

Abstract
The bicyclic fatty acid lubiprostone (formerly known as SPI-0211) activates two types of anion channels in A6 cells. Both channel types are rarely, if ever, observed in untreated cells. The first channel type was activated at low concentrations of lubiprostone (<100 nM) in >80% of cell-attached patches and had a unit conductance of ∼3–4 pS. The second channel type required higher concentrations (>100 nM) of lubiprostone to activate, was observed in ∼30% of patches, and had a unit conductance of 8–9 pS. The properties of the first type of channel were consistent with ClC-2 and the second with CFTR. ClC-2's unit current strongly inwardly rectified that could be best fit by models of the channel with multiple energy barrier and multiple anion binding sites in the conductance pore. The open probability and mean open time of ClC-2 was voltage dependent, decreasing dramatically as the patches were depolarized. The order of anion selectivity for ClC-2 was Cl > Br > NO3 > I > SCN, where SCN is thiocyanate. ClC-2 was a “double-barreled” channel favoring even numbers of levels over odd numbers as if the channel protein had two conductance pathways that opened independently of one another. The channel could be, at least, partially blocked by glibenclamide. The properties of the channel in A6 cells were indistinguishable from ClC-2 channels stably transfected in HEK293 cells. CFTR in the patches had a selectivity of Cl > Br ≫ NO3 ≅ SCN ≅ I. It outwardly rectified as expected for a single-site anion channel. Because of its properties, ClC-2 is uniquely suitable to promote anion secretion with little anion reabsorption. CFTR, on the other hand, could promote either reabsorption or secretion depending on the anion driving forces.
Keywords: ClC–2, CFTR, single-channel recording, lubiprostone, SPI-0211
epithelial tissues are designed to form a barrier between two body compartments and to facilitate transport from one compartment to the other, thereby, regulating the composition of the compartments. In particular, most epithelia regulate the salt and water balance of their luminal compartment. Regulation of salt balance involves active transport of sodium (Na+) and chloride (Cl−), but, in eukaryotic organisms, water movement across epithelia is driven osmotically. That is, water movement depends on prior transport of osmotic equivalents, usually salt. Therefore, to regulate water balance requires the ability to regulate both salt reabsorption and salt secretion. An enormous amount of research (some performed by us) has examined the reabsorptive phase of the balance process, particularly that which involves reabsorption of Na+ through apical epithelial Na channels (ENaC) (8, 37, 57, 74, 82, 99).
But even for reabsorption, Na+ uptake is only part of the salt reabsorption equation: there must also be a pathway for the movement of anions. In the past, the Cl− channel protein, cystic fibrosis transmembrane conductance regulator (CFTR), which is defective in patients with cystic fibrosis, has been suggested as the pathway for anion movement in many epithelia. CFTR can certainly act as an entry pathway for Cl− entry into epithelial cells in parallel with Na+ entry through ENaC; however, fluid balance requires appropriate balance between salt reabsorption and salt secretion, but regulation of secretion has not been studied as extensively as reabsorption.
In salt-transporting epithelial cells, Cl− movement across the basolateral membrane occurs via an energy-dependent Na+-K+-2Cl− cotransporter and possibly Cl− channels (like the Cl− channel that leads to defective reabsorption in one form of Bartter's Syndrome). Cl− movement across the apical (luminal) membrane occurs via specific Cl− channels. CFTR may be involved in secretion since when CFTR is defective there appears to be excess reabsorption in the lung and intestine. CFTR is widely distributed in tissues (43, 69) and is found in the apical membrane of epithelial cells (96), including the intestine (17, 49, 93), T84 intestinal cell lines (113), and lung (12). Nonetheless, in tissues that also transport Na+, CFTR is not particularly well suited to promote secretion (19), and, except for pathophysiological circumstances, other chloride channels may be more important for stimulated secretion. In particular, the Cl− channel, ClC-2, has been suggested as a route for chloride movement across the luminal membrane of several different epithelia. ClC-2 is a member of the ClC Cl− channel family, which consists of at least nine members that are widely distributed in nature (27, 65, 84, 111, 115). ClC-2 is found in the intestine (3, 17, 51, 72, 86), gastric parietal cells (77, 103), liver (98), lung (9, 27, 33, 101, 104), retina (38), and parotid acinar cells (95). However, the properties of ClC-2 appear different depending on the conditions and tissue in which the channels are examined. In the present study, we examined ClC-2 at a single-channel level in the A6 epithelial cell model and in HEK293 cells and examined the effect of a pharmacological agent, lubiprostone (which has been reported to open Cl− channels), on anion channels in A6 cells.
Some investigators have suggested that the lubiprostone-mediated Cl− flux is mediated by the ClC-2 Cl− channel (15, 31). ClC-2-mediated Cl− transport has been studied in two human intestinal cell lines: Caco-2 (86) and T84 (3). Caco-2 cells contain ClC-2 protein, which contributes to Cl− secretion (86). Patch-clamp studies of ClC-2 in T84 cells also suggest a role in Cl− transport (3); however, immunostaining suggested that only a small subset of cells in the cultures contained ClC-2 when grown on glass coverslips (31, 72). Therefore, a second objective of this study was to determine whether one of the anion channels activated by lubiprostone in A6 cells is ClC-2 by examining the biophysical properties of the channels at a single-channel level.
This study shows that lubiprostone even at low doses is a potent activator of ClC-2, but at much higher doses, it also opens CFTR (to a lesser extent). These findings suggest a possible physiological role for ClC-2 in epithelial fluid transport.
METHODS
Cell culture.
A highly transporting clone, 2F3, of the Xenopus laevis distal nephron epithelial cell line (A6) was a gift from Dr. Thomas Kleyman and was maintained by standard tissue culture techniques as previously described (6) with a culture medium consisting of a 50% (vol/vol) mix of DMEM and Ham's F-12 medium adjusted to amphibian tonicity plus 0.6% penicillin-1.0% streptomycin, 5% (vol/vol) fetal bovine serum, 1.5 μM aldosterone, 1 mM glutamine, and 25 mM NaHCO3. For patch-clamp experiments, A6 cells were plated at confluent density on permeable, glutaraldehyde-fixed, collagen-coated Millipore-CM filters (Millipore, Billerica, MA) attached to the bottoms of small Lucite rings (53). This sided preparation allowed patch pipette access to the apical membrane and physical separation of the apical and basolateral bath compartments (5, 6, 21, 22, 53, 59, 60, 105–107, 120). Just prior to recording, monolayers were washed with a solution containing 96 mM NaCl, 3.4 mM KCl, 0.8 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES, or 10 mM Tris, adjusted to pH 7.4 with HCl or NaOH.
HEK293 stably transfected with human ClC-2 were a generous gift of John Cuppoletti and Danuta Malinowska. Untransfected HEK-293 cells were obtained from American Type Culture Collection (Bethesda, MD). Cells were grown under conditions described previously (31).
Lubiprostone was dissolved in spectroscopic grade DMSO to prepare a 1 mM stock solution that was kept at 4°C until diluted so that drug could be added to the apical bathing solution at various drug concentrations with a final DMSO concentration of 0.1% vol/vol. All cells were treated. Patch recording sometimes commenced prior to drug addition and continued after addition. In these cases, DMSO was added as a control for subsequent drug addition. Otherwise, cells were pretreated for 5 to 15 min with appropriate concentrations of drug before recording.
Transepithelial electrical measurements.
The transepithelial voltage (Vte) and resistance (Rte) across cell monolayers were measured with an epithelial volt-ohmmeter (World Precision Instruments, Sarasota, FL). The transepithelial current Ite was calculated according to Ohm's law Ite = Vte/Rte and normalized by the surface area of the insert. Amiloride (10 μM) was added to the apical side of the inserts at the end of each experiment, and amiloride-sensitive sodium current was calculated as the difference between total current and amiloride-insensitive current.
Patch-clamp recording and analysis.
A6 distal nephron cells were visualized with Hoffman modulation optics (Modulation Optics, Greenvale, NY) mounted on a Nikon Diaphot-TMD inverted microscope. Unless otherwise noted, patch pipette and extracellular bath solutions consisted of a physiological amphibian saline containing (in mM): 95 NaCl, 3.4 KCl, 0.8 CaCl2, 0.8 MgCl2, and 10 HEPES or 10 Tris. The solutions were titrated with 0.1 N NaOH or HCl to a pH of 7.3–7.4. Experiments were performed at room temperature. Unitary channel events were measured using an Axopatch 1-D (Axon Instruments, Sunnyvale, CA). Data were acquired using a 902LPF 8-pole Bessel filter (Frequency Devices, Haverhill, MA), TL-2 acquisition hardware and Axotape software (Axon Instruments). By convention, outward flux of Cl−, an anion, produces inward current (pipette to cell) and is represented as downward transitions in single-channel records (inward flux produces outward current-upward deflections). The total number of functional channels (N) in the patch was determined by observing the number of transitions in the single-channel recordings. As a measure of channel activity, the number of channels times the open probability, NPo, was also calculated. With an estimate of N and a measure of NPo, we also were able to estimate open probability, Po.
Cell-attached patches were used for all experiments. The voltage conventions for patch potentials in epithelial monolayers require special consideration. For high-resistance epithelial monolayers, with the electrical ground in the basolateral compartment, the potential to which channels in the patch membrane are exposed are not simple displacements from the apical membrane potential, but rather, the potential of the pipette is the sum of the applied potential from the patch-clamp amplifier and the transepithelial potential (see Fig. 1). Under typical recording conditions the transepithelial potential depends on the tightness of the monolayer. In the presence of lubiprostone, the potential can be as high as −80 mV (apical potential with respect to the basolateral solution at ground), and, therefore, the potentials in the current-voltage relationships can be offset to the right by as much as 80 mV. The offset will vary depending on the transepithelial voltage, which in turn depends on the transepithelial current and the transepithelial resistance (the primary determinant of which is the shunt or edge resistance).
Fig. 1.

Schematic diagram of cell-attached recording from A6 epithelial monolayer. This figure shows a schematic of the typical recording situation for the experiments in this paper with A6 cells grown on permeable supports and electrical ground in the basolateral solution and the patch pipette applied to the apical surface of the cell. The potentials applied to the ion channels in cell-attached single-channel patches from isolated cells are typically offset by the membrane potential. In a tight epithelial monolayer with the electrical ground in the basolateral solution, the patch potential is actually offset by the entire transepithelial potential, VT. That is, the potential recorded by the patch amplifier is actually the displacement of the patch potential, VP, from the transepithelial potential. This implies that current-voltage relationships are shifted to the right by the magnitude of the transepithelial potential, which after lubiprostone can be as large as 80 mV (compare Figs. 2 and 6B). VA and VBL are the apical and basolateral membrane potentials, respectively. GND is bath ground.
Data analysis.
We used modified constant-field approaches first described by Goldman (48) and later by Hodgkin and Katz (56) to describe the permeability of anions through ClC-2 and CFTR channels. In general, their approach gave the ionic flux (as current) and ion permeability across a cell membrane as described by the following equation:
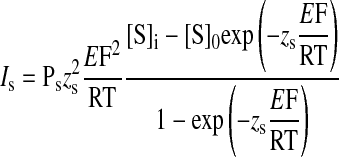 |
where Is is the current and PS is the permeability of ion S, [S]i and [S]o are the internal and external concentrations of ion S, z is the valence, and E is the potential difference across the cell membrane, and R, T, and F are constants.
For channels that rectify in a manner opposite that predicted by the above equation, more complicated models that are based on Eyring rate theory are necessary to fit the data. We used three different models: a single-barrier model (109), a two-barrier, one-well model (79), or a three-barrier, two-well model (7, 14, 54). We fit the data to these models using a Marquardt-Levenberg minimization algorithm as provided by the software companion to (97). To avoid local minimums, we fit each model 200 different times with randomly selected initial parameters and determined the least square minimum.
Regardless of which method was used model the current, selectivity was determined by the Goldman voltage equation with known concentrations of ions on each side of the membrane and with the reversal potential, ER, given by
 |
where Px and Py and [X] and [Y] are the permeabilities and concentrations of X and Y, respectively.
Western blot analysis.
Membrane-enriched fractions of proteins were obtained from 2F3 cells by using a lysis buffer (2 mM EGTA; 50 mM Tris-base; 320 mM sucrose, and 2.3% SDS). The protein concentration was assessed by a Bradford assay (Pierce Chemicals, Orlando, FL) so that equal protein could be loaded in each lane. The proteins were incubated for 10 min at 70°C in the presence of detergent (SDS) and reducing agents (β-mercaptoethanol), loaded in a 4–15% Tris·HCl precast polyacrylamide gel, and electroblotted to a 0.45-μm nitrocellulose membrane via the Trans-Blot SD Semi-Dry Electrophoretic system with a voltage of 15 V for 45 min in Towbin transfer buffer (25 mM Tris-base, 192 mM glycine, 20% methanol, pH 8.3). The proteins on the membrane were blocked with 5% nonfat dry milk in Tween 20 PBS for 2 h at room temperature. As a control, the primary antibody was preabsorbed with the antigenic peptide at a concentration of 2 μg/ml overnight at 4°C. The membrane was incubated in the solutions either with the primary (ClC-2 or CFTR) antibody at a final concentration of 0.37 μg/ml overnight at 4°C and then was extensively washed with PBS and incubated with the secondary antibody conjugated with horseradish peroxidase at a concentration of 1:3,000 for 1 h at room temperature. Finally, the membrane was washed with PBS and tested by chemiluminescence. ClC-2 antibody detects a single band at 97 kDa and was obtained from U.S. Biologicals, Swampscott, MA (Clcn2, chloride channel: C5837-05E). This antibody has been previously used and characterized by others (24–26, 88, 89); the CFTR antibody detects bands at 170 kDa and lower molecular weight degradation or trafficking products. The antibody was obtained from Santa Cruz Biotechnology, Santa Cruz, CA (CFTR, N-20: sc-8909) and has been previously characterized in Ref. 73.
Measurement of intracellular cAMP.
Confluent 2F3 cells grown on permeable supports were treated according to the following conditions for 15 min: no treatment control; 20 μM forskolin-positive control; or 10 nM, 50 nM, 100 nM, or 1 μM RU-0211. Cells were lysed with 0.1 N HCl, scraped, and sonicated. cAMP levels were measured by using the acetylated version of the colorimetric cAMP Direct Immunoassay Kit (Calbiochem online catalog no. 116811). A polyclonal antibody to cAMP was added to the standards and samples. After a 2-h incubation, plates were washed and substrate was added to produce yellow color change. Color density was read on a microplate reader at 405 nM, with concentration of cAMP inversely proportional to color intensity. The standard curve was used to quantify the concentration of cAMP in each condition. Results were normalized to the amount of protein by conducting a BCA assay and presented as picomoles cAMP per gram protein. Error in the measurement were derived from the standard error of the standard curve.
Chemicals.
Amiloride, diphenylcarboxylic acid, and glibenclamide were purchased from Calbiochem or Sigma Chemical (St. Louis, MO). Lubiprostone was obtained from Sucampo Pharmaceuticals (Sanda, Japan) as frozen aliquots of 2 mM solutions in 100% DMSO. DMSO, also obtained as frozen aliquots from Sucampo Pharmaceuticals, was used to dilute the lubiprostone and when testing for vehicle effects.
Statistics.
Data are reported as mean values ± SE. In general, cell-attached patch experiments were conducted in a paired fashion: data from each patch membrane served as the control for an experimental manipulation. In this case, the average change in Po for a group of patches, compared before and after an experimental treatment, was analyzed by a paired t-test or ANOVA for multiple comparisons. For unpaired experiments, traditional t-test or ANOVA was used when the data appeared to be normally distributed; otherwise an appropriate nonparametric test was used. Results were considered significant if P < 0.05. Statistical analysis was performed with SigmaPlot and SigmaStat software (Jandel Scientific, San Rafael, CA).
RESULTS
Lubiprostone increases transepithelial current and voltage in A6 cells.
We first tested the ability of lubiprostone to alter transepithelial electrical parameters. Adding even low concentrations of lubiprostone significantly increased transepithelial voltage and current (Fig. 2). Lubiprostone did not produce a significant difference in amiloride-sensitive voltage and current (presumably sodium transport) although there might have been a small increase in current. On the other hand, lubiprostone caused a significant increase in amiloride-insensitive voltage and current. The increase in current produced by even the lowest concentrations of lubiprostone is particularly striking. Lubiprostone (10 nM) significantly decreases the transepithelial resistance from 2.41 ± 0.127 Ωcm2 to 1.44 ± 0.0397 Ωcm2 (mean ± SE; n = 12). Addition of higher concentrations of lubiprostone further decrease resistance until at 1 μM the resistance is 1.08 ± 0.0462 Ωcm2 (mean ± SE; n = 6). The lubiprostone-mediated decrease in resistance is not amiloride sensitive.
Fig. 2.

Lubiprostone increases transepithelial current and voltage in A6 cells. A: effect of different concentrations of lubiprostone on total, amiloride-sensitive, and amiloride-insensitive transepithelial voltage. B: effect on transepithelial current. Adding even low concentrations of lubiprostone significantly increased transepithelial voltage and current. Lubiprostone did not produce a significant difference in amiloride-sensitive voltage and current (presumably sodium transport), although there might have been a small increase in current. On the other hand, lubiprostone caused a significant increase in amiloride-insensitive voltage and current. The increase in current produced by even the lowest concentrations of lubiprostone is particularly striking.
Lubiprostone activates Cl− channels in A6 cells.
We formed cell-attached patches on A6 cells with either a pipette solution containing 96 mM NaCl plus 3 μM amiloride or saline in which all the Na+ had been replaced with N-methyl-d-glucamine (neither saline contained K+). This meant that single-channel currents could not be due to ENaC in the first case, or any cation channel in the second case. Under these conditions, we generally saw little, if any, single-channel activity (Figs. 3A and 4A). The all-points amplitude histograms for the entire recording period below the records confirm that the patches reside predominantly in a zero-current state with no channel activity (Fig. 3, B and C and Fig. 4, B and C). After a control recording period, we slowly added 100 nM lubiprostone to the apical bath and after 5–15 min we observed channel activity in over 80% of the patches we examined. The channels were of two different types. The first type of channel, which we suggest is ClC-2 (Fig. 3D), had inward current at zero pipette potential (because of the large lubiprostone-mediated increase in the transepithelial voltage) and increasingly larger inward currents and increasing open probability at all more negative potentials. The second type of channel is the CFTR Cl− channel that has previously been described in A6 cells (71, 80, 81) and that has currents that usually reverse near or slightly positive to zero (no potential applied to the pipette) and characteristic flickery openings at positive pipette potentials (Fig. 4D). The all-points histograms below the records show that both types of channels spend a significant amount of time in one or more open states (Fig. 3, E and F and Fig. 4, E and F).
Fig. 3.

Lubiprostone activates small-conductance Cl− channels in A6 cells. A: typical single-channel records from a cell-attached patch before application of lubiprostone with no obvious channel activity (patch potentials to the left of the traces are the potential displacement from the apical membrane potential-about −40 mM). The all-points amplitude histograms (0 mV at B; −60 mV at C) confirm that the patch is predominantly in a zero-current state (marked with an arrow to designate the closed state). D: after addition of 100 nM lubiprostone channel activity is easily observed and the all-points histograms (0 mV at E; −60 mV at F) reflect the fact that there are 2 current levels that were not previously visible (marked as open 1 and open 2). In this figure (and all subsequent figures), the arrows mark the level of the closed state.
Fig. 4.

Lubiprostone activates CFTR in A6 cells. A: typical single-channel records from a cell-attached patch before application of lubiprostone with no obvious channel activity (patch potentials to the left of the traces are the potential displacement from the apical membrane potential-about −40 mM). The all-points amplitude histograms (0 mV at B; −60 mV at C) confirm that the patch is predominantly in a zero-current state (marked with an arrow to designate the closed state). D: after addition of 100 nM lubiprostone channel activity is easily observed and the all-points histograms (0 mV at E; −60 mV at F) reflect the fact that there are 2 current levels that were not previously visible. The characteristics of the channel is consistent with the channel being CFTR. In this figure, arrows mark the level of the closed state.
ClC-2 and CFTR proteins are detectable by Western blotting in A6 cells.
We used conventional Western blotting methods on lysates of A6 cells and blotted for ClC-2 and CFTR using commercially available antibodies. Figure 5 shows that both proteins are easily detectable in A6 cells. CFTR had been previously reported (71, 80, 81), but ClC-2 has not been shown before in A6 cells although it has been described in several other epithelia (2, 10, 17, 18, 23, 29, 34, 35, 41, 72, 85, 87, 114).
Fig. 5.

ClC-2 and CFTR proteins are detectable by Western blotting in A6 cells. We used conventional Western blotting methods (see methods) on lysates of A6 cells and blotted for ClC-2 and CFTR. Equal amounts of cellular protein were loaded in each lane (determined by Bradford assay). Both channels proteins are easily detectable in A6 cells. CFTR had been previously reported (67), but ClC-2 has not been shown before in A6 cells (although it has been described in several other epithelia). The ClC-2 antibody detects a single band at 97 kDa and was obtained from U.S. Biologicals, Swampscott, MA (Clcn2, chloride channel: C5837-05E); the CFTR antibody detects bands that correspond to CFTR at 170 kDa and CFTR processing and degradation products at lower molecular weight. The antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA; CFTR, N-20: sc-8909). The bands were competed by antigenic peptide obtained from the distributors.
The two channels have different properties.
Examination of single-channel records in cell-attached patches after exposure to 100 nM lubiprostone over a large range of voltages shows that the two channel types have distinctive and different biophysical characteristics. The channel we have identified as ClC-2 (Fig. 6A) has long open times at hyperpolarizing potentials, but both the current and the open probability decrease as the membrane is depolarized. At positive membrane potentials channel openings are small and difficult to detect. In contrast, in a lubiprostone-treated cell-attached patch containing CFTR (Fig. 7A), single-channel currents at positive potentials are easily detectable. At more negative potentials, the current becomes smaller until it reverses near zero (in a cell-attached patch, zero refers to no applied potential; that is, the patch membrane is near the transepithelial potential of the monolayer, probably −30 to −40 mV in this case). At potentials more negative than the reversal potential, channel openings are still easy to see, but the openings are now interrupted by fast closures. Such flickery, burstlike events are characteristic of CFTR in A6 cells.
Fig. 6.

Single-channel records show an anion channel with characteristic properties. Cells were pretreated with 100 nM lubiprostone for 5–15 min before forming a cell-attached seal. A: single-channel records were recorded from a complete series of voltages displacements from the membrane potential (0 mV displacement) for the 3-pS anion channel. More than 80% of the treated patches had this type of channel. This recording was made with amiloride in the pipette so that the recording could not be confounded by epithelial Na channel (ENaC) currents (which are generally present in the patches). The open probability and the unit current of the channel are large at hyperpolarized potentials and very small at depolarized potentials. In this figure, the arrows mark the level of the closed state. B: currents for ClC-2 strongly inwardly rectify. Such rectification is consistent with a multi-ion pore with greater access to the inner mouth of the pore than the outer mouth. The line through the data points is a fit of current to a multiple-barrier Eyring rate theory model (see text for details). The ratio of inward current to outward current is 1.37 ± 0.10, implying that the flux ratio for the channel is >1 and will secrete Cl− much better than reabsorb Cl−. The channel reverses at a positive potential because the current-voltage relationship is shifted to the right by the magnitude of the transepithelial voltage (see Fig. 1).
Fig. 7.

Single-channel records also show an anion channel with properties like CFTR. A: cells were pretreated with 100 nM lubiprostone for 5–15 min before forming a cell-attached seal. Single-channel records were recorded from a complete series of voltages displacements from the membrane potential (0 displacement) for the 8-pS anion channel. This recording was made with N-methyl-d-glucose replacing all cations in the pipette so that the recording could not be confounded by ENaC or any other cation channel currents. The open probability does not depend on voltage. The unit current of the channel is large at depolarized potentials and smaller at hyperpolarized potentials. The channel has the typical flickery closure at hyperpolarizing potentials that are characteristic of CFTR. In this figure, the arrows mark the level of the closed state. B: the current-voltage relationship for CFTR rectifies in a way expected for a single-site anion channel with more flux from the side that has a higher concentration of Cl− (outward current/inward flux is greater than inward current/outward flux). The current through CFTR channels can be fit well with the Goldman-Hodgkin-Katz current equation. The fit provides estimates for CFTR channel permeability (6.74 ± 0.525 × 10−6 cm/s), the transepithelial potential (−41 ± 8.4 mV apical bath negative), and the intracellular Cl− activity (38 ± 8.9. mM).
The rectification of the two channel types is different.
Some of these properties are reflected in the current voltage relationships for the channels (Fig. 6B). The single-channel currents for ClC-2 strongly inwardly rectify. Given that the concentration of Cl− is greater outside the cell (96 mM) than inside (∼20–30 mM), one might expect inward flux of Cl− (outward current) to be greater than outward flux (inward current) when, in fact, it is the reverse. This implies that the permeability for Cl− is smaller for Cl− entry than for Cl− exit. Such a property is consistent with a pore that has one or more barriers to ion movement and possible binding sites within the pore that will produce greater access to the inner mouth of the pore than the outer mouth. Such a pore would be expected to favor outward flux over inward; that is, the flux ratio should vary as some higher power of Cl− concentration and voltage. We fit the current-voltage data to several models that contained one or more energy barriers and energy wells (as described in methods). The fit to two of the models is shown in Fig. 8 with best-fit values given in Table 1. As is obvious, the fit to a single energy barrier, although the model can produce strong inward rectification, fits the data very poorly. The fit of either model 2 (2 barriers and 1 well) or model 3 (3 barriers and 2 wells) produces a fit that appears the same. However, model 2 has low-energy barriers and predicts an extremely high intracellular chloride concentration. The low-energy barriers seem inconsistent with the small unit conductance of ClC-2. Therefore, we tend to favor model 3 or a model with even more barriers and wells.
Fig. 8.

Models for the current-voltage relationships for ClC-2. The single-channel currents for ClC-2 strongly inwardly rectify. Such a property is consistent with a pore that has one or more barriers to ion movement and possible binding sites within the pore that will produce greater access to the inner mouth of the pore than the outer mouth. We fit the current-voltage data to several models that contained one or more energy barriers and energy wells (as described in methods). The fit to 3 of the models is shown in Fig. 8. As is obvious, the fit to a single energy barrier B1, although the model can produce strong inward rectification, fits the data very poorly. The fit of either model 2 (2 barriers, B1 and B2, and 1 well, W1) or model 3 (3 barriers, B1–B3, and 2 wells, W1 and W2) produces a fit that appears the same. However, model 2 has low-energy barriers and predicts an extremely high intracellular chloride concentration (see Table 1). The low-energy barriers seem inconsistent with the small unit conductance of ClC-2. Therefore, we tend to favor model 3 or a model with even more barriers and wells.
Table 1.
Best fit parameters for Eyring models of CLC2 current-voltage relationships
|
Model 1 (1 barrier) |
Model 2 (2 barriers, 1 well)
|
Model 3 (3 barriers, 2 wells)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔG, KJ/mol | δ | ΔG, KJ/mol | δ | ΔG, KJ/mol | δ | ||||
| Barrier 1 | 24.4 | 0.5 | 6.6 | 0.68 | 62.6 | 0.052 | |||
| Well 1 | −7.8 | 0.22 | −6.8 | 0.5 | |||||
| Barrier 2 | 9.5 | 0.098 | 58.5 | 0.047 | |||||
| Well 2 | −0.1 | 0.18 | |||||||
| Barrier 3 | 66.3 | 0.22 | |||||||
| [Cl]i | 37 mM | 50 mM | 24 mM | ||||||
ΔG, height or depth of the energy barrier or well; δ, electrical distance from the outside of the membrane to the barrier or well.
The reversal potential for ClC-2 in Fig. 6B appears more positive than might be expected for a Cl− channel. Of course, it is necessary to remember that channels in cell-attached patches on cells in a tight epithelial monolayer also measure the transepithelial potential (Fig. 1); i.e., the voltages in the current-voltage relationship are the displacement of the patch potential from the transepithelial potential. For example, +20 mV pipette potential is 20 mV more positive than the transepithelial potential. Since lubiprostone activates transepithelial Cl− transport, the transepithelial potential can be quite large if the monolayer is high resistance. The transepithelial potential will shift the current-voltage relationship to the right by up to 90 mV (see Fig. 2). This is approximately the deviation of the reversal potential from zero in Fig. 6B. Therefore, the apparent reversal potential will depend on variability in transepithelial potential: if the monolayer is intact the reversal potential may be far to the right; if there is even a very slight amount of edge damage the reversal potential will be close to zero. To test this possibility we reasoned that if we found a patch that had both CFTR and ClC-2, they should both reverse at the same potential whether it is very positive or close to zero. Figure 9 shows single-channel records and the current-voltage relationships for just such a patch with the two types of channels reversing at the same positive potential.
Fig. 9.

The transepithelial potential shifts the single-channel current-voltage relationships to the right. The reversal potential for ClC-2 in Fig. 6B appears more positive than might be expected for a Cl− channel, but channels in cell-attached patches on cells in a tight epithelial monolayer also measure the transepithelial potential (Fig. 1); i.e., the voltages in the current-voltage relationship are the displacement of the patch potential from the transepithelial potential. Since lubiprostone activates transepithelial Cl− transport, the transepithelial potential can be quite large if the monolayer is high resistance. The transepithelial potential will shift the current-voltage relationship to the right by up to 90 mV (see Fig. 2). This is approximately the deviation of the reversal potential from zero in Fig. 6B. Therefore, the apparent reversal potential will depend on variability in transepithelial potential: if the monolayer is intact the reversal potential may be far to the right; if there is even a very slight amount of edge damage the reversal potential will be close to zero. To test this possibility we reasoned that if we found a patch that had both CFTR and ClC-2, they should both reverse at the same potential whether it is very positive or close to zero. The current-voltage relationship above shows single-channel records (A) and the current-voltage relationships (B) for just such a patch with the 2 types of channels reversing at the same positive potential.
The current-voltage relationship for CFTR (Fig. 7B) rectifies in a way expected for a single-site anion channel with more flux from the side that has a higher concentration of Cl− (outward current/inward flux is greater than inward current/outward flux). The current through CFTR channels can be fit well with the Goldman-Hodgkin-Katz current equation. The fit provides estimates for CFTR channel permeability (6.74 ± 0.525 × 10−6 cm/s), the transepithelial potential (−41 ± 8.4 mV apical compartment negative), and the intracellular Cl− activity (38 ± 8.9. mM consistent with the value of 24 ± 11. mM predicted by model 3 for ClC-2).
Lubiprostone activates ClC-2 at much lower dose than CFTR.
We were somewhat surprised that lubiprostone activated CFTR but thought that the activation might be associated only with the higher doses of lubiprostone. Therefore, we examined the lubiprostone dose-response relationship for ClC-2 and CFTR by examining the frequency with which we observed specific channels after treatment with different concentrations of lubiprostone. In Fig. 10, we then plotted the percentage of patches with either ClC-2 or CFTR vs. dose and fitted the data to the following dose equation:
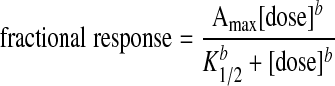 |
where Amax is the maximum fraction of patches with channels, K1/2 is the half-activating dose, and b is the slope of the dose relationship (the Hill coefficient). As is obvious from Fig. 10, much higher doses of lubiprostone are required to activate CFTR. The K1/2 for ClC-2 is 69 ± 18.8 nM whereas for CFTR the K1/2 is 791 ± 273 nM. These K1/2 values are significantly different from one another (P < 0.01). The maximal responses for ClC-2 and CFTR are 69 ± 7.8 and 61 ± 5.4% of patches with channels, respectively, and the slope coefficient for ClC-2 and CFTR is 1.5 ± 0.37 and 0.84 ± 0.13, respectively (all values are means ± SE). These results show that it requires at least 10-fold more lubiprostone to activate CFTR than ClC-2 and is consistent with using lubiprostone as an activator of ClC-2. We were interested in the mechanism by which lubiprostone activated either ClC-2 or CFTR. Traditionally, CFTR is activated by increases in intracellular cAMP. However, an increase in cAMP does not appear to be how lubiprostone increases channel activity (neither ClC-2 or CFTR) since when we measured cAMP (Fig. 11), we found that lubiprostone did not increase cAMP even at high dosages even though the positive control, forskolin, produced a robust cAMP increase.
Fig. 10.

Lubiprostone activates ClC-2 at much lower doses than CFTR. This figure shows the frequency of ClC-2 and CFTR in patches in response to lubiprostone measured as the percentage of patches that contained one channel or the other. The frequency of ClC-2 (•) and CFTR (○) were fitted (solid lines) to the Hill equation (see results). This figure shows that much higher doses of lubiprostone are required to activate CFTR. The half-activating dose for ClC-2 is 69 ± 18.8 nM whereas for CFTR the dose is 791 ± 273 nM. The maximal response for ClC-2 and CFTR is 69 ± 7.8 and 61 ± 5.4% of patches with channels, respectively, and the slope coefficient for ClC-2 and CFTR is 1.5 ± 0.37 and 0.84 ± 0.13, respectively (all values are means ± SE).
Fig. 11.

Lubiprostone does not increase intracellular cAMP. Lubiprostone did not increase cAMP even at high dosages even though the positive control, forskolin, produced a robust cAMP increase.
The open probability of ClC-2 decreases with depolarizing voltages.
Figure 4 showed that ClC-2 single-channel currents strongly rectify as the apical membrane was made more positive; however, besides the smaller than expected outward current, the frequency of ClC-2 openings also decreased. That is, the open probability of ClC-2 decreased at positive potentials. Figure 12 shows the voltage sensitivity of the channel open probability. The decrease in open probability can be fit by a Boltzman relationship with half-maximal activation at 170 ± 8.4 mV and the open probability decreasing e-fold for every 43 ± 8.4 mV of depolarization. We were able to determine the open probability by first measuring NPo and then using the fact that at very hyperpolarized potentials the open probability is large enough that even recording for a relatively short period of time allows an accurate count of the number of channels in the patch.
Fig. 12.

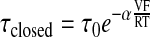 |
The decrease in open probability was due to both a voltage-dependent decrease in channel mean open time and an increase in mean closed time. To obtain a better idea of the voltage dependence of channel opening and closing, we took advantage of the fact that the mean duration in either the open or closed state changes according to the following expression:
 |
where τ(V) is the mean duration (open or closed time) at a voltage V; τ(0) is the mean duration (open or closed time) of the channel at 0 mV; and α is the voltage dependence of the channel. One way to interpret α is as the fraction of the membrane field detected by charged sites on the channel although for ClC-2 the interpretation may be more complicated (75). The value for α can be obtained by determining the slope of the relationship between log τ(V) and voltage or by fitting the relationship between τ(V) and voltage to the expression above. Figure 12, B and C shows the voltage dependence of mean open and closed times for positive potentials. We examined patches that had no more than two channels (at hyperpolarized potentials) and then held the patches at positive potentials since the open probability was so low that we did not observe overlapping open events. Unbiased open time analysis is possible if only one current level is observed in a patch even if the patch contains more than one channel. However, our estimates of closed time are biased if the patches contain more than one channel. However, the voltage dependence of mean closed time is not affected by counting since there is the same bias at all potentials. With no applied potential, the mean open time is 874 ± 22.6 ms and the mean open time decreases with increasing voltage as if the voltage dependence for channel closing senses 76 ± 5.0% (α = 0.76 ± 0.05) of the transepithelial membrane potential. With no applied potential, the mean closed time is 2,700 ± 377 ms and the mean closed time increases with increasing voltage as if the voltage sensor for opening the channel senses 23 ± 3.4% of the transepithelial membrane potential. Thus the change in the open time reduces the inward flux of Cl− over and above the inward rectification of the channel. In contrast to ClC-2, the gating of CFTR is quite complicated but has been extensively studied (4, 16, 20, 47, 50, 58, 70, 100, 121); therefore, we did not examine the gating other than to qualitatively verify that it was the same as previously reported.
ClC-2 is a “double-barreled” channel.
At least two other members of the ClC family of Cl− channel proteins form dimers and, thus, have two ion-conductance pathways for each channel protein that open and close independently of one another (92, 123). Despite the independence, the fact that each protein molecule in effect contains two channels means the patches will appear to contain even numbers of channels and patches. Since counting the number of channels in a patch involves counting the number of current levels and since the two halves of the dimers open independently of one another, it might still be possible to observe patches with what appear to be odd numbers of channels. This would occur when the recording period was so short or the open probability was so low that there was a low probability of seeing both conductance pathways open at the same time. We have discussed the problem of determining whether a channel contains two conducting pathways in previous work (68). We recorded from ClC-2 channels at negative potentials (maximum open probability) for several minutes and counted the apparent number of channels (as the difference between the minimum and maximum number of current levels). Figure 13 shows a histogram examining the frequency of observing different number of levels for 22 patches. The histogram shows that even numbers of levels are strongly favored over odd numbers of levels, implying that ClC-2 like ClC-0 is also a “double-barreled” Cl− channel. There were no zero-level (empty) patches in the sample, reflecting the fact that in the presence of lubiprostone virtually all patches have ClC-2 activity (sometimes both ClC-2 and CFTR channels). The frequency of channels in even-numbered patches is well fit (r = 0.87) by a binomial distribution with an open probability of 0.355 ± 0.0962. The probability that the patches with odd numbers of channels could have come from the same distribution is 4.7 × 10−6, supporting the idea that the channel has two conducting pores.
Fig. 13.

ClC-2 is a dimeric channel. We recorded from ClC-2 channels at negative potentials (maximum open probability) for several minutes and counted the apparent number of channels (as the difference between the minimum and maximum number of current levels). A histogram examining the frequency of observing different number of levels for 46 patches shows that even number of levels are strongly favored over odd numbers of levels, implying that ClC-2, like ClC-0, is also a dimeric Cl− channel. There were few zero-level (empty) patches in the sample, reflecting the fact that in the presence of lubiprostone virtually all patches have activity (sometimes both types of channels). If ClC-2 channels were inserted into the membrane as monomers, then one would expect (based on a binomial distribution) levels for 1 and 3 channels comparable to 2 and 4 channels. In fact, the observed number of patches containing either 1 or 3 channels (black bars) is far below the predicted levels (light gray bars). The fact that we see some patches with what appears to be 3 channels probably means that, even when open probability is high, we have underestimated the number of channels in the patch.
ClC-2 and CFTR are more permeable to Cl− than other anions.
Part of the biophysical characterization of ion channels is a determination of the selectivity of the channels for different ions. Examination of selectivity of single ion channels in cell-attached patches involves filling the pipettes with salines containing alternative anions and then comparing in the same batch of cells the reversal potential for the anions. In our experiments, we filled the pipettes with either 96 mM NaCl, NaBr, or NaNO3 or 115 mM NaI or NaSCN, where SCN is thiocyanate. Figure 14 shows the single-channel currents for inward and outward currents for Cl−, Br−, and NO3−. Outward Br− current is relatively easy to detect, but outward NO3− current is difficult to observe. Figure 15 shows current-voltage relationships for the different ions near the reversal potentials. The currents were fit to the Goldman-Hodgkin-Katz current equation assuming one permeable coefficient for outward current (flux of anion from the pipette into the cell) and another for inward current (see methods). It is obvious from the reversal potentials that the order of permeability for ClC-2 is Cl− > Br− > NO3− > I− > SCN− and for CFTR is Cl− > Br− ≫ NO3− = I− = SCN−. Table 2 gives values for the permeability ratios and estimates of the specific permeabilities for the ions that had significant outward current. The position of NO3− in the sequence for ClC-2 is ambiguous because little outward current was recorded and, therefore, the reversal potential was poorly determined. This implies that NO3− could be lower (but not higher) in the sequence of anions. For CFTR, we observed only small outward currents for SCN− and no outward current for NO3−, or I−, even at the most depolarizing potentials (up to +250 mV), implying that the permeability of these latter two ions is <1% of Cl− permeability.
Fig. 14.

ClC-2 and CFTR are more permeable to Cl− than other anions. In our experiments, we filled the pipettes with either 96 mM NaCl (A), NaBr (B), or NaNO3 (C) (plus amiloride to ensure that there were no cation channels) or 115 mM NaI or NaSCN and recorded single-channel currents for inward and outward currents for Cl−, Br−, and NO3−. Outward bromide current is relatively easy to detect, but outward NO3− current is difficult to observe. In this figure, the arrows mark the level of the closed state.
Fig. 15.

Current-voltage relationships for the different ions near the reversal potentials. The CFTR currents were fit to the Goldman-Hodgkin-Katz current equation and the ClC-2 channels were fit to a 3 barrier-2 well model. It is obvious from the reversal potentials that the order of permeability for ClC-2 is Cl− > Br− > NO3− (data not shown) > I− > SCN− and for CFTR is Cl− > Br− ≫ NO3− = I− = SCN−. Table 1 gives values for the permeability ratios for the ions that had significant outward current. The position of NO3− in the sequence for ClC-2 is ambiguous because little outward current was recorded and, therefore, the reversal potential was poorly determined. This implies that NO3− could be lower (but not higher) in the sequence of anions. For CFTR, we observed only small outward currents for SCN− and no outward current for NO3−, or I−, even at the most depolarizing potentials (up to +250 mV), implying that the permeability of these latter 2 ions is <1% of Cl− permeability.
Table 2.
Selectivity of ion channels in A6 cells stimulated by lubiprostone
| Ion | ClC-2 Channel Pion/PCl | CFTR Channel Pion/PCl |
|---|---|---|
| Cl− | 1.00 | 1.00 |
| Br− | 0.766 | 0.261 |
| NO3− | 0.546 | very small |
| I− | 0.00124 | very small |
| SCN− | 0.00771 | 0.0442 |
Permeability ratios were calculated from the shifts in the reversal potentials in Fig. 9 for different ions in the external (pipette) solution. Permeabilities were calculated from the fits to the current-voltage relationships for inward and outward currents. Pion/PCl is the ratio of the permeability of the ion in the first column to the chloride permeability. SCN, thiocyanate.
Thiocyanate and iodide are ClC-2 channel blockers.
If inward flux of anions through ClC-2 is independent of outward flux, then application of any anion regardless of its permeability to the external surface of the channel should have no effect on the current mediated by the ion on the internal surface of the channel. In other words, even though I− and SCN− have a very low permeability (little outward current), their application should not affect efflux of Cl− (inward current). Figure 16 shows that this is definitely not the case. Both I− and SCN− reduce the mean open time of the channel for inward currents when open times are usually relatively long. At −60 mV, the mean open and closed times with external and internal Cl− were 211 ± 2.94 ms and 549 ± 4.47 ms, respectively. At the same potential, the mean open and closed times in the presence of I− were 12.2 ± 4.24 and 85.4 ± 0.464 ms, respectively, and in the presence of SCN− were 18.2 ± 2.46 and 108 ± 0.199 ms, respectively. Such “trans-side” block has been extensively described for multi-ion cation channels (42, 54, 118) and is consistent with either model 2 or model 3 described above.
Fig. 16.

I− and SCN− block ClC-2 channels. Both I− and SCN− reduce the mean open time of the channel for inward currents when open times are usually relatively long. At −60 mV, the mean open and closed times with external and internal Cl− (top trace) were 211 ± 2.94 ms and 549 ± 4.47 ms, respectively. At the same potential, the mean open and closed times in the presence of I− were 12.2 ± 4.24 ms and 85.4 ± 0.464 ms (bottom trace with expanded section) and in the presence of SCN− were 18.2 ± 2.46 ms and 108 ± 0.199 ms (middle trace with expanded section). Such “trans-side” block has been extensively described for multi-ion cation channels (38, 50, 111). In this figure, the arrows mark the level of the closed state.
Lubiprostone activates ClC-2 in stably transfected HEK293 cells.
There are few descriptions of the single-channel properties of ClC-2 channels. Only one study in cortical astrocytes (92) provided data that could be compared with ours, and the quantitative and qualitative comparisons supported our conclusion that we were examining ClC-2. However, to further support our conclusion, we examined the properties of the predominant anion channels in HEK293 cells stably transfected with human ClC-2 (31). The patches we formed on HEK cells were not as stable or as high resistance as those on A6 cells; however, the anion channel activated by lubiprostone had channel kinetics (Fig. 17A) and a current-voltage relationship (Fig. 17B) that were essentially indistinguishable from the channels in A6 cells we had identified as ClC-2 channels.
Fig. 17.

Lubiprostone activates ClC-2 in stably transfected HEK293 cells. We examined the properties of the predominant anion channels in HEK293 cells stably transfected with human ClC-2 (27) (A). The patches we formed on HEK cells were not as stable or as high resistance as those on A6 cells; however, the anion channel activated by lubiprostone had channel kinetics that were essentially indistinguishable from the channels in A6 cells we had identified as ClC-2 channels (Figs. 1 and 4). B: the current-voltage relationship of ClC-2 channels in stably transfected HEK293 cells. The current-voltage relationship of the anion channel activated by lubiprostone was essentially the same as that of the channels in A6 cells we had identified as ClC-2 channels (Fig. 6A). In this figure, the arrows mark the level of the closed state.
Other channel blockers.
Specific anion channel blockers have been notoriously difficult to find. Several inhibitors of CFTR have been described, but most of these tend to be hydrophobic anions that are likely to interact with many different molecules. We examined two such blockers: diphenylcarboxylic acid and glibenclamide. Both of these agents block CFTR and we confirmed this block in our experiments (data not shown). Unfortunately, they also both block ClC-2 (the effect of glibenclamide on ClC-2 is shown in Fig. 18). Therefore, these blockers do not appear useful for distinguishing the two channel types in whole cell experiments. We further examined the effect of glibenclamide on ClC-2 to determine the nature of the block. Glibenclamide appears to be an open channel blocker since it reduces the mean open time (Fig. 19B) as judged from the open-interval histograms (mean open time at −60 mV before glibenclamide is 60 ± 2.9 ms, which is reduced to 32 ± 2.2 ms) and induces a second component in the closed-interval histogram (Fig. 19A). The closed-interval histogram before glibenclamide is best fit (as judged by a χ2 comparison) by a single exponential distribution with a mean closed time of 278 ± 3.3 ms, but after glibenclamide, the histogram is best fit by two exponential distributions with mean times of 233 ± 2.55 and 37 ± 5.6 ms. The second component (with the shorter mean duration) should represent the mean duration of the glibenclamide-blocked state. Figure 19C shows the open probability vs. time after exposure of the whole cell to glibenclamide. The block is relatively slow (the time constant for block is 466 ± 290 s), presumably reflecting the time necessary for glibenclamide to enter the cell, and, even at long times, the block does not appear to be complete reflecting the fact that glibenclamide is not a particularly effective blocker of ClC-2. Nonetheless, the reduction in Po is statistically significant (P < 0.001 by Mann-Whitney rank sum test).
Fig. 18.

Glibenclamide blocks ClC-2. A: ClC-2 induced by 100 nM lubiprostone before (top) or after glibenclamide (0.1 mM), ordinarily considered a CFTR blocker. The all-point amplitude histograms below shows that glibenclamide significantly reduces the open state and increases the occupancy of the closed state (compare histograms in B and C). In this figure, the arrows mark the level of the closed state.
Fig. 19.

Glibenclamide appears to block open ClC-2 channels. Glibenclamide appears to be an open channel blocker since it reduces the mean open time as judged from the open-interval histograms (before glibenclamide in light gray; after in dark gray). The mean open time at −60 mV before glibenclamide is 60 ± 2.9 ms, which is reduced to 32 ± 2.2 ms and induces a second component in the closed-interval histogram (before glibenclamide in gray; after in dark gray). The closed-interval histogram before glibenclamide is best fit (as judged by a χ2 comparison) by a single exponential distribution with a mean closed time of 278 ± 3.3 ms, but after glibenclamide, the histogram is best fit by 2 exponential distributions with mean times of 233 ± 2.55 and 37 ± 5.6 ms. The second component (with the shorter mean duration) should represent the mean duration of the glibenclamide blocked state. C: open probability vs. time after exposure of the whole cell to glibenclamide. The block is relatively slow (the time constant for block is 466 ± 290 s) presumably reflecting the time necessary for glibenclamide to enter the cell and, even at long times, the block does not appear to be complete reflecting the fact that glibenclamide is not a particularly effective blocker of ClC-2. Nonetheless, the reduction in open probability (Po) is statistically significant (P < 0.001 by Mann-Whitney rank-sum test).
The other blocker we examined was cadmium (Cd2+) since it has been reported to be a specific blocker of ClC-2 (23, 29, 66, 67, 76, 92, 123). The presumptive mechanism by which Cd2+ produces it block is by interaction with a critical cysteine residue in ClC-2. This suggests that Cd2+ may not be very specific and since it has previously always been bath applied the route and mechanism of block is not completely clear. In our experiments, we included 1 μM Cd2+ in the pipette and did not reduce the incidence of ClC-2 in the patches (∼70–80% of patches). If we pretreat the entire monolayer with cadmium for 30 min, we see no ClC-2 channels, or for that matter, any other channels (119). This suggests a generally toxic effect of cadmium due to its sulfhydryl reactivity.
DISCUSSION
The main objectives of this study were to determine clearly which channels were activated by lubiprostone in a model epithelial cell line and to provide a biophysical characterization of these channels at a single-channel level. For comparison, the main results are summarized in Table 3.
Table 3.
Comparison of the properties of ion channels activated by lubiprostone
| Property | ClC-2 | CFTR | Notes |
|---|---|---|---|
| Unit conductance | 3–4 pS | 7–9 pS | maximum conductance (outward current for CFTR; inward current for ClC-2) |
| Voltage dependence | Po decreases with depolarization | none | |
| Selectivity | Cl>Br>NO3>I>SCN | Cl>Br>>NO3=I=SCN | |
| Blockers | DPC, glibenclamide, I−, SCN− | DPC, glibenclamide | reduces open time |
| Rectification | inward | outward | |
| Other | “double-barreled” | burst behavior at negative potentials | |
| Frequency in treated patches | >80% | about 30% |
Po, open probability.
Lubiprostone activation of ClC-2 channels.
There are surprisingly few examples of single-channel records from ClC-2 channels in native, untransfected cells and only limited examples in any preparations. There has been some disagreement about the properties of the unitary channels underlying ClC-2 (39, 62, 77, 116). At least part of the variability likely has to do with the low conductance of ClC-2 single channels, the disparate conditions under which recordings of ClC-2 activity are made, and the fact that ClC-2 gating is apparently sensitive to changes in intracellular Cl−, pH, hormonal agents, voltage protocols, and tonicity (3, 9, 18, 23, 28, 32, 33, 35, 40, 46, 55, 63, 78, 90, 91, 94, 98, 123). Inside-out patch recordings of ClC-2 dimers expressed in Xenopus oocytes produces channels with a single-channel conductance of ∼3 pS that behaved as a double-barrel-like channel, although the pore stoichiometry of the homomeric channel was not determined (116). ClC-2 expressed in NIH 3T3 cells (13) had similar properties (3–4 pS) to those in oocytes and an inwardly rectifying single-channel conductance. The channels observed in one of the few examples of single-channel recordings from native cells (rat cortical astrocytes) have similar properties to those observed in oocytes and ClC-2-transfected cells (92). These properties are in contrast to a ClC-2 variant from gastric parietal cells, ClC-2G, which when expressed in Xenopus oocytes is activated by protein kinase A and acid pH with a linear current-voltage curve and a conductance of 29 pS. These properties were measured at high Cl− concentration (800 mM CsCl) (77, 104). The same laboratory has also shown that ClC-2 channels are activated by arachidonic acid and the drug omeprazole in lung and a recombinant expression system (33, 110). The source of this difference in behavior is unclear, but it is interesting to note that in whole cell recordings, even when the steady-state current-voltage relationship rectifies, the current shortly after a voltage step is close to linear (39, 45, 101, 111). Therefore, differences may well have to do with the protocols for recording in different laboratories.
In our work, the predominant lubiprostone-induced single Cl− channels had a low unit conductance (3–4 pS), the conductance inwardly rectified, and the open probability was voltage dependent with increased open probability at hyperpolarized potentials. The channel appeared to be a dimer with even numbers of channels in patches. These characteristics are similar to those of channels in cells transfected with ClC-2 and associated with native channels (as described above). In fact, the channels in A6 cells are virtually indistinguishable from those of ClC-2 stably expressed in HEK293, which we and others (31) have shown are activated by lubiprostone. The selectivity sequence for anions of the channels in A6 cells is also the same as that reported by others for ClC-2 (39, 45, 101, 111) and the “double-barreled” nature of the channels is similar to channels we have previously described in rat distal nephron principal cells that are stimulated by PGE2. We therefore conclude that the lubiprostone-induced channels are, indeed, ClC-2. Our results are interesting because, in unstimulated A6 cells, the frequency of observing ClC-2 channels was extremely low (no more than 1 or 2% of successful patches); however, after treatment with lubiprostone virtually every patch (>80%) had activity that appeared to be ClC-2. We verified that the activity was ClC-2 in the patches by examining the current-voltage relationship and voltage sensitivity.
Comparison of lubiprostone-activated channels in A6 cells with channels in other epithelia.
We observed that lubiprostone at high concentrations (more than 50 times higher than the concentration necessary to activate ClC-2) activated CFTR. As mentioned above, CFTR channels have been previously reported by us in A6 cells (71). The properties of the CFTR channels activated by lubiprostone are identical to those previously described by us as cAMP-activated channels although we have shown that lubiprostone is not activating CFTR by increasing intracellular cAMP. They also have properties essentially identical to those described for single CFTR channels in many other epithelial preparations (for reviews, see Refs. 36, 52, 61). In T84 cells, another cell line that expresses both CFTR and ClC-2, lubiprostone did not activate CFTR. We do not fully understand why CFTR should be activated in A6 cells but not in T84 or in HEK cells transfected with CFTR and ClC-2. One possibility is that T84 cells (and the transfected HEK cells), unlike A6 cells, are considered exclusively Cl− secretory cells, whereas A6 cells are a model for more generalized salt transport and salt homeostasis. Therefore, A6 cells require the ability to activate both anion (and cation) secretion and reabsorption. Their more extensive signaling pathways may lead to activation of channels that could promote secretion or reabsorption. Alternatively, we have previously shown that prostaglandins can activate CFTR in A6 cells (68). Lubiprostone is structurally similar to prostaglandin, but does not interact with mammalian prostaglandin receptors; however, prostaglandins are metabolized to prostones. If part of the stimulation of CFTR by PGE2 (11) is not due to the prostaglandins themselves, but rather to their metabolic products, then the activation of CFTR in A6 cells might not be surprising.
Blocking ClC-2 channels.
It is unfortunate that there are no effective small-molecule inhibitors of ClC-2 or CFTR. Glibenclamide has been used as a selective blocker of CFTR to the point of some investigators using the drug as diagnostic for the contribution of CFTR anion flux. However, on the basis of our observation, glibenclamide is almost as good at blocking ClC-2 as CFTR. Therefore, substantial care must be exercised in the use glibenclamide, diphenylcarboxylic acid, or other small anionic blockers to determine the contributions of CFTR and ClC-2 to the anion flux in epithelial tissues (83). Cd2+ has been used as a blocker of ClC-2, but Cd2+ can hardly be considered selective since it blocks many transporters owing to its strong interaction with cysteine residues. In fact, when applied selectively to the isolated apical membrane (inside the patch pipette), Cd2+ does not alter the properties of ClC-2 or the frequency with which the channel is observed. Thus any modifiable cysteines that alter ClC-2 activity are not directly accessible from the apical surface of the channel. More promising inhibitors are recently described small peptide toxins derived from scorpion venom that appear to be substantially more selective blockers of ClC-2 (44, 112, 122).
Implications for epithelial fluid balance.
A major question concerns the physiological role of the two channels we have identified in A6 cells and their role in normal epithelial function. A second question involves the functional implications of lubiprostone activation of the channels. The role of CFTR in epithelial transport has been discussed at great length in the context of cystic fibrosis. The general consensus is that CFTR promotes Cl− secretion (30, 64, 102, 108), but several investigators who have carefully examined the conditions present in normal epithelial tissues have suggested that CFTR could just as easily promote Cl− reabsorption (19). In our experiments, inward flux of Cl− (outward current) is larger than outward flux, and the potential at which current reverses from inward to outward is very near the membrane potential of the cell, suggesting that CFTR could easily switch from secreting to reabsorbing with very minor changes in apical membrane potential or intracellular or extracellular Cl− concentration. When CFTR is in a reabsorptive mode it will reabsorb more Cl− for a given driving force than when it is secreting.
The properties of ClC-2 are more complex. As a strongly rectifying channel, ClC-2 has a much greater capacity to secrete Cl− than reabsorb Cl−. Thus, even if the apical membrane potential were more positive than the reversal potential, there would be only limited Cl− entry. Moreover, ClC-2 channels seem even more specialized to promote Cl− (and salt and water) secretion. Not only does the single-channel conductance rectify to reduce Cl− reabsorption, but the open probability of the channel decreases with depolarization to further reduce the amount of Cl− that is reabsorbed at depolarizing potentials and promote the amount that is secreted. It is interesting to note that it is ClC-2 that is the predominant Cl− channel in the fetal lung when it is most avidly secreting salt and water. The specific properties of ClC-2 that strongly promote secretion explain why lubiprostone has been associated with increased Cl− secretion and concomitant salt and water secretion (15, 31) into the gut and its approved use as an agent for treating chronic idiopathic constipation (1, 117).
Conclusions.
These studies show that lubiprostone is a potent activator of ClC-2 Cl− channels in A6 cells, but, to a much lesser extent, it also activated CFTR. Therefore lubiprostone activation of Cl− transport in the human intestine (15) likely occurs through activation of the ClC-2 Cl− channel. These properties suggest that lubiprostone is an excellent candidate for the clinical treatment of gastrointestinal syndromes involving reduced water content of the intestinal contents. These studies also suggest that, beyond pharmacological activation, ClC-2 Cl− channels may play a physiological role in Cl− transport in the intestine and other epithelial tissues. Further studies are needed to define the specific mechanism by which lubiprostone activates ClC-2 and CFTR Cl− channels.
GRANTS
This work was partially supported by P30 DK-064399, P01 DK-061521, and R37 DK-37963 to D. C. Eaton. Functional studies were partially supported by a grant from Sucampo Pharmaceuticals, and Takeda Pharmaceutical Company, Ltd.
DISCLOSURES
D. C. Eaton serves as a consultant to and R. Ueno is an employee and shareholder of Sucampo Pharmaceuticals, Inc.
Acknowledgments
We thank B. J. Duke for assistance with tissue culture and Semra Ramosevac for the Western blot in Figure 3. We also thank J. Cuppoletti and D. Malinowska for the kind gift of HEK293 cells stably transfected with human ClC-2.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Anonymous. Lubiprostone: RU 0211, SPI 0211. Drugs R D 6: 245–248, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Arreola J, Park K, Melvin JE, Begenisich T. Three distinct chloride channels control anion movements in rat parotid acinar cells. J Physiol 490: 351–362, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bali M, Lipecka J, Edelman A, Fritsch J. Regulation of ClC-2 chloride channels in T84 cells by TGF-alpha. Am J Physiol Cell Physiol 280: C1588–C1598, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Bear CE, Li C, Galley K, Wang Y, Garami E, Ramjeesingh M. Coupling of ATP hydrolysis with channel gating by purified, reconstituted CFTR. J Bioenerg Biomembr 29: 465–473, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Becchetti A, Kemendy AE, Stockand JD, Sariban-Sohraby S, Eaton DC. Methylation increases the open probability of the epithelial sodium channel in A6 epithelia. J Biol Chem 275: 16550–16559, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Becchetti A, Malik B, Yue G, Duchatelle P, Al Khalili O, Kleyman TR, Eaton DC. Phosphatase inhibitors increase the open probability of ENaC in A6 cells. Am J Physiol Renal Physiol 283: F1030–F1045, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Begenisich T, Cahalan MD. Sodium channel permeation in squid axons. II: non-independence and current-voltage relations. J Physiol 307: 243–257, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 291: F714–F721, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Blaisdell CJ, Edmonds RD, Wang XT, Guggino S, Zeitlin PL. pH-regulated chloride secretion in fetal lung epithelia. Am J Physiol Lung Cell Mol Physiol 278: L1248–L1255, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Blaisdell CJ, Morales MM, Andrade AC, Bamford P, Wasicko M, Welling P. Inhibition of ClC–2 chloride channel expression interrupts expansion of fetal lung cysts. Am J Physiol Lung Cell Mol Physiol 286: L420–L426, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Boie Y, Rushmore TH, Darmon-Goodwin A, Grygorczyk R, Slipetz DM, Metters KM, Abramovitz M. Cloning and expression of a cDNA for the human prostanoid IP receptor. J Biol Chem 269: 12173–12178, 1994. [PubMed] [Google Scholar]
- 12.Boucher RC New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23: 146–158, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Britton FC, Wang GL, Huang ZM, Ye L, Horowitz B, Hume JR, Duan D. Functional characterization of novel alternatively spliced ClC-2 chloride channel variants in the heart. J Biol Chem 280: 25871–25880, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Cahalan MD, Begenisich T. Sodium channel permeation in squid axons. I: reversal potential experiments. J Physiol 307: 217–242, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camilleri M, Bharucha AE, Ueno R, Burton D, Thomforde GM, Baxter K, McKinzie S, Zinsmeister AR. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol 290: G942–G947, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Carson MR, Travis SM, Welsh MJ. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J Biol Chem 270: 1711–1717, 1995. [DOI] [PubMed] [Google Scholar]
- 17.Catalan M, Cornejo I, Figueroa CD, Niemeyer MI, Sepulveda FV, Cid LP. ClC-2 in guinea pig colon: mRNA, immunolabeling, and functional evidence for surface epithelium localization. Am J Physiol Gastrointest Liver Physiol 283: G1004–G1013, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Catalan M, Niemeyer MI, Cid LP, Sepulveda FV. Basolateral ClC-2 chloride channels in surface colon epithelium: regulation by a direct effect of intracellular chloride. Gastroenterology 126: 1104–1114, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Chalfant ML, Coupaye-Gerard B, Kleyman TR. Distinct regulation of Na+ reabsorption and Cl− secretion by arginine vasopressin in the amphibian cell line A6. Am J Physiol Cell Physiol 264: C1480–C1488, 1993. [DOI] [PubMed] [Google Scholar]
- 20.Champigny G, Imler JL, Puchelle E, Dalemans W, Gribkoff V, Hinnrasky J, Dott K, Barbry P, Pavirani A, Lazdunski M. A change in gating mode leading to increased intrinsic Cl− channel activity compensates for defective processing in a cystic fibrosis mutant corresponding to a mild form of the disease. EMBO J 14: 2417–2423, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen XJ, Eaton DC, Jain L. β-Adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol 282: L609–L620, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287: L366–L373, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Chu S, Blaisdell CJ, Bamford P, Ferro TJ. Interferon-gamma regulates ClC-2 chloride channel in lung epithelial cells. Biochem Biophys Res Commun 324: 31–39, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Chu S, Blaisdell CJ, Liu MZ, Zeitlin PL. Perinatal regulation of the ClC-2 chloride channel in lung is mediated by Sp1 and Sp3. Am J Physiol Lung Cell Mol Physiol 276: L614–L624, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Chu S, Murray CB, Liu MM, Zeitlin PL. A short CIC-2 mRNA transcript is produced by exon skipping. Nucleic Acids Res 24: 3453–3457, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chu S, Zeitlin PL. Alternative mRNA splice variants of the rat ClC-2 chloride channel gene are expressed in lung: genomic sequence and organization of ClC-2. Nucleic Acids Res 25: 4153–4159, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cid LP, Montrose-Rafizadeh C, Smith DI, Guggino WB, Cutting GR. Cloning of a putative human voltage-gated chloride channel (CIC-2) cDNA widely expressed in human tissues. Hum Mol Genet 4: 407–413, 1995. [DOI] [PubMed] [Google Scholar]
- 28.Cid LP, Niemeyer MI, Ramirez A, Sepulveda FV. Splice variants of a ClC-2 chloride channel with differing functional characteristics. Am J Physiol Cell Physiol 279: C1198–C1210, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Comes N, Abad E, Morales M, Borras T, Gual A, Gasull X. Identification and functional characterization of ClC-2 chloride channels in trabecular meshwork cells. Exp Eye Res 83: 877–889, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol 10: 38–47, 1994. [DOI] [PubMed] [Google Scholar]
- 31.Cuppoletti J, Malinowska DH, Tewari KP, Li QJ, Sherry AM, Patchen ML, Ueno R. SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol Cell Physiol 287: C1173–C1183, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Cuppoletti J, Tewari KP, Sherry AM, Ferrante CJ, Malinowska DH. Sites of protein kinase A activation of the human ClC-2 Cl(-) channel. J Biol Chem 279: 21849–21856, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Cuppoletti J, Tewari KP, Sherry AM, Kupert EY, Malinowska DH. ClC2 Cl− channels in human lung epithelia: activation by arachidonic acid, amidation, and acid-activated omeprazole. Am J Physiol Cell Physiol 281: C46–C54, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Davies N, Akhtar S, Turner HC, Candia OA, To CH, Guggenheim JA. Chloride channel gene expression in the rabbit cornea. Mol Vis 10: 1028–1037, 2004. [PubMed] [Google Scholar]
- 35.De Santiago JA, Nehrke K, Arreola J. Quantitative analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J Gen Physiol 126: 591–603, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Devidas S, Guggino WB. The cystic fibrosis transmembrane conductance regulator and ATP. Curr Opin Cell Biol 9: 547–552, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Ecelbarger CA, Tiwari S. Sodium transporters in the distal nephron and disease implications. Curr Hypertens Rep 8: 158–165, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Enz R, Ross BJ, Cutting GR. Expression of the voltage-gated chloride channel ClC-2 in rod bipolar cells of the rat retina. J Neurosci 19: 9841–9847, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fahlke C Ion permeation and selectivity in ClC-type chloride channels. Am J Physiol Renal Physiol 280: F748–F757, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Fava M, Ferroni S, Nobile M. Osmosensitivity of an inwardly rectifying chloride current revealed by whole-cell and perforated-patch recordings in cultured rat cortical astrocytes. FEBS Lett 492: 78–83, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Flach CF, Lange S, Jennische E, Lonnroth I. Cholera toxin induces expression of ion channels and carriers in rat small intestinal mucosa. FEBS Lett 561: 122–126, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Fukushima Y Blocking kinetics of the anomalous potassium rectifier of tunicate egg studied by single channel recording. J Physiol 331: 311–331, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuller CM, Benos DJ. CFTR! Am J Physiol Cell Physiol 263: C267–C286, 1992. [DOI] [PubMed] [Google Scholar]
- 44.Fuller MD, Zhang ZR, Cui G, Kubanek J, McCarty NA. Inhibition of CFTR channels by a peptide toxin of scorpion venom. Am J Physiol Cell Physiol 287: C1328–C1341, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Furukawa T, Ogura T, Katayama Y, Hiraoka M. Characteristics of rabbit ClC-2 current expressed in Xenopus oocytes and its contribution to volume regulation. Am J Physiol Cell Physiol 274: C500–C512, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Furukawa T, Ogura T, Zheng YJ, Tsuchiya H, Nakaya H, Katayama Y, Inagaki N. Phosphorylation and functional regulation of ClC-2 chloride channels expressed in Xenopus oocytes by M cyclin-dependent protein kinase. J Physiol 540: 883–893, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gadsby DC, Hwang TC, Baukrowitz T, Nagel G, Horie M, Nairn AC. Regulation of CFTR channel gating. Jpn J Physiol 44, Suppl 2: S183–S192, 1994. [PubMed] [Google Scholar]
- 48.Goldman DE Potential, impedance, and rectification in membranes. J Gen Physiol 27: 37–60, 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greger R Role of CFTR in the colon. Annu Rev Physiol 62: 467–491, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Gunderson KL, Kopito RR. Conformational states of CFTR associated with channel gating: the role ATP binding and hydrolysis. Cell 82: 231–239, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Gyomorey K, Yeger H, Ackerley C, Garami E, Bear CE. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am J Physiol Cell Physiol 279: C1787–C1794, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Hanrahan JW, Tabcharani JA, Becq F, Mathews CJ, Augustinas O, Jensen TJ, Chang XB, Riordan JR. Function and dysfunction of the CFTR chloride channel. Soc Gen Physiol Ser 50: 125–137, 1995. [PubMed] [Google Scholar]
- 53.Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol 290: L710–L722, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Hille B, Schwarze W. Potassium channels as multi-ion single-file pores. J Gen Physiol 72: 409–442, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinzpeter A, Lipecka J, Brouillard F, Baudoin-Legros M, Dadlez M, Edelman A, Fritsch J. Association between Hsp90 and the ClC-2 chloride channel upregulates channel function. Am J Physiol Cell Physiol 290: C45–C56, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Hodgkin AL, Katz B. The effect of temperature on the electrical activity of the giant axon of the squid. J Physiol 109: 240–249, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hummler E Epithelial sodium channel, salt intake, and hypertension. Curr Hypertens Rep 5: 11–18, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Hwang TC, Nagel G, Nairn AC, Gadsby DC. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc Natl Acad Sci USA 91: 4698–4702, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain L, Chen XJ, Brown LA, Eaton DC. Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am J Physiol Lung Cell Mol Physiol 274: L475–L484, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Jain L, Chen XJ, Malik B, Al-Khalili OK, Eaton DC. Antisense oligonucleotides against the α-subunit of ENaC decrease lung epithelial cation-channel activity. Am J Physiol Lung Cell Mol Physiol 276: L1046–L1051, 1999. [DOI] [PubMed] [Google Scholar]
- 61.Jentsch TJ, Gunther W. Chloride channels: an emerging molecular picture. Bioessays 19: 117–126, 1997. [DOI] [PubMed] [Google Scholar]
- 62.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev 82: 503–568, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Jiang B, Hattori N, Liu B, Kitagawa K, Inagaki C. Expression of swelling- and/or pH-regulated chloride channels (ClC-2, 3, 4 and 5) in human leukemic and normal immune cells. Life Sci 70: 1383–1394, 2002. [DOI] [PubMed] [Google Scholar]
- 64.Jiang C, Finkbeiner WE, Widdicombe JH, McCray PB Jr, Miller SS. Altered fluid transport across airway epithelium in cystic fibrosis. Science 262: 424–427, 1993. [DOI] [PubMed] [Google Scholar]
- 65.Joo NS, Clarke LL, Han BH, Forte LR, Kim HD. Cloning of ClC-2 chloride channel from murine duodenum and its presence in CFTR knockout mice. Biochim Biophys Acta 1446: 431–437, 1999. [DOI] [PubMed] [Google Scholar]
- 66.Kajita H, Omori K, Matsuda H. The chloride channel ClC-2 contributes to the inwardly rectifying Cl− conductance in cultured porcine choroid plexus epithelial cells. J Physiol 523: 313–324, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kajita H, Whitwell C, Brown PD. Properties of the inward-rectifying Cl− channel in rat choroid plexus: regulation by intracellular messengers and inhibition by divalent cations. Pflügers Arch 440: 933–940, 2000. [DOI] [PubMed] [Google Scholar]
- 68.Kokko KE, Matsumoto PS, Zhang ZR, Ling BN, Eaton DC. Prostaglandin E2 increases 7-pS Cl− channel density in the apical membrane of A6 distal nephron cells. Am J Physiol Cell Physiol 273: C548–C557, 1997. [DOI] [PubMed] [Google Scholar]
- 69.Kunzelmann K The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Rev Physiol Biochem Pharmacol 137: 1–70, 1999. [DOI] [PubMed] [Google Scholar]
- 70.Larsen EH, Price EM, Gabriel SE, Stutts MJ, Boucher RC. Clusters of Cl− channels in CFTR-expressing Sf9 cells switch spontaneously between slow and fast gating modes. Pflügers Arch 432: 528–537, 1996. [DOI] [PubMed] [Google Scholar]
- 71.Ling BN, Zuckerman JB, Lin C, Harte BJ, McNulty KA, Smith PR, Gomez LM, Worrell RT, Eaton DC, Kleyman TR. Expression of the cystic fibrosis phenotype in a renal amphibian epithelial cell line. J Biol Chem 272: 594–600, 1997. [DOI] [PubMed] [Google Scholar]
- 72.Lipecka J, Bali M, Thomas A, Fanen P, Edelman A, Fritsch J. Distribution of ClC-2 chloride channel in rat and human epithelial tissues. Am J Physiol Cell Physiol 282: C805–C816, 2002. [DOI] [PubMed] [Google Scholar]
- 73.Liu X, Jiang Q, Mansfield SG, Puttaraju M, Zhang Y, Zhou W, Cohn JA, Garcia-Blanco MA, Mitchell LG, Engelhardt JF. Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat Biotechnol 20: 47–52, 2002. [DOI] [PubMed] [Google Scholar]
- 74.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol 16: 3182–3187, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Maduke M, Miller C, Mindell JA. A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu Rev Biophys Biomol Struct 29: 411–438, 2000. [DOI] [PubMed] [Google Scholar]
- 76.Makara JK, Rappert A, Matthias K, Steinhauser C, Spat A, Kettenmann H. Astrocytes from mouse brain slices express ClC-2-mediated Cl− currents regulated during development and after injury. Mol Cell Neurosci 23: 521–530, 2003. [DOI] [PubMed] [Google Scholar]
- 77.Malinowska DH, Kupert EY, Bahinski A, Sherry AM, Cuppoletti J. Cloning, functional expression, and characterization of a PKA-activated gastric Cl− channel. Am J Physiol Cell Physiol 268: C191–C200, 1995. [DOI] [PubMed] [Google Scholar]
- 78.Malinowska DH, Sherry AM, Tewari KP, Cuppoletti J. Gastric parietal cell secretory membrane contains PKA- and acid-activated Kir2.1 K+ channels. Am J Physiol Cell Physiol 286: C495–C506, 2004. [DOI] [PubMed] [Google Scholar]
- 79.Mansoura MK, Dawson DC. 2 Barrier-1 site pore: an interactive spreadsheet model for two permeating ions. Comput Biol Med 28: 255–273, 1998. [DOI] [PubMed] [Google Scholar]
- 80.Marunaka Y, Eaton DC. Different types of chloride channels in a distal nephron cell line. Biophys J 55: 160a, 1989. [Google Scholar]
- 81.Marunaka Y, Eaton DC. Chloride channels in the apical membrane of a distal nephron A6 cell line. Am J Physiol Cell Physiol 258: C352–C368, 1990. [DOI] [PubMed] [Google Scholar]
- 82.Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc 2: 206–213, 2005. [DOI] [PubMed] [Google Scholar]
- 83.McCarty NA Permeation through the CFTR chloride channel. J Exp Biol 203: 1947–1962, 2000. [DOI] [PubMed] [Google Scholar]
- 84.Mindell JA, Maduke M. ClC chloride channels. Genome Biol 2: REVIEWS3003, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moeser AJ, Haskell MM, Shifflett DE, Little D, Schultz BD, Blikslager AT. ClC-2 chloride secretion mediates prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Gastroenterology 127: 802–815, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Mohammad-Panah R, Gyomorey K, Rommens J, Choudhury M, Li C, Wang Y, Bear CE. ClC-2 contributes to native chloride secretion by a human intestinal cell line, Caco-2. J Biol Chem 276: 8306–8313, 2001. [DOI] [PubMed] [Google Scholar]
- 87.Mummery JL, Killey J, Linsdell P. Expression of the chloride channel ClC–K in human airway epithelial cells. Can J Physiol Pharmacol 83: 1123–1128, 2005. [DOI] [PubMed] [Google Scholar]
- 88.Murray CB, Chu S, Zeitlin PL. Gestational and tissue-specific regulation of C1C-2 chloride channel expression. Am J Physiol Lung Cell Mol Physiol 271: L829–L837, 1996. [DOI] [PubMed] [Google Scholar]
- 89.Murray CB, Morales MM, Flotte TR, Grath-Morrow SA, Guggino WB, Zeitlin PL. CIC-2: a developmentally dependent chloride channel expressed in the fetal lung and downregulated after birth. Am J Respir Cell Mol Biol 12: 597–604, 1995. [DOI] [PubMed] [Google Scholar]
- 90.Nascimento DS, Reis CU, Goldenberg RC, Ortiga-Carvalho TM, Pazos-Moura CC, Guggino SE, Guggino WB, Morales MM. Estrogen modulates ClC-2 chloride channel gene expression in rat kidney. Pflügers Arch 446: 593–599, 2003. [DOI] [PubMed] [Google Scholar]
- 91.Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE, Melvin JE. Loss of hyperpolarization-activated Cl(-) current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem 277: 23604–23611, 2002. [DOI] [PubMed] [Google Scholar]
- 92.Nobile M, Pusch M, Rapisarda C, Ferroni S. Single-channel analysis of a ClC-2-like chloride conductance in cultured rat cortical astrocytes. FEBS Lett 479: 10–14, 2000. [DOI] [PubMed] [Google Scholar]
- 93.O'Donnell EK, Sedlacek RL, Singh AK, Schultz BD. Inhibition of enterotoxin-induced porcine colonic secretion by diarylsulfonylureas in vitro. Am J Physiol Gastrointest Liver Physiol 279: G1104–G1112, 2000. [DOI] [PubMed] [Google Scholar]
- 94.Palmada M, Dieter M, Boehmer C, Waldegger S, Lang F. Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem Biophys Res Commun 321: 1001–1006, 2004. [DOI] [PubMed] [Google Scholar]
- 95.Park K, Arreola J, Begenisich T, Melvin JE. Comparison of voltage-activated Cl− channels in rat parotid acinar cells with ClC-2 in a mammalian expression system. J Membr Biol 163: 87–95, 1998. [DOI] [PubMed] [Google Scholar]
- 96.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev 79: S215–S255, 1999. [DOI] [PubMed] [Google Scholar]
- 97.Press W, Teukolsky S, Vetterling V, Flannery B. Modeling of data In: Numerical Recipes: The Art of Scientific Computing (3rd ed.). West Nyack, NY: Cambridge University Press, 2008, chapt. 15, p. 773–836.
- 98.Roman RM, Smith RL, Feranchak AP, Clayton GH, Doctor RB, Fitz JG. ClC-2 chloride channels contribute to HTC cell volume homeostasis. Am J Physiol Gastrointest Liver Physiol 280: G344–G353, 2001. [DOI] [PubMed] [Google Scholar]
- 99.Rossier BC Hormonal regulation of the epithelial sodium channel ENaC: N or P(o)? J Gen Physiol 120: 67–70, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schultz BD, Bridges RJ, Frizzell RA. Lack of conventional ATPase properties in CFTR chloride channel gating. J Membr Biol 151: 63–75, 1996. [DOI] [PubMed] [Google Scholar]
- 101.Schwiebert EM, Cid-Soto LP, Stafford D, Carter M, Blaisdell CJ, Zeitlin PL, Guggino WB, Cutting GR. Analysis of ClC-2 channels as an alternative pathway for chloride conduction in cystic fibrosis airway cells. Proc Natl Acad Sci USA 95: 3879–3884, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shen BQ, Mrsny RJ, Finkbeiner WE, Widdicombe JH. Role of CFTR in chloride secretion across human tracheal epithelium. Am J Physiol Lung Cell Mol Physiol 269: L561–L566, 1995. [DOI] [PubMed] [Google Scholar]
- 103.Sherry AM, Malinowska DH, Morris RE, Ciraolo GM, Cuppoletti J. Localization of ClC-2 Cl− channels in rabbit gastric mucosa. Am J Physiol Cell Physiol 280: C1599–C1606, 2001. [DOI] [PubMed] [Google Scholar]
- 104.Sherry AM, Stroffekova K, Knapp LM, Kupert EY, Cuppoletti J, Malinowska DH. Characterization of the human pH- and PKA-activated ClC-2G2α Cl− channel. Am J Physiol Cell Physiol 273: C384–C393, 1997. [DOI] [PubMed] [Google Scholar]
- 105.Stockand JD, Al-Baldawi NF, Al Khalili OK, Worrell RT, Eaton DC. S-adenosyl-l-homocysteine hydrolase regulates aldosterone-induced Na+ transport. J Biol Chem 274: 3842–3850, 1999. [DOI] [PubMed] [Google Scholar]
- 106.Stockand JD, Bao HF, Schenck J, Malik B, Middleton P, Schlanger LE, Eaton DC. Differential effects of protein kinase C on the levels of epithelial Na+ channel subunit proteins. J Biol Chem 275: 25760–25765, 2000. [DOI] [PubMed] [Google Scholar]
- 107.Stockand JD, Spier B, Worrell RT, Yue G, Al-Baldawi NF, Eaton DC. Regulation of Na+ reabsorption by the aldosterone-induced small G protein K-Ras2A. J Biol Chem 274: 35449–35455, 1999. [DOI] [PubMed] [Google Scholar]
- 108.Stutts MJ, Fitz JG, Paradiso AM, Boucher RC. Multiple modes of regulation of airway epithelial chloride secretion by extracellular ATP. Am J Physiol Cell Physiol 267: C1442–C1451, 1994. [DOI] [PubMed] [Google Scholar]
- 109.Tanaka A, Tokimasa T. Theoretical background for inward rectification. Tokai J Exp Clin Med 24: 147–153, 1999. [PubMed] [Google Scholar]
- 110.Tewari KP, Malinowska DH, Sherry AM, Cuppoletti J. PKA and arachidonic acid activation of human recombinant ClC-2 chloride channels. Am J Physiol Cell Physiol 279: C40–C50, 2000. [DOI] [PubMed] [Google Scholar]
- 111.Thiemann A, Gründer S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 356: 57–60, 1992. [DOI] [PubMed] [Google Scholar]
- 112.Thompson CH, Fields DM, Olivetti PR, Fuller MD, Zhang ZR, Kubanek J, McCarty NA. Inhibition of ClC-2 chloride channels by a peptide component or components of scorpion venom. J Membr Biol 208: 65–76, 2005. [DOI] [PubMed] [Google Scholar]
- 113.Tousson A, Fuller CM, Benos DJ. Apical recruitment of CFTR in T-84 cells is dependent on cAMP and microtubules but not Ca2+ or microfilaments. J Cell Sci 109: 1325–1334, 1996. [DOI] [PubMed] [Google Scholar]
- 114.Vij N, Zeitlin PL. Regulation of the ClC-2 lung epithelial chloride channel by glycosylation of SP1. Am J Respir Cell Mol Biol 34: 754–759, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Waldegger S, Jentsch TJ. From tonus to tonicity: physiology of CLC chloride channels. J Am Soc Nephrol 11: 1331–1339, 2000. [DOI] [PubMed] [Google Scholar]
- 116.Weinreich F, Jentsch TJ. Pores formed by single subunits in mixed dimers of different CLC chloride channels. J Biol Chem 276: 2347–2353, 2001. [DOI] [PubMed] [Google Scholar]
- 117.Winpenny JP Lubiprostone. IDrugs 8: 416–422, 2005. [PubMed] [Google Scholar]
- 118.Woodhull AM Ionic blockage of sodium channels in nerve. J Gen Physiol 61: 687–708, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yu L, Eaton DC, Helms MN. Effect of divalent heavy metals on epithelial na channels in A6 cells. Am J Physiol Renal Physiol 293: F236–F244, 2007. [DOI] [PubMed] [Google Scholar]
- 120.Yue G, Malik B, Yue G, Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem 277: 11965–11969, 2002. [DOI] [PubMed] [Google Scholar]
- 121.Zhang ZR, McDonough SI, McCarty NA. Interaction between permeation and gating in a putative pore domain mutant in the cystic fibrosis transmembrane conductance regulator. Biophys J 79: 298–313, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang ZR, Song B, McCarty NA. State-dependent chemical reactivity of an engineered cysteine reveals conformational changes in the outer vestibule of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 280: 41997–42003, 2005. [DOI] [PubMed] [Google Scholar]
- 123.Zuniga L, Niemeyer MI, Varela D, Catalan M, Cid LP, Sepulveda FV. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J Physiol 555: 671–682, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]