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 226610-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerosaletti, K., and P. Concannon. 2004. Independent roles for nibrin and Mre11-Rad50 in the activation and function of Atm. J. Biol. Chem. 27938813-38819. [DOI] [PubMed] [Google Scholar]
- 11.Cerosaletti, K., J. Wright, and P. Concannon. 2006. Active role for nibrin in the kinetics of Atm activation. Mol. Cell. Biol. 261691-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cerosaletti, K. M., and P. Concannon. 2003. Nibrin forkhead-associated domain and breast cancer C-terminal domain are both required for nuclear focus formation and phosphorylation. J. Biol. Chem. 27821944-21951. [DOI] [PubMed] [Google Scholar]
- 13.Cerosaletti, K. M., A. Desai-Mehta, T. C. Yeo, M. Kraakman-Van Der Zwet, M. Z. Zdzienicka, and P. Concannon. 2000. Retroviral expression of the NBS1 gene in cultured Nijmegen breakage syndrome cells restores normal radiation sensitivity and nuclear focus formation. Mutagenesis 15281-286. [DOI] [PubMed] [Google Scholar]
- 14.Corbin-Lickfett, K. A., and E. Bridge. 2003. Adenovirus E4-34kDa requires active proteasomes to promote late gene expression. Virology 315234-244. [DOI] [PubMed] [Google Scholar]
- 15.D'Amours, D., and S. P. Jackson. 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell. Biol. 3317-327. [DOI] [PubMed] [Google Scholar]
- 16.Desai-Mehta, A., K. M. Cerosaletti, and P. Concannon. 2001. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol. Cell. Biol. 212184-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Difilippantonio, S., A. Celeste, O. Fernandez-Capetillo, H. T. Chen, B. Reina San Martin, F. Van Laethem, Y. P. Yang, G. V. Petukhova, M. Eckhaus, L. Feigenbaum, K. Manova, M. Kruhlak, R. D. Camerini-Otero, S. Sharan, M. Nussenzweig, and A. Nussenzweig. 2005. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 7675-685. [DOI] [PubMed] [Google Scholar]
- 18.Difilippantonio, S., A. Celeste, M. J. Kruhlak, Y. Lee, M. J. Difilippantonio, L. Feigenbaum, S. P. Jackson, P. J. McKinnon, and A. Nussenzweig. 2007. Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J. Exp. Med. 2041003-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans, J. D., and P. Hearing. 2003. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 775295-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 796207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falck, J., J. Coates, and S. P. Jackson. 2005. Conserved modes of recruitment of ATM, ATR, and DNA-PKcs to sites of DNA damage. Nature 434605-611. [DOI] [PubMed] [Google Scholar]
- 22.Gatei, M., D. Young, K. M. Cerosaletti, A. Desai-Mehta, K. Spring, S. Kozlov, M. F. Lavin, R. A. Gatti, P. Concannon, and K. Khanna. 2000. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 25115-119. [DOI] [PubMed] [Google Scholar]
- 23.Ginsberg, H. S., L. L. Moldawer, and G. A. Prince. 1999. Role of the type 5 adenovirus gene encoding the early region 1B 55-kDa protein in pulmonary pathogenesis. Proc. Natl. Acad. Sci. USA 9610409-10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halbert, D. N., J. R. Cutt, and T. Shenk. 1985. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J. Virol. 56250-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang, M.-H., and P. Hearing. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 632605-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayaram, S., and E. Bridge. 2005. Genome concatenation contributes to the late gene expression defect of an adenovirus E4 mutant. Virology 342286-296. [DOI] [PubMed] [Google Scholar]
- 27.Jazayeri, A., J. Falck, C. Lukas, J. Bartek, G. C. Smith, J. Lukas, and S. P. Jackson. 2006. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 837-45. [DOI] [PubMed] [Google Scholar]
- 28.Kastan, M. B., and J. Bartek. 2004. Cell-cycle checkpoints and cancer. Nature 432316-323. [DOI] [PubMed] [Google Scholar]
- 29.Kastan, M. B., and D. S. Lim. 2000. The many substrates and functions of ATM. Nat. Rev. Mol. Cell. Biol. 1179-186. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi, J. 2004. Molecular mechanism of the recruitment of NBS1/hMRE11/hRAD50 complex to DNA double-strand breaks: NBS1 binds to γ-H2AX through FHA/BRCT domain. J. Radiat. Res. 45473-478. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi, J., H. Tauchi, S. Sakamoto, A. Nakamura, K. Morishima, S. Matsuura, T. Kobayashi, K. Tamai, K. Tanimoto, and K. Komatsu. 2002. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 121846-1851. [DOI] [PubMed] [Google Scholar]
- 32.Lavin, M. F. 2007. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 267749-7758. [DOI] [PubMed] [Google Scholar]
- 33.Lee, J. H., R. Ghirlando, V. Bhaskara, M. R. Hoffmeyer, J. Gu, and T. T. Paull. 2003. Regulation of Mre11/Rad50 by Nbs1: effects on nucleotide-dependent DNA binding and association with ataxia-telangiectasia-like disorder mutant complexes. J. Biol. Chem. 27845171-45181. [DOI] [PubMed] [Google Scholar]
- 34.Lee, J. H., and T. T. Paull. 2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 267741-7748. [DOI] [PubMed] [Google Scholar]
- 35.Lee, J. H., and T. T. Paull. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308551-554. [DOI] [PubMed] [Google Scholar]
- 36.Lee, J. H., and T. T. Paull. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 30493-96. [DOI] [PubMed] [Google Scholar]
- 37.Lim, D. S., S. T. Kim, B. Xu, R. S. Maser, J. Lin, J. H. Petrini, and M. B. Kastan. 2000. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 404613-617. [DOI] [PubMed] [Google Scholar]
- 38.Limbo, O., C. Chahwan, Y. Yamada, R. A. de Bruin, C. Wittenberg, and P. Russell. 2007. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28134-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukas, C., F. Melander, M. Stucki, J. Falck, S. Bekker-Jensen, M. Goldberg, Y. Lerenthal, S. P. Jackson, J. Bartek, and J. Lukas. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 232674-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathew, S. S., and E. Bridge. 2008. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 37411-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathew, S. S., and E. Bridge. 2007. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365346-355. [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka, S., B. A. Ballif, A. Smogorzewska, E. R. McDonald III, K. E. Hurov, J. Luo, C. E. Bakalarski, Z. Zhao, N. Solimini, Y. Lerenthal, Y. Shiloh, S. P. Gygi, and S. J. Elledge. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 3161160-1166. [DOI] [PubMed] [Google Scholar]
- 43.McGowan, C. H., and P. Russell. 2004. The DNA damage response: sensing and signaling. Curr. Opin. Cell Biol. 16629-633. [DOI] [PubMed] [Google Scholar]
- 44.McSharry, B. P., H. G. Burgert, D. P. Owen, R. J. Stanton, V. Prod'homme, M. Sester, K. Koebernick, V. Groh, T. Spies, S. Cox, A. M. Little, E. C. Wang, P. Tomasec, and G. W. Wilkinson. 2008. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 824585-4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreau, S., J. R. Ferguson, and L. S. Symington. 1999. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 19556-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nghiem, P., P. K. Park, Y. Kim, C. Vaziri, and S. L. Schreiber. 2001. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 989092-9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paull, T. T., and M. Gellert. 1999. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 131276-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrini, J. H., and T. H. Stracker. 2003. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 13458-462. [DOI] [PubMed] [Google Scholar]
- 49.Robinson, A. J., J. W. Bodnar, D. H. Coombs, and G. D. Pearson. 1979. Replicating adenovirus 2 DNA molecules contain terminal protein. Virology 96143-158. [DOI] [PubMed] [Google Scholar]
- 50.Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, and R. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 594375-4382. [PubMed] [Google Scholar]
- 51.Sartori, A. A., C. Lukas, J. Coates, M. Mistrik, S. Fu, J. Bartek, R. Baer, J. Lukas, and S. P. Jackson. 2007. Human CtIP promotes DNA end resection. Nature 450509-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shepard, R. N., and D. A. Ornelles. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 789924-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3155-168. [DOI] [PubMed] [Google Scholar]
- 54.Stiff, T., C. Reis, G. K. Alderton, L. Woodbine, M. O'Driscoll, and P. A. Jeggo. 2005. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 24199-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418348-352. [DOI] [PubMed] [Google Scholar]
- 56.Stracker, T. H., D. V. Lee, C. T. Carson, F. D. Araujo, D. A. Ornelles, and M. D. Weitzman. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 796664-6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stracker, T. H., M. Morales, S. S. Couto, H. Hussein, and J. H. Petrini. 2007. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature 447218-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stracker, T. H., J. W. Theunissen, M. Morales, and J. H. Petrini. 2004. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair 3845-854. [DOI] [PubMed] [Google Scholar]
- 59.Stucki, M., and S. P. Jackson. 2006. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 5534-543. [DOI] [PubMed] [Google Scholar]
- 60.Tauber, B., and T. Dobner. 2001. Molecular regulation and biological function of adenovirus early genes: the E4 ORFs. Gene 2781-23. [DOI] [PubMed] [Google Scholar]
- 61.Usui, T., H. Ogawa, and J. H. Petrini. 2001. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell 71255-1266. [DOI] [PubMed] [Google Scholar]
- 62.Uziel, T., Y. Lerenthal, L. Moyal, Y. Andegeko, L. Mittelman, and Y. Shiloh. 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 225612-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vo, A. T., F. Zhu, X. Wu, F. Yuan, Y. Gao, L. Gu, G. M. Li, T. H. Lee, and C. Her. 2005. hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair. EMBO Rep. 6438-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warming, S., N. Costantino, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiden, M. D., and H. S. Ginsberg. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 91153-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weinberg, D. H., and G. Ketner. 1983. A cell line that supports the growth of a defective early region 4 deletion mutant of human adenovirus type 2. Proc. Natl. Acad. Sci. USA 805383-5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weitzman, M. D. 2005. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 101106-1117. [DOI] [PubMed] [Google Scholar]
- 68.Weitzman, M. D., and D. A. Ornelles. 2005. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 247686-7696. [DOI] [PubMed] [Google Scholar]
- 69.Williams, R. S., J. S. Williams, and J. A. Tainer. 2007. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 85509-520. [DOI] [PubMed] [Google Scholar]
- 70.Woo, J. L., and A. J. Berk. 2007. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 81575-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu, X., V. Ranganathan, D. S. Weisman, W. F. Heine, D. N. Ciccone, T. B. O'Neill, K. E. Crick, K. A. Pierce, W. S. Lane, G. Rathbun, D. M. Livingston, and D. T. Weaver. 2000. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 405477-482. [DOI] [PubMed] [Google Scholar]
- 72.You, Z., C. Chahwan, J. Bailis, T. Hunter, and P. Russell. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 255363-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao, S., W. Renthal, and E. Y. Lee. 2002. Functional analysis of FHA and BRCT domains of NBS1 in chromatin association and DNA damage responses. Nucleic Acids Res. 304815-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]