

Abstract
Replication of poliovirus RNA takes place on the cytoplasmic surface of membranous vesicles that form after infection of the host cell. It is generally accepted that RNA polymerase 3Dpol interacts with membranes in a complex with viral protein 3AB, which binds to membranes by means of a hydrophobic anchor sequence that is located near the C-terminus of the 3A domain. In this study, we used fluorescence and fluorescence quenching methods to define the topography of the anchor sequence in context of 3A and 3AB proteins inserted in model membranes. Mutants with a single tryptophan near the center of the anchor sequence but lacking Trp elsewhere in 3A/3AB were constructed which, after the emergence of suppressor mutations, replicated well in HeLa cells. When a peptide containing the mutant anchor sequence was incorporated in model membrane vesicles, measurements of Trp depth within the lipid bilayer indicated formation of a transmembrane topography. However, rather than the 22 residue length predicted from hydrophobicity considerations, the transmembrane segment had an effective length of 16 residues, such that Gln64 likely formed the N-terminal boundary. Analogous experiments using full length proteins bound to pre-formed model membrane vesicles showed that the anchor sequence formed a mixture of transmembrane and non-transmembrane topographies in the 3A protein but adopted only the non-transmembrane configuration in the context of 3AB protein. Studies of the function of 3A/3AB inserted into model membrane vesicles showed that membrane-bound 3AB is highly efficient in stimulating the activity of 3Dpol in vitro while membrane-bound 3A totally lacks this activity. Moreover, in vitro uridylylation reactions showed that membrane-bound 3AB is not a substrate for 3Dpol but free VPg released by cleavage of 3AB with proteinase 3CDpro could be uridylylated.
After poliovirus (PV) enters the host cell its plus strand RNA genome is translated into a polyprotein that contains one structural (P1) and two nonstructural domains (P2, P3; Fig. 1A). There is an efficient and regulated cascade of protein processing that produces cleavage products with function distinct from those of the precursor proteins (Fig. 1A). A variety of precursor and mature proteins are released from the PV polyprotein by cleavage with proteinases 2Apro, 3Cpro/3CDpro. The proteins of the P1 domain (VP1-VP4) assemble to form the capsid while those derived from P2 (2Apro, 2B, 2BC, 2CATPase) are primarily responsible for the biochemical and structural changes that occur in the infected cell. The proteins of the P3 domain are those most directly involved in RNA synthesis. These include two important and relatively stable precursors, proteinase 3CDpro and 3AB, which were shown to specifically interact with a cloverleaf-like structure at the 5′ end of the viral RNA in a reaction that required for RNA replication. The two precursors are slowly processed to the mature polypeptides, 3A, 3B, 3Cpro, and 3Dpol (1).
Figure 1.

Genomic organization of poliovirus, processing of the polyprotein and amino acid sequences of protein 3AB. (A) The single-stranded RNA genome is covalently linked to the viral-encoded protein VPg at the 5′ end of the non-translated region (5′NTR). The linkage of the 5′ terminal UMP to the hydroxyl group of tyrosine in VPg is shown on the left. The 5′NTR consists of two cis-acting domains. The cloverleaf is involved in genome replication, and the internal ribosomal entry site (IRES), controls translation of a 247 kDa polyprotein (open box). The polyprotein is processed by the virus encoded proteinases 2Apro and 3Cpro/3CDpro into structural proteins (P1) and nonstructural (P2 and P3), the latter specifying the proteins involved in replication. Triangles indicate cleavages by 3Cpro/3CDpro and circles cleavage by 2Apro. Filled symbols indicate fast cleavages and open symbols indicate slow cleavages. The open diamond marks the VP0 capsid maturation cleavage. The 3′NTR contains a structured heteropolymeric region and is polyadenylylated. The open arrow indicates one of two suppressor mutations resulting from the W42F/F69W mutations in 3AB (see Fig. 1B). (B). Presentation of 3AB and its cleavage product 3A. Shown is the amino acid sequence of 3AB with the Q/G cleavage site between 3A and 3B(VPg). The hydrophobic anchor domain of 3A, starting at amino acid 59, is indicated in a box, which is enlarged below. The shaded area represents the TM segment as defined in this report. Downward or upward arrows indicate two separate sets of mutations. Filled arrows depict mutations engineered into the protein, open arrows the suppressor mutations that rapidly emerged during the first cycle of virus replication. Anchor peptide-1 and anchor peptide-2 delineate the peptides used for membrane binding studies.
Shortly after infection of susceptible cells with PV, dramatic changes take place in the host cell. There is a striking rearrangement of the cellular membranes and vesicles accumulate in the cytoplasm (2–4). These vesicles are mostly derived from the endoplasmic reticulum, Golgi apparatus, and lysosomes of the host cell. Recent studies indicate that many of the vesicles are autophagosomes, which carry virus-specific proteins, for example 2BC and 3A (5). The viral nonstructurual proteins and their precursors, as well as some cellular proteins and the viral RNA assemble to form a replication complex on the cytoplasmic surface of the vesicles that appear as rosette-like structures (3, 6). Such structures are believed to provide a suitable environment for RNA replication by increasing the local concentrations of viral proteins or by providing a scaffold for the assembly of the members of the complex. Some of the viral nonstructural proteins (2B, 2CATPase and 3A) associate with the replication complex via their own membrane binding domains while others are recruited to the complex by protein/protein or protein/RNA interactions (1, 7). The PV RNA polymerase 3Dpol is an example of a soluble protein that interacts with another protein, 3AB, whose hydrophobic domain, located in 3A, anchors the complex to membranes (8, 9).
3AB is a small basic protein that contains a hydrophobic and membrane-binding region near the C-terminus of its 3A domain. It was shown to possess the biochemical characteristics of an integral membrane protein (10). Yeast two hybrid and biochemical analyses have indicated that the 3AB protein strongly interacts with 3Dpol and the sequences primarily responsible for this interaction are located in the 3B domain of the protein (9, 11). Four amino acids in a hydrophobic patch on the surface of 3Dpol were recently identified as binding partners of 3AB (8). Protein 3AB also has the propensity to dimerize and form higher oligomers in solution in the absence of detergent. Both the N-terminal region and the hydrophobic domain of 3A were found to be involved in these interactions (9, 12). In vitro studies have suggested that 3AB has multiple functions in RNA replication: (i) 3AB is a nonspecific RNA binding protein but in a complex with proteinase 3CDpro it specifically binds to both the 5′ cloverleaf and the 3′NTR of the PV genome (13–15) (ii) 3AB stimulates the elongation activity of 3Dpol on a poly(A) template with an oligonucleotide primer (13–19) (iii) 3AB stimulates the autoprocessing of 3CDpro (20) (iv) the membrane bound form of 3AB is required for processing by 3CDpro (16). In vitro the N-terminal domain of 3AB can be glycosylated but the biological significance of this observation is not known (21).
Proteolytic cleavage of 3AB by 3Cpro/3CDpro yields 3A and 3B (VPg). The 3A protein is 87 amino acids long (Fig. 1B), consisting of a soluble cytosolic N-terminus (58 residues) (22), which exists as a symmetric dimer (12), a 22-residue long hydrophobic domain (aa 59–81) followed by 7 additional residues at the C-terminus. Part of the hydrophobic domain has been subdivided into two domains (I: aa 64–72 and II: aa 73–80), the latter of which was shown to be the primary determinant of membrane binding in the context of 3AB (10). The first 15 amino acids of the hydrophobic domain (aa 59–73) were predicted to form an amphipathic helix (10). Amino acid changes in the hydrophobic domain of 3A were shown to result in impaired RNA replication and virus death (23). The site of action of enviroxime, an antiviral drug which blocks PV RNA replication, maps to the 3A sequences, supporting a critical role for 3A in the replication complex (24). The 3A protein itself possesses additional functions beside those that are in the context of 3AB. In vivo 3A inhibits ER-to Golgi membrane and secretory protein traffic and induces specific translocation of different members of the ARF family (ADP-ribosylation factor) to membranes (25–28). Both a mammalian and a yeast two-hybrid system showed that 3A multimerizes and interacts with 2B and 2CATPase (9, 29) (Yin, Paul and Wimmer, unpublished observations). Studies with an in vitro translation/replication system indicated that a mutation in 3A (M97A or M97T) affected only plus strand synthesis and not minus strand RNA synthesis (30).
The second product derived from 3AB is VPg, a small basic peptide consisting of 22 amino acids, which is covalently linked to the 5′ end of the genomic RNA (31). PV RNA synthesis is initiated by the covalent linkage of a UMP to the hydroxyl group of a tyrosine in VPg yielding VPgpU(pU), the primer for both minus and plus strand RNA synthesis (32). In vitro, both poly(A) and a small RNA hairpin cre(2C), located in the coding sequences of 2CATPase, function as templates for the uridylylation reaction (32, 33). It has been proposed that in vivo the VPgpU synthesized on the cre(2C) RNA is used for plus strand RNA synthesis while the poly(A) tail of the genomic RNA serves as template for VPgpU synthesis prior to minus strand synthesis (32, 34). This claim, however, has been recently challenged (35).
Studies of 3AB and 3A function in vitro have been hampered by their low solubility in aqueous solutions, and purified preparations of these proteins always contain detergent. However, it has been shown before that detergents are detrimental to PV RNA replication. The uridylylation of VPg is totally blocked when crude replication complexes, isolated from poliovirus-infected cells, or in vitro translation/RNA replication reactions, are treated with detergent (36, 37). In addition, 3AB can be processed by 3CDpro only when membrane-bound and not in the presence of detergents (16).
Therefore, studies of 3A and 3AB in model membrane vesicles might be of more relevance than studies in detergents. However, although the membrane association of 3A and 3AB has been known for years, the precise arrangement of these proteins in the hydrophobic environment of a lipid bilayer is obscure. In this study, we exploited the fluorescence properties of a Trp residue placed in the center of the hydrophobic anchor sequence to determine the topology of the anchor sequence when incorporated into model membrane vesicles, using both a peptide containing the anchor sequence and intact 3A and 3AB proteins. Our results indicate that the anchor sequence has the intrinsic ability to form a transmembrane (TM) topography, and adopts a mixture of a transmembrane and non-transmembrane topography in the context of the intact 3A protein. The hydrophobic anchor of intact polypeptide 3AB, however, assumes only a non-transmembrane configuration. We also tested some of the in vitro activities of 3AB bound to synthetic model membrane vesicles. The protein was highly active in stimulating the elongation activity of the RNA polymerase using a poly(A) template and an oligo dT15 primer. On the other hand, membrane bound 3AB did not serve as a substrate for uridylylation on a cre(2C) template. However, the VPg released from 3AB proteolytic cleavage with 3CDpro could be uridylylated. These results suggest that while 3AB itself may not be a substrate for uridylylation, it can serve as the precursor from which VPg is released and is subsequently uridylylated.
EXPERIMENTAL PROCEDURES
Plasmids
All PV cDNA sequences were derived from plasmid pT7PVM (38). The sequences listed for plasmids or oligonucleotides refer to the full length plus strand PV sequences.
pRibo(+) PVM: Plasmid pRibo(+) PVM was constructed by inserting a cis-acting hammerhead ribozyme at the 5′ end of the poliovirus genome to increase the efficiency of replication in vitro (39). To obtain the ribozyme sequence, the following primers were used: RIBO1:5′ GCCGGCGGCCGCCTCGGCCTAATACGACTCACTATAGGGTGTTTTAA-3′; RIBO 2: 5′ ATCAGTTAAAACACCCTATAGTGAGTCGTATTAGGCCGAGGCGGCCGCCG GC-3′; RIBO3:5′ CTGATGAGGCCGAAAGGCCGAAAACCCGGTATCCCGGGTTCTTAAAAC-3′, RIBO 4: 5′ GAGCTGTTTTAAGAACCCGGGATACCGGGTTTTCGGCCTTTCGGCCTC-3′, RIBO 5: 5′ AGCTCTGGGGTTGTACCCACCCCAGAGGCCCACGTG3′; RIBO6:5′ CACGTGGGCCTCTGGGGTGGGACAACCCCA-3′. These oligonucleotides were purified on a 10% polyacrylamide gel, and were phosphorylated by T4 polynucleotide kinase (Epicentre, Madison, WI). Then, oligo pairs 1 and 2, 3 and 4, and 5 and 6 were annealed, ligated and inserted into the SmaI site of the pUC19 vector. The resulting plasmid was digested by SfiI and PmlI, and the fragment was inserted between the same restriction sites of pT7PVM.
3A mutants: Plasmids harboring mutations within the 3A-coding sequence were constructed by site-directed mutagenesis using the overlapping PCR method. The oligonucleotides used are summarized in Table 1. Fragments containing the mutations were introduced into pRibo(+) PVM by exchanging the MluI-BglII fragment of the plasmid.
3A and 3AB expression plasmids: Coding sequences of 3A and 3AB were amplified using 5′-GGACCACTCCAGTATAAAGAC-3′ and 5′-CCGAGCTCTACTGGTGTCCAGCAAACAGTT-3′ (for 3A) or 5′-CCGAGCTCTATTGTACCTT-TGCTGTCCGAA-3′ (for 3AB) as primers. The PCR products were digested by SacI, treated with T4 polynucleotide kinase and inserted between the PshAI and SacI sites of pET42b(+) vector (Novagen, San Diego, CA).
Table 1.
List of Constructs
| No. | Position (Sense) | Sequence | Construct |
|---|---|---|---|
| 1 | 5213–5250 (+) | GATTACTGTGAGAAGAAGGGTTTCATAGTTAACATCAC | W42F |
| 2 | 5219–5254 (−) | GCTGGTGATGTTAACTATGAAACCCTTCTTCTCACA | W42F |
| 3 | 5297–5333 (+) | CTACAAGCGGTGACAACCTGGGCCGCAGTGGCTGGAG | F69W |
| 4 | 5292–5328 (−) | GCCACTGCGGCCCAGGTTGTCACCGCTTGTAGAATTG | F69W |
| 5 | 5309–5341 (+) | ACAACCTTCGCCGCATGGGCTGGAGTTGTCTAT | V72W |
| 6 | 5309–5341 (−) | ATAGACAACTCCAGCCCATGCGGCGAAGGTTGT | V72W |
| 7 | 5312–5344 (+) | ACCTTCGCCGCAGTGTGGGGAGTTGTCTATGTC | A73W |
| 8 | 5312–5344 (−) | GACATAGACAACTCCCCACACTGCGGCGAAGGT | A73W |
| 9 | 5315–5347 (+) | TTCGCCGCAGTGGCTTGGGTTGTCTATGTCATG | G74W |
| 10 | 5315–5347 (−) | CATGACATAGACAACCCAAGCCACTGCGGCGAA | G74W |
| 11 | 5318–5350 (+) | GCCGCAGTGGCTGGATGGGTCTATGTCATGTAT | V75W |
| 12 | 5318–5350 (−) | ATACATGACATAGACCCATCCAGCCACTGCGGC | V75W |
Note: Changed nucleotides are underlined
In vitro transcription and transfection of poliovirus RNA
All plasmids were linearized by EcoRI prior to the transcription reactions. Full-genomic poliovirus RNA was transcribed by T7 RNA polymerase (Epicentre) according to the manufacturer’s instructions. The transcripts were purified by phenol/chloroform extraction followed by ethanol precipitation. HeLa cells in 6-well plates were transfected with in vitro-transcribed poliovirus RNA using Transmessenger (Qiagen, Valencia, CA) as a transfection reagent, according to the manufacturer’s instruction.
One-step growth curves and plaque assays
HeLa cell monolayers in 35-mm culture dishes were washed with Dulbecco’s minimal essential medium (DMEM) and inoculated with each virus at a multiplicity of infection of 10. After adsorption for 30 min at room temperature, the cells were extensively washed to remove unbound virus, and then incubated at 37°C or 39°C. The dishes were harvested at 0, 2, 4, 6, 8, 12, and 24 h post-infection, and subjected to three cycles of freezing/thawing. The viral titers of the supernatants were determined by plaque assay, as described previously (40).
Purification of 3Dpol and 3CDpro
Purification of 3Dpol and 3CDpro (active-site mutant) were performed as described previously (41, 42). Recombinant 3CDpro, which harbors a cleavage-site mutation (Q183A) was generously provided by C. Cameron.
Purification of 3A and 3AB
E. coli BL21 strain harboring a 3A- or 3AB-expression plasmid was grown in M9 media at 37°C. When the optical density at 600 nm reached 0.8, 0.5 mM IPTG (isopropylthio-β-galactoside) was added to the culture and protein expression was induced at 37°C for 3.5 hr. The cell pellet was washed once with PBS, pH 7.2 (Gibco, Rockville, MD) and stored at −80°C. The pellet was suspended in PBS containing 5% glycerol (v/v), 1 μg/ml of leupeptin and 1 μg/ml of pepstatin A. Cells were lysed by passing through a French Pressure Cell four times and sonicated for 30s to reduce viscosity. After the addition of PMSF (phenylmethansulfonyl fluoride) to a final concentration of 1 mM, the lysate was centrifuged at 9,000 rpm in Ty50.2 rotor at 4°C for 20 min (Beckman L7-65 centrifuge). The supernatant was further centrifuged at 34,000 rpm for 1h in the same rotor. Then, the pellet was suspended in PBS containing 5% glycerol, 1 μg/ml of leupeptin, 1 μg/ml of pepstatin A, 1 mM PMSF, 1 M NaCl and 2% octylglucoside (w/v) and incubated at 4°C for 16 h to solubilize the membrane proteins. The lysate was centrifuged at 34,000 rpm in Ty50.2 rotor at 4°C for 1h. The supernatant was collected and applied to a Glutathione Sepharose column which was equilibrated with PBS containing 5% glycerol and 0.8% octylglucoside. Bound proteins were eluted in 50 mM Tris, 100 mM NaCl, 5 mM CaCl2, 0.8% octylglucoside, 10 mM reduced glutathione. Proteins were concentrated and the buffer exchanged to elution buffer without glutathione using a 10 kDa Amicon Ultra Filter (Millipore) Purified GST-3A or GST-3AB was digested by 0.01 U of Factor Xa (Novagen) per mg protein for 4 h at 4°C. After digestion, the buffer was exchanged to PBS containing 5% glycerol and 0.8% octylglucoside using a PD-10 desalting column (Amersham, Piscataway, NJ), and applied to a Glutathione Sepharose column (4 ml) connected with a Benzamidine Sepharose column (2 ml) (Amersham). The flow through was collected and concentrated using a 5 kD Amicon Ultra Filter (Millipore, Billerica, MA). Before freezing at −80°C, 1 mM DTT was added.
Peptides and Lipids
Synthetic PV VPg was a generous gift of the late J. H. van Boom. Acetyl-NRAMTILQAVTTWVAVAGVVYVMYK-NH2 (anchor peptide 1), and peptide Acetyl-NRAMTILQAVTTFAAWAGVVYVMYK-NH2 (anchor peptide 2), were purchased from Anaspec Inc. (San Jose, CA). Peptide purity was checked using MALDI-TOF (Proteomics Center, Stony Brook University) and found to be >85% pure in each case. The concentration of the peptides was calculated by absorbance spectroscopy using a Beckman DU-610 spectrophotometer, using ε for Trp of 5560 M−1cm−1 at 280 nm. Phosphatidylcholines (1, 2 diacyl-sn-glycero-3-phosphocholines): diC16:1Δ9cPC; diC18:1Δ9cPC (dioleoylphosphatidylcholine, DOPC); diC20:1Δ11cPC; diC22:1Δ13cPC, (dierucoylphosphatidylcholine, DEuPC); and diC24:1Δ15cPC, were purchased from Avanti Polar Lipids (Alabaster, AL). The lipids were stored in chloroform at −20°C. 10-doxylnonadecane (10-DN) was custom synthesized by Molecular Probes (Eugene, OR). It was stored as a 4.9 mM stock solution in ethanol at −20 °C.
Preparation of Model Membrane Vesicles Containing Protein
For experiments in which 3Am or 3ABm protein were reconstituted into model membrane vesicles to form proteoliposomes, lipid vesicles were prepared using the ethanol-dilution method (43, 44). Lipids dissolved in chloroform were dried under a stream of N2 gas, followed by drying under high vacuum for 1h. The dried lipid samples were dissolved in a minimum volume (10 μl) of ethanol, and then diluted to the final volume (typically 800 μl), using PBS, pH 7 (10 mM sodium phosphate and 150 mM NaCl, adjusted to pH 7.0) with constant vortexing. The samples were then allowed to equilibrate for 30–60 min at room temperature. Then aliquots of protein (20 μl from 0.5 mg/ml protein stocks) were added to the pre-formed vesicles, and incubated for an additional 1 h before fluorescence was measured. Samples contained final concentrations of 12.5 μg/ml protein and 500 μM lipid.
Since octylglucoside was rapidly diluted to far below its cmc in this protocol, it should not have affected vesicle integrity. Confirming this, a protocol in which the protein was diluted into buffer before addition of vesicles gave similar fluorescence and fluorescence quenching results to those with the protocol described above (data not shown).
Other experiments in which different amounts of preformed vesicles were titrated into 3Am or 3ABm protein that had been previously diluted with buffer to about a 2 μM concentration showed that binding to vesicles was maximal at 200 μM DOPC. This was determined both from the extent of Trp λmax shift upon lipid binding (from λmax in buffer of 335 nm to final values of 331 nm for 3ABm and 328–329nm for 3Am), and by the extent of quenching of Trp fluorescence by 10 mol% 10-DN which was incorporated into the vesicles (maximum degree of quenching 40–50% of initial fluorescence (i.e. in solution)).
Peptide Incorporation into Vesicles
In previous studies hydrophobic polypeptides were incorporated into ethanol dilution vesicles by mixing lipids and peptides in ethanol, and then forming vesicles as described above (43, 44). Due to the insolubility of the anchor peptides in ethanol, the procedure was modified to use dimethylsulfoxide (DMSO). In the modified procedure, peptide dissolved in DMSO was mixed with the lipids dissolved in chloroform and then dried under N2 gas and high vacuum. The dried lipid/peptide mixtures were dissolved in 10μl DMSO and then diluted with PBS, pH 7.0 as described above. Control experiments with previously studied hydrophobic peptides that form transmembrane α-helices (45, 46) showed that substitution of DMSO for ethanol did not alter transmembrane topography upon incorporation into lipid vesicles (data not shown). Samples contained final concentrations of 2 μM peptide and 500 μM lipid.
Fluorescence Measurements
Fluorescence data was obtained on SPEX τ2 Fluorolog spectrofluorometer operating in steady-state condition at room temperature. Excitation slits of 2.5 mm and emission slits of 5 mm was used for all measurements (band pass of 4.5 and 9.0 nm, respectively). Fluorescence emission spectra were measured in the range 300–375 nm with an excitation wavelength of 280 nm, unless otherwise noted. Fluorescence from background samples containing lipid with no peptide or protein was subtracted from the reported values.
Acrylamide-Quenching Measurements
To quantify acrylamide quenching, fluorescence intensity and emission spectra were first measured in samples containing model membrane incorporated peptide/protein or background samples, prepared as described above. Then a 50 μl aliquot of acrylamide from a 4 M stock solution dissolved in water was added. After a brief incubation (5 min), the fluorescence was remeasured. Fluorescence intensity before and after acrylamide addition was measured using an excitation wavelength of 295 nm and emission wavelength of 340 nm. This excitation wavelength was chosen to reduce acrylamide absorbance (and the resulting inner-filter effect). Fluorescence intensity was corrected both for the dilution from addition of acrylamide and for inner-filter effect (47). Emission spectra in the presence of acrylamide were measured with excitation wavelength of 280 nm because samples had stronger fluorescence intensity at that excitation wavelength, despite the inner filter effect. Controls showed that the emission spectra have similar wavelength maximum for either of the excitation wavelength (47).
10-Doxylnonadecane Quenching Measurements
To measure the efficiency of 10-doxylnonadecane (10-DN) quenching, the fluorescence of samples containing model membrane incorporated peptide/protein or background samples prepared in the absence of 10-DN was compared that in samples containing 10-DN. To prepare the later, samples were prepared as noted above, except that either 10% mol (for DOPC) and 12% mol (for DEuPC) of the lipid was replaced with equivalent mol % of 10-DN. Fluorescence intensity was measured using an excitation wavelength of 280 nm and emission wavelength of 330 nm. Emission spectra were measured with excitation wavelength of 280 nm.
Calculation of Acrylamide/10-DN Quenching Ratio
The acrylamide/10-DN quenching ratio (Q-ratio) was used to calculate the Trp depth in the bilayer (47). The ratio was calculated using the formula Q-ratio = [(Fo/Facrylamide)−1]/[(Fo/F10-DN)−1], where Fo is the fluorescence of samples with no quencher and Facrylamide and F10-DN are the fluorescence intensities in presence of acrylamide or 10-DN, respectively. Q-ratio varies inversely with the depth of a Trp in the membrane (47).
Stimulation of 3Dpol elongation by 3A(B)
3AB-proteoliposomes were prepared by mixing 10 μg 3AB with 1 ml of 500 μM DOPC liposomes prepared as described above followed by incubation for 1 hr at room temperature. The mixture was centrifuged for 1 hr at 60,000 RPM in a TLA-100 rotor and the pellet was resuspendend in 10 μl of PBS pH 7.2. Primer-dependent elongation reactions were carried out at 30°C for 1 h in 25 μl of reaction mixture containing 50 mM Hepes-KOH (pH 8.0), 3 mM magnesium acetate, 10 mM DTT, 10 μM UTP, 0.2 μCi of [32P] UTP, 0.5 μg of poly(A), 120 ng of oligo(dT)15, 3.5 ng of 3Dpol, and appropriate amounts of 3A- or 3AB-proteoliposomes. Reaction mixtures were spotted onto DE52 filter membranes. The membranes were washed three times for 8 min in 5% dibasic sodium phosphate, washed briefly in water, then rinsed twice for 2 min in absolute ethanol. The membranes were dried and radioactivity was measured by scintillation counting.
Uridylylation Assay
3AB-proteoliposomes were prepared as described above. Uridylylation reactions were carried out at 30°C for 1 h in 20 μl of a reaction mixture containing 50 mM Hepes-KOH (pH 7.5), 8% glycerol (v/v), 3.5 mM magnesium acetate, 10 μM unlabeled UTP, 0.1 μCi of [32P]-UTP, 0.5 μg of cre(2C) RNA, 1 μg of 3Dpol, and varying amounts of 3CDpro and 3AB-proteoliposomes. In control reactions, 2 μg of synthetic VPg was used instead of 3AB-proteoliposomes. After the incubation 5 μl of 5x loading buffer was added, and the reaction mixtures were analyzed by Tricine-SDS PAGE [12% acrylamide (w/v)], followed by autoradiography.
RESULTS
Construction of 3A/3AB mutants suitable for membrane-association studies
To analyze the properties of the hydrophobic domain when 3A and 3AB are membrane associated, we adopted the method of measuring Trp fluorescence. For these fluorescence experiments, a Trp is most useful when placed at the center of the hydrophobic sequence (43–46). In that case, Trp location can distinguish whether the hydrophobic segment within which it is located forms a transmembrane helix, in which case the Trp locates at the bilayer center, or forms a non-transmembrane structure, in which case the Trp locates near the bilayer surface (43–47). Because the hydrophobic anchor domain of 3A does not contain Trp, a single Trp residue was introduced by a substitution mutation into a position we thought to be at the center of the hydrophobic region (F69W; Fig. 1B). To render the spectroscopic measurements specific to the hydrophobic sequence, the Trp residue in the hydrophilic region of 3A was replaced with a Phe residue (W42F; Fig. 1B). It was then determined to what extent these mutations influence the function of 3A/3AB by assaying the ability of corresponding variant RNAs to produce cytopathogenic effects (CPE) on HeLa cell monolayers.
Whereas the W42F mutation alone did not influence the replication phenotype of the PV variant (compare Table 2-A with Table 2-B), the single F69W mutation slightly delayed the appearance of CPE, an observation suggesting impaired replication (Table 2-C). In combination, however, the two mutations greatly debilitated the virus as they delayed the appearance of CPE to 7 days (Table 2-D). Delayed CPE suggested the emergence of either reversions and/or suppressor mutations, allowing the highly debilitated genome to survive. Virus isolated after 7 days from HeLa cells showing extensive CPE was plaque purified and its genome was sequenced. This revealed two unexpected suppressor mutations. One mutation (A70V) mapped to the 3A hydrophobic region next to the new Trp residue (Fig. 1B), the other to the membrane-associated viral protein 2B (I47V; Fig. 1A). To investigate whether the 2B mutation is essential for mutant survival or an irrelevant spontaneous mutation, mutant cDNA with wt 2B but containing the W42F/F69W/A70V mutations in 3A was constructed, and the corresponding transcript RNAs were transfected into HeLa cells. The replication phenotype of this triple mutant was quasi-infectious and the variant isolated after 7 days had acquired the 2B I47V suppressor mutation (Table 2-E). This observation suggested that the second suppressor mutation is essential for adequate replication of the poliovirus variant. On the other hand, when HeLa cells were transfected with transcript RNAs containing only the 2B I47V mutation, no change in the replication phenotype was observed (Table 2-F). A construct containing all three mutations in 3A and the mutation in 2B (referred to in the following as 3A/2B mutant, Table 2-G)) yielded a variant with replication properties similar to the variant (Table 2-D) recovered from mutant W42F/F69W (data not shown). Therefore, the combination of the four mutations is sufficient to produce a well-replicating virus.
Table 2.
Characterization of mutant polioviruses
| Parental Virus | Time for complete CPE | Mutations in recovered viruses | |
|---|---|---|---|
| A | wt | 24 h | N.D. |
| B | W42F | 24 h | W42F |
| C | F69W | 40 h | F69W |
| D | W42F/F69W | 7 days | W42F/F69W/A70V + 2B I47V |
| E | W42F/F69W/A70V | 7 days | W42F/F69W/A70V + 2B I47V |
| F | 2B I47V | 26 h | N.D. |
| G | W42F/F69W/A70V + 2B I47V | 48 h | N.D. |
| H | W42F/V72W | 48 h | T14K/W42F/V72W*
P17A/W42F/V72W* |
| I | W42F/A73W | 72 h | W42F/A73C |
| W42F/G74W | 48 h | W42F/G74C*
W42F/G74S* |
|
| K | W42F/V75W | 60 h | W42F/V75C |
All mutations shown map to polypeptide 3A with the exception of I47V, which maps to polypeptide 2B (see Fig. 1). CPE: cytopathogenic effect; N.D.: not determined;
Two genetic variants that emerged independently.
Interestingly, the 2B I47V mutation maps to a hydrophobic domain of the 2B protein, an observation indicating that the hydrophobic domains of 3A/3AB and 2B/2BC functionally interact during the replicative cycle of poliovirus. Similar results were obtained by Towner et al, who introduced mutations within the hydrophobic region of 3A and found a compensatory mutation within the first hydrophobic domain of the 2B protein (48).
The replication kinetics of the 2B/3A mutant virus, assayed by one-step growth curve experiments in HeLa cell monolayers, revealed slightly slower growth phenotypes as compared to wt PV (Fig. 2), as would be expected from the delay of the appearance of CPE. However, the maximum titer at 37°C approached that of wt PV (Fig. 2) whereas at 39°C, a small temperature sensitive phenotype ts was apparent (Fig. 2). Nevertheless, these growth phenotypes indicated satisfactory function of 3A/3AB in the context of the poliovirus genome and thus the 3A/3AB (W42F/F69W/A70V) polypeptides were used in the following studies.
Figure 2.
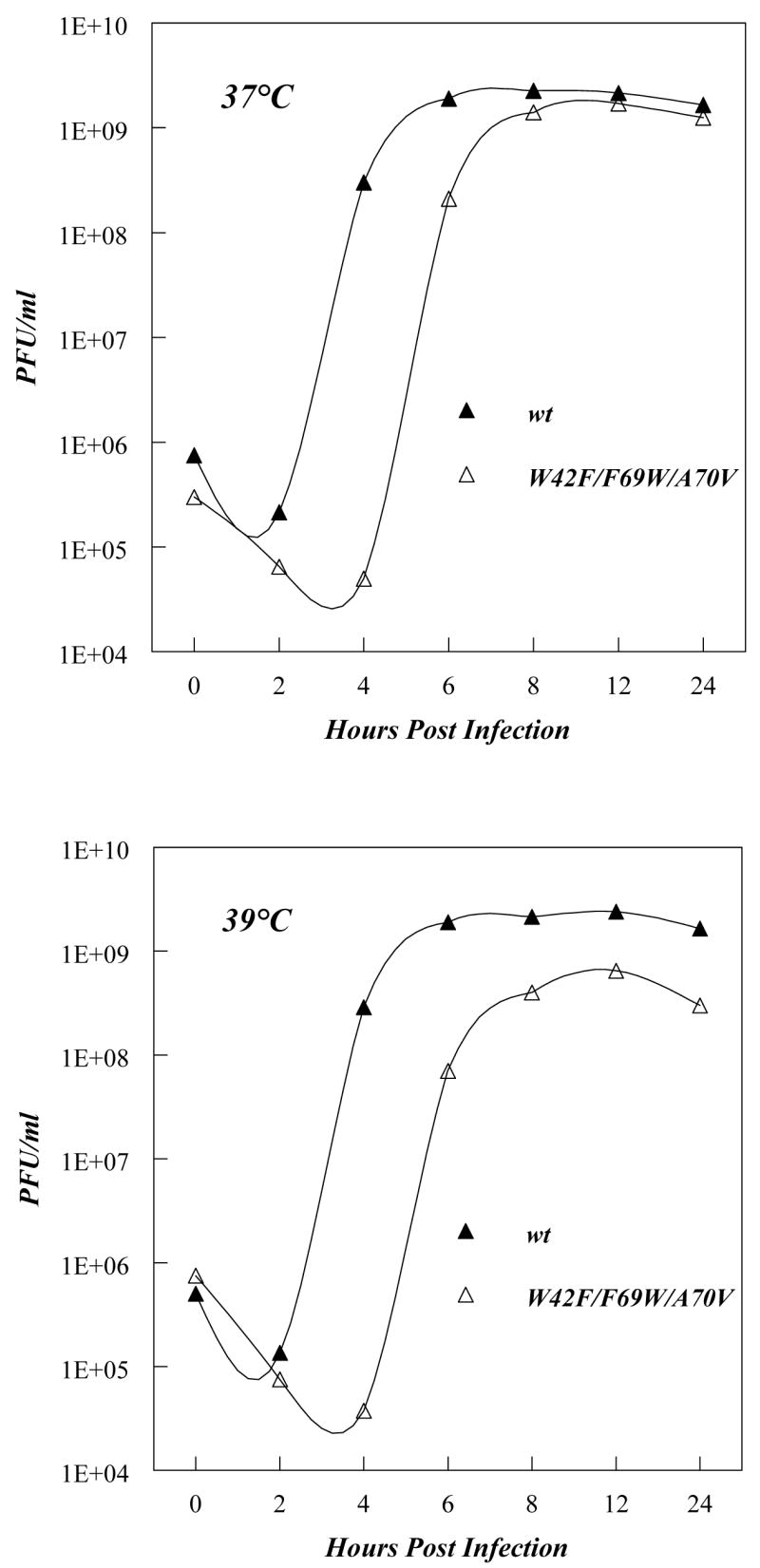
One step growth curves of wt poliovirus and a 3A mutant (W42F/F69W/A70V). Growth curves and plaque assays were carried out as described in Experimental Procedures. Upper panel: growth at 37 °C. Lower panel: growth at 39 °C. Note that this 3A mutant also has a suppressor mutation (I47V) within the first hydrophobic domain of the 2B-coding sequence (Fig. 1A). The Y-axis shows pfu/ml in exponential units (base 10).
Ability of the polio 3A hydrophobic sequence to span lipid bilayers in TM form: Behavior of the anchor peptide-1
A peptide corresponding to the hydrophobic anchor sequence (underlined) of protein 3A carrying the Trp-Val mutation at positions 69–70 plus the flanking charged residues (acetyl58RAMTILQAVTTWVAVAGVVYVMYK81amide) (Fig. 1B) was used to study the ability of the hydrophobic sequence to span lipid bilayers. Fluorescence methods we developed previously were used to define the topography of the bilayer-inserted sequences (43–47, 49). The first assay involved measurement of Trp emission λmax to evaluate approximate Trp depth. When a 20 residue long hydrophobic sequence with a Trp near the center of its hydrophobic sequence adopts a TM (transmembrane) orientation, the Trp locates near the bilayer center, and gives highly blue shifted emission (λmax = 315–325 nm) (43, 44). If the sequence adopts a non-TM topography located close to the surface of the bilayer Trp λmax red shifts strongly (335–340 nm) (43, 44). Additional information was derived from the Trp λmax vs. bilayer width curve. A highly hydrophobic peptide with a Trp at the center of the hydrophobic sequence exhibits a minimum λmax value at the maximum width bilayer in which the peptide maintains a fully TM structure. In wider bilayers, due to negative mismatch (i.e. bilayer width exceeding hydrophobic helix length) there is formation of an appreciable fraction of a non-TM, surface topography which gives more red shifted Trp emission (43, 44, 47). The bilayer widths in which Trp emission red shifts can reveal the length of the TM sequence (43, 44).
To measure Trp depth directly, a dual fluorescence quenching method was used (47). In this method the ratio of quenching of Trp by acrylamide, which resides in the aqueous solution, to quenching by the membrane-inserted quencher 10-doxylnonadecane is measured. This quenching ratio (Q-ratio) responds nearly linearly to Trp depth in the bilayer, such that a low quenching ratio (<0.15) indicates a deeply located Trp near the center of the bilayer, while a high Q-ratio (>1) is indicative of a Trp near the bilayer surface (45, 47).
Figure 3 and Table 3 show values for anchor peptide-1 Trp λmax and how it is affected by bilayer width (varied by using lipids with different length acyl chains). Even in vesicles in which the λmax was most highly blue-shift, e.g. those vesicles composed of DOPC (diC18:1PC), anchor peptide-1 exhibited a somewhat red shifted λmax (332 nm), although the λmax red shift increased considerably (to 339 nm) in wider bilayers (Fig. 3, filled triangles). The somewhat red shifted λmax observed in DOPC vesicles indicates that the Trp was not located at the bilayer center in these vesicles. However, there was at least some formation of a TM configuration in DOPC vesicles, as the λmax even more red-shifted in wider bilayers, under which conditions a non-TM form would predominate. When quenching was measured, results in agreement with the λmax data were obtained. The Q-ratio was 0.35 in DOPC (C18:1 acyl chains) and 0.82 in DEuPC (C22:1 acyl chains) (Table 3). This behavior indicates that anchor peptide-1 forms, at least to some degree, a TM structure when incorporated into DOPC bilayers and adopts a shallow, non-TM topography in wider bilayers. (Circular dichroism experiments indicate that the membrane-inserted peptide is highly α-helical, as expected (data not shown).
Figure 3.

Effect of lipid bilayer width upon Trp emission λmax for anchor peptide-1 and anchor peptide-2. Anchor peptide-1 (filled triangles) or anchor peptide-2 (open triangles) were reconstituted into model membrane vesicles composed of monounsaturated PCs with different acyl-chain lengths. Samples contained 2 μM peptide incorporated into 500 μM lipid dispersed in PBS at pH 7.0. The values shown are the average of six samples. The λmax values are generally reproducible to ±1 nm.
Table 3.
Quenching of Membrane-Inserted Anchor Peptide-1 and Anchor Peptide-2 by Acrylamide and 10-Doxylnonadecane.
| Peptide | Fo/F Acrylamide | Fo/F 10-DN | Q-Ratio | λmax (nm) |
|---|---|---|---|---|
| Peptide inserted into DOPC vesicles (C18:1 acyl chains) a | ||||
| Anchor Peptide -1 | 1.31 ± 0.02 | 1.88 ± 0.01 | 0.352 ± 0.02 | 332± 0.2 |
| Anchor Peptide-2 | 1.25 ± 0.01 | 2.33 ± 0.14 | 0.192 ± 0.21 | 327± 0.25 |
| Peptide inserted into DEuPC vesicles (C22:1 acyl chains) | ||||
| Anchor Peptide-1 | 2.11 ± 0.11 | 2.29 ± 0.13 | 0.82 ± 0.15 | 339.8± 0.4 |
| Anchor Peptide-2 | 1.91 ± 0.02 | 1.99 ± 0.04 | 0.92 ± 0.05 | 340± 0.3 |
Fo/F is the ratio of fluorescence in the absence of quencher to the that in the presence of quencher. The ratio of quenching by acrylamide to that by 10-DN, the Q-ratio, is defined in Experimental Procedures. Average values and standard deviations derived from six samples are shown.
The fluorescence properties of anchor peptide-1 incorporated into DOPC vesicles do not distinguish between the peptide forming a homogeneous configuration, in which the Trp has a single intermediate depth, and a case in which the peptide exists in a mixture of two populations: one with a TM topography, and thus a deep Trp depth, and another population with a non-TM topography having a Trp near the bilayer surface. The effect of quenchers on the λmax of Trp emission can distinguish between these alternatives. When deep and shallow Trp populations co-exist, intermediate λmax values are observed because the two populations have overlapping blue and red-shifted spectra. In such samples, there is selective acrylamide quenching of the shallow Trp population because it is closer to the acrylamide than the deep Trp. This induces a blue shift in λmax, because the deeper Trp population is only weakly quenched. In contrast, 10-DN quenching induces red shifts due to selective quenching of deep Trp, which is closer to 10-DN than shallow Trp. In such heterogeneous populations, these shifts can result in a λmax value in the presence of acrylamide that differs from that in the presence of 10-DN by 5–15 nm. In populations with a homogeneous Trp depth, the quencher-induced shifts are much smaller (45, 47). For anchor peptide-1 in DOPC vesicles we observed very little difference between Trp λmax in the presence of acrylamide and 10-DN (1.5 nm) indicating that it adopts a homogeneous state in which the Trp is not at the center of the bilayer.
The observation that the Trp was not located at the bilayer center suggested that the TM was not 22 residues long. This was supported by the observation that emission was significantly more red-shifted in diC20:1PC bilayers than in DOPC vesicles. This is indicative of a partial loss of TM topography in diC20:1PC bilayers and previous studies shows that this behavior corresponds to that expected for a TM sequence <18-19 residues long (43, 44)(and see below).
Recovery of recombinant polioviruses harboring mutations at different positions within the hydrophobic domain of 3A
To confirm that TM sequence was shorter than expected, and that as a result the center of the transmembrane sequence was at a position closer to the C-terminus of the hydrophobic sequence than originally estimated, we decided to introduce a Trp at a position closer to the C-terminus of the hydrophobic sequence than Phe69. In order to determine at what positions Trp could be introduced while maintaining PV viability, several mutant cDNA clones containing both a Trp substitution in the hydrophobic sequence and the W42F mutation in the hydrophilic region were constructed (W42F/V72W, W42F/A73W, W42F/G74W, and W42F/V75W). Transfection of RNA transcripts of the clones into HeLa cells yielded viruses in all cases. However, genome sequence analysis of the recovered variants revealed that PV does not tolerate a Trp in positions 73, 74 and 75 (Table 2-I, 2-J, and 2-K, respectively). That is, similar to the case studied in the mutational analysis described above, the transcript RNAs were quasi-infectious (50) such that recovered viruses contained Trp residues substituted with either with a Cys (W73C, W74C, W75C) or Ser (W74S). In mutant W42F/V72W the Trp residue was retained, although two independent variants containing one suppressor mutation each in the hydrophilic region of 3A emerged. In one plaque-purified virus, a Thr at position 14 was changed to Lys (Fig. 1B; Table 2-H), and in another virus, a Pro at position 17 was changed to Ala (Table 2-H). We found no additional suppressor mutations when we analyzed the sequences of the entire coding region of the non-structural proteins of the recovered viruses containing the T14K/W42F/V72W or P17A/W42F/V72W mutations. The growth kinetics of the T14K/W42F/V72W and P17A/W42F/V72W variants at 37° C and 39° C are presented in Fig. 4. Similar to the previous W42F/F69W/A70V variant, these viruses did not exhibit a pronounced ts phenotype (Fig. 4), and they grew moderately well at 37°C although somewhat slower than wt PV. We conclude that the 3A/3AB polypeptides containing the T14K/W42F/V72W and P17A/W42F/V72W mutations are functional in the context of the replicating PV genome. Somewhat arbitrarily, we selected the T14K/W42F/V72W variant for further studies.
Figure 4.
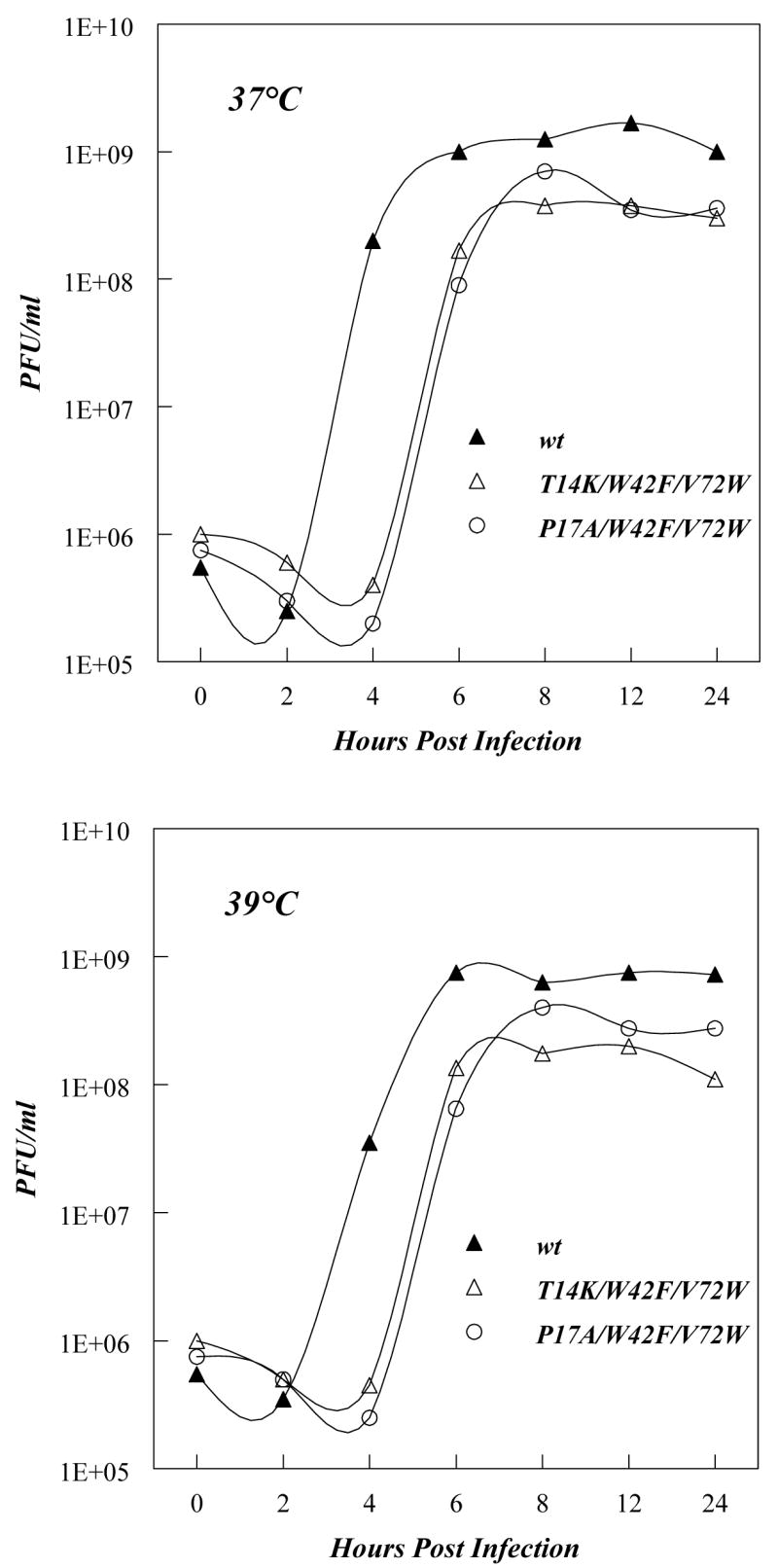
One step growth curves of wt poliovirus and two 3A mutants (T14K/W42F/V72W and P17K/W42F/V72W). Growth curves and plaque assays were carried out as described in Experimental Procedures. Upper panel: growth at 37 °C. Lower panel: growth at 39 °C.
Behavior of anchor peptide-2 in model membranes
Since viruses containing Trp at position 72 grew well in HeLa cells, a synthetic peptide carrying a Trp at position 72 (anchor peptide-2; acetyl-58RAMTILQAVTTFAAWAGVVYVMYK81-NH2) was synthesized and subjected to the fluorescence analysis. In DOPC vesicles, the values of both λmax and Q ratio for the Trp of anchor peptide-2 were just slightly above those expected for a Trp exactly at the bilayer center, and this indicated a significantly deeper Trp depth than measured for anchor peptide-1, and (Fig. 3 and Table 3). These results show that anchor peptide-2 predominantly forms the TM state in DOPC bilayers, and confirm that the Trp in anchor peptide-1 was not located at the bilayer center.
The fact that the Trp of anchor peptide-2 is close to the bilayer center, while the Trp of anchor peptide-1 is not as close to the bilayer center, indicates residue 72 is closer to the center of the TM sequence than residue 69. The TM sequence cannot end beyond Lys81, which is the last residue in the peptide, and it is far more likely to end at residue Tyr80. If residue 72 is at the bilayer center of the TM sequence, the TM sequence must start at, or very near, Gln64, which would mean the TM sequence is 16 residues long (Fig. 1B; shaded area in the hydrophobic domain).
This conclusion is supported by the shape of the Trp λmax vs. bilayer width curve, which shows that the minimum λmax value (observed in lipids forming bilayers with widths that are close to the point where there is no mismatch between TM helix length and bilayer width) occurs in liposomes formed from lipids with acyl chains that are 17–18 carbon atoms long (Fig. 3). We previously found that 19–20 residue long TM sequences exhibit a λmax minimum in liposomes composed of lipids with acyl chains that are 20 carbon atoms long (43). Since bilayer width increases by about 1.8 angstroms per acyl chain carbon atom (0.9 angstroms per lipid molecule) (51), and the length of a helix increases by 1.5 angstroms per residue, the difference between the acyl chain lengths at which a 16 and 19–20 residue-long TM helix experience no mismatch should be about that 3 carbon atoms, close to what is observed.
Fluorescence studies with intact polio 3Am and 3ABm proteins
We next wished to study the membrane configuration of the anchor sequence in the context of the intact proteins 3A and 3AB. Since the mutant virus containing three mutations within the 3A sequence (T14K/W42F/V72W) grows similarly compared to wt virus (Fig. 4), indicating that 3A/3AB was functional, we proceeded to purify the mutant 3A and 3AB proteins with these mutations (referred to as 3Am and 3ABm) for further studies. The purified 3Am or 3ABm proteins were then incorporated into preformed liposomes as described in the Experimental Procedure section. This process mimics insertion in vivo (see Discussion).
Trp fluorescence was again used to characterize the topography of the hydrophobic anchor sequence within the membrane-incorporated full length proteins. The behavior of the anchor sequence might differ in peptide and intact 3AB protein because intact 3AB protein has large hydrophilic N- and C-terminal domains which should not be able to cross membranes. This would prevent the formation of a TM state by the anchor sequence upon binding of 3AB to membranes. In contrast, the 3A protein has only a short C-terminal hydrophilic sequence that might be able cross membranes (Fig. 1B), and so the anchor sequence in 3A protein might form a TM state when added to membranes. Table 4 shows that Trp λmax values support these hypotheses. In DOPC vesicles, the 3Am protein exhibited a more blue-shifted λmax (328 nm) and lower Q-ratio (0.39) than was observed for the 3ABm protein (λmax 331 nm, Q-ratio 0.67). This suggests that a significant fraction of the hydrophobic sequence of the 3Am protein formed a state with a deep Trp, i.e. a TM configuration, while the hydrophobic sequence of the 3ABm protein formed a greater degree of a shallow, non-TM state in which Trp is near the surface of the bilayer. The latter conclusion is confirmed by the observation that the 3ABm protein incorporated into wider DEuPC bilayers, in which a non-TM form would be expected to be present to a significant degree because the length of the hydrophobic segment is too short to span hte DEuPC bilayer, showed λmax and Q ratios (λmax 332 nm and Q-ratio = 0.69, Table 4) that were very similar to those in DOPC bilayers. As noted above, this insensitivity to mismatch between hydrophobic helix length and bilayer width is characteristic of a sequence that is fully non-TM. In contrast, 3Am protein in DEuPC vesicles showed a higher λmax and Q-ratio (λmax 331 nm and Q-ratio = 0.62, Table 4) than it did in DOPC vesicles. Furthermore, λmax and Q-ratios for 3Am in DEuPC vesicles were similar to those for 3ABm protein in DEuPC vesicles, indicating that negative mismatch affects the topography of the 3Am hydrophobic segment, such that it converts from the TM to non-TM state, as expected. This type of sensitivity to mismatch is indicative of a TM state in the absence of mismatch (i.e. in the DOPC vesicles). Thus, a number of internally consistent fluorescence parameters support the TM assignment for the topography of the hydrophobic segment in the 3Am protein (at least for some population of 3Am molecules, see below), and non-TM topography in 3ABm protein.
Table 4.
Quenching of Membrane-Inserted Polio 3AB and Polio 3A Protein by Acrylamide and 10-Doxylnonadecane.
| Protein | Fo/F Acrylamide | Fo/F 10-DN | Q-Ratio | λmax (nm) |
|---|---|---|---|---|
| Protein inserted into DOPC vesicles (C18:1 acyl chains) a | ||||
| Polio 3AB | 1.84 ± 0.12 | 2.26 ± 0.17 | 0.67 ± 0.20 | 330.8 ± 0.4 |
| Polio 3A | 1.45 ± 0.03 | 2.16 ± 0.04 | 0.39 ± 0.04 | 328 ± 0.3 |
| Protein inserted into DEuPC vesicles (C22:1 acyl chains) | ||||
| Polio 3AB | 1.40 ± 0.03 | 1.57 ± 0.02 | 0.69 ± 0.03 | 331.7 ± 0.3 |
| Polio 3A | 1.54 ± 0.03 | 1.88 ± 0.04 | 0.62 ± 0.05 | 330.8 ± 0.2 |
Fo/F is the ratio of fluorescence in the absence of quencher to the that in the presence of quencher. The ratio of quenching by acrylamide to that by 10-DN, the Q-ratio, is defined in Experimental Procedures. Average values and standard deviations derived from six samples are shown.
However, there are some differences between the fluorescence behavior of the hydrophobic sequences in the isolated peptides and intact protein. One difference is that the Trp depth in DOPC for 3Am protein is significantly shallower than for the isolated peptide as judged by Q-ratio. This suggests that in the case of the intact protein not all of the hydrophobic sequence is in a TM state. The second difference is that in the DEuPC vesicles, in which the non-TM state predominates, Trp fluorescence is more red-shifted and Q ratios are much higher in the isolated anchor peptide than in the intact protein. This suggests that the hydrophilic portion of the 3Am domain affects how deeply the hydrophobic anchor sequence penetrates the bilayer in the non-TM state.
Stimulation of 3Dpol polymerase activity by 3AB-associated proteoliposomes
The experiments above showed that 3A and 3AB could interact efficiently with model membrane vesicles, and that in the model membrane bound states, they have different topographies. This raised the question of whether interaction of 3A or 3AB (=3A(B)) with model membrane would affect their functional properties, and whether 3A and 3AB would differ in function once membrane-bound.
It was previously shown that purified 3AB dispersed in detergent strongly stimulates the elongation activity of the poliovirus RNA polymerase 3Dpol in vitro on a poly(A) template with a dT15 primer, while 3A is not able to do so (14, 16–19). Since 3Dpol interacts with 3AB primarily via the VPg segment of the polypeptide, and because there is no detectable interaction between 3A and 3D (9), the stimulatory activity of 3AB on 3Dpol appears to be dependent on the presence of the 3B domain. To test whether membrane insertion of 3A(B) would affect their ability to stimulate the activity of 3Dpol we performed elongation reactions using 3A- or 3AB-proteoliposomes. For these studies we used 3A and 3AB proteins that contained wild type amino acid sequences. Reconstituted proteoliposomes (Experimental Procedures) containing bound 3A or 3AB protein were pelleted by ultracentrifugation and resuspended in a small amount of buffer. Liposomes without any protein were also prepared and used as controls. The values obtained with empty lipid vesicles (lacking protein) were subtracted from those obtained with the 3A- or 3AB- proteoliposomes. We observed that 3AB proteoliposomes stimulate the elongation activity of 3Dpol in a dose dependent manner (Fig. 5). In contrast, 3A-proteoliposomes did not have such stimulatory activity within the range of protein concentrations used in this study (Fig. 5). It should be noted that there was no significant stimulatory activity when increasing amounts of empty liposomes were used (data not shown).
Figure 5.

Stimulation of the elongation activity of 3Dpol by 3A- or 3AB-proteoliposomes. Primer-dependent elongation reactions were performed in the presence of 3A-(filled triangle) or 3AB (open triangles)-proteoliposomes, and 32[P]UMP incorporated into polymer was measured by a scintillation counter. Values were corrected for using the same amount of empty vesicles.
Uridylylation of VPg Derived from Membrane-bound 3AB
Although the in vitro uridylylation of synthetic VPg to VPgpU and VPgpUpU has been studied extensively the exact nature of the substrate used in this reaction in vivo remains unknown (1). Uridylylation of VPg, which is strictly template dependent, is catalyzed by 3Dpol and it occurs at a Tyr, the third amino acid of VPg (Fig 1A;(1, 32). The RNA template can be either poly(A) or cre, a cis acting replication element mapping to the reading frame of polypeptide 2CATPase (1, 32). 3AB has been suggested to be the precursor for uridylylation (52), but under standard in vitro reaction conditions purified 3AB (in detergent) does not serve as substrate for uridylylation (A. Paul and E. Wimmer, unpublished results). Interestingly, a very recent study by Richards et al. describes the uridylylation of 3AB in detergent-containing solutions by 3Dpol on an oligo A15 template but not on longer poly(A) templates (53). (This template length-dependent difference has been attributed to a lack of oligomerization of the polymerase on the small RNA template (53). In contrast, VPg itself and minor cleavage intermediates in polyprotein processing (3BC and 3BCD; not shown in Fig. 1A) can be readily uridylylated in an in vitro assay consisting of an RNA template, 3Dpol, 3CDpro (as cofactor) and UTP (Pathak and Cameron, unpublished observations; Paul and Wimmer, unpublished observations). All active substrates for in vitro uridylylation (VPg, 3BC, 3BCD) have in common a free N-terminus of which Tyr is the third residue.
Inclusion of 3AB into uridylylation assays of purified proteins has been difficult owing to the water-insoluble property of the polypeptide. Furthermore, 3AB is cleaved by 3Cpro or 3CDpro only when bound to membranes (16), an observation suggesting that the cleavage site between the two domains 3A and 3B, and possibly the Tyr residue three residues downstream from the cleavage site, may not be accessible for enzyme recognition when the protein is not membrane bound. Therefore, in vitro-reconstituted liposome-bound 3AB seemed a suitable model substrate to study whether 3AB in the membranous environment can be uridylylated. The reaction partners in the uridylylation assay were similar to those of VPg uridylylation described previously, including the cre element as RNA template (see Experimental Procedures).
In the presence of a 3CDpro derivative in which the proteinase activity was eliminated by an active-site mutation, no uridylylation of 3AB in liposomes was observed (data not shown). However, when proteolytically-active 3CDpro proteinase was used as cofactor with 300 ng or 3 μg of 3AB per reaction, the synthesis of VPg-pU was detected (Fig. 6A; lanes 12–18). It should be noted that this 3CDpro cofactor carried a 3C*3D cleavage site mutation to permit isolation of the uncleaved 3CDpro polypeptide. Controls showed VPg uridylylation was not dependent upon the presence of proteolytically active 3CDpro (Fig. 6A, lanes 2 and 20). The results suggest that 3AB itself is not the template for uridylylation, but once it is cleaved into 3A and VPg, free VPg is subsequently uridylylated.
Figure 6.

Uridylylation reactions using 3AB-proteoliposomes. A. VPgpUpU synthesis as a function of 3AB and 3CDpro concentrations. Uridylylation reactions were carried out as described in Experimental Procedures on a cre(2C) template RNAs using proteoliposomes containing 3 ng, 30 ng, 300 ng or 3 μg of 3AB, as indicated on the figure. At each 3AB concentration proteolytically active 3CDpro (cleavage-site mutant to prevent cleavage between 3Cpro and 3Dpol) (lanes 4–18) were included in the reactions, at concentrations 0.35 μg, 0.7 μg, 1.4 μg or 2.8 μg, from left to right in each set of experiments. The reaction shown on lane 3 contained 3AB (3 μg) and inactive proteinase (0.7 μg). As a control the uridylylation of synthetic VPg was measured in the presence of 0.7 μg of inactive 3CDpro (lanes 2 and 20). B. VPgpUpU synthesis requires proteolytically active 3CDpro. Reactions were carried out using 3 μg 3AB in proteoliposomes alone (lane 1) or with different concentrations of proteolytically active or inactive 3CDpro for 1h (0.35 μg, 0.7 μg, 1.4 μg or 2.8 μg per reaction from left to right (lanes 2–9)). As a control synthetic VPg peptide (2 μg) was used as substrate in the uridylylation reaction with 0.7 μg of active 3CDpro (lane 14). Uridylylation reactions were also carried out with pre-incubation of 3AB-liposomes with active 3CDpro for 2 hr (lanes 10–13), or using purified 3AB in detergent (0.8% octylglucoside), (lanes 15–18), again with different concentrations of active 3CDpro (0.35 μg, 0.7 μg, 1.4 μg or 2.8 μg per reaction, from left to right). Note the loss of activity at very high 3CDpro concentrations, the reason for this is unclear, but we have also observed this in other experiments.
Figure 6B shows that when the membrane-bound 3AB was preincubated with active 3CDpro in vitro the yield of VPg-pU was very small (lanes 6–9) compared to that obtained with a synthetic VPg peptide (lane 14), even after prolonged incubation of 3AB with 3CDpro (lanes 10–13). The reason for this most likely is that 3AB is a relatively stable protein in vivo and a poor substrate for 3CDpro in vitro (16).
Uridylylation reactions were also carried out using 3AB in detergent-containing solutions (0.8% octylglucoside). However, we could not detect any VPg-pU even if 3AB was preincubated with 3CDpro for 2 h (Fig. 6B; lanes 15–18). This result supports previous observation suggesting that only membrane-bound 3AB can be cleaved by 3CDpro (16).
DISCUSSION
PV proteins change function during the PV life cycle
Since plus strand RNA viruses have a limited genome size they have adopted an ingenious way to utilize their polyprotein for diverse and changing requirements during the viral life cycle: the precursor proteins obtained by incomplete processing of the polyprotein possess different functions than the mature cleavage products. Proteins 3AB and 3A exemplify such a relationship between a precursor and a mature polypeptide. Although they share the 3A sequence their functions are quite different. It is generally accepted that one of the important functions of 3AB is to anchor VPg to the membranes via the hydrophobic sequences located in its 3A domain. 3AB then interacts with the RNA polymerase primarily through its VPg domain and recruits it to the replication complex. In this context it should be noted that poliovirus 3Dpol does not posses any membrane binding domains, unlike some other viral RNA polymerases such as the RDRP of HCV (NS5B) (54).
Tryptophan mutations in 3A highlight intra and intermolecular interaction between PV proteins
In this study, we used tryptophan-specific fluorescence measurements to determine the topography of the 3A and 3AB proteins inserted into pre-formed model membrane vesicles. In order to make fluorescence measurements specific to the 3A-hydrophobic domain, we placed a Trp at position 69 within the hydrophobic domain of 3A (F69W) and replaced a Trp residue in the hydrophilic region of 3A with a Phe residue (W42F). The effects of these mutations upon viral replication are interesting. Although it has been reported that the substitution of the well-conserved Trp at position 42 to Arg resulted in a lethal phenotype (55) and aberrant protein processing, the Trp to Phe substitution yielded a viable virus (Table 2-B). Other previous studies have shown that a Phe 69 to His substitution leads to a quasi-infectious virus, which acquired a second-site mutation at the adjacent amino acid at position 70 to either Val or Thr (56). We found a similar second-site mutation at position 70 to Val in the virus recovered from the W42F/F69W mutant. Furthermore, we observed a suppressor mutation within the first hydrophobic domain of 2B (I47V) in the virus recovered from the W42F/F69W clone. This result suggested that the hydrophobic domains of 3A/3AB and 2B/2BC functionally interact during poliovirus replication, in agreement with previous studies by Towner et al. (48). Interestingly, no second-site reversions were observed either in 3A or in 2B in the virus derived from the W42F or F69W single mutants (Table 2-B and 2-C, respectively). One possible explanation of this finding is that the mutation in the N-terminal hydrophilic region of 3A affects the structure of the C-terminal hydrophobic domain, which results in inefficient interaction of the hydrophobic domain of 3A with itself or with those of other viral proteins (e.g. 2B). Interestingly, we found second-site mutations within the hydrophilic domain of 3A (T14K or P17A) when we introduced a Trp mutation at position 72. These results support the hypothesis that the hydrophilic and the hydrophobic domains of 3A structurally affect each other.
Topography and membrane-interactions of the 3A hydrophobic anchor domain in intact 3A and 3AB proteins
Based on the information above, single-Trp mutants were designed to test the configuration of membrane-associate 3A and 3AB proteins. The model membrane interaction of the isolated hydrophobic sequence of 3A, and that 3A and 3AB proteins with single Trp were then used to evaluate the topography in membranes. The studies using peptides containing the hydrophobic sequence within the 3A protein clearly demonstrated that it had the capacity to form a TM configuration, but that did not answer whether it does so in intact protein. To do that we devised a membrane-interaction protocol that would mimic the post-translational interaction of 3AB and 3A with membranes that occurs in vivo. This method resulted in a membrane insertion that was about as efficient as that used to incorporate peptide into membranes, as demonstrated by the observation that membrane-bound 10-DN quenched peptide and intact protein fluorescence to similar degrees (compare Tables 3 and 4).
Using this protocol we found that the hydrophobic anchor domain of 3A adopts a membrane-bound non-TM configuration in the context of the 3AB protein, wherein both the 3A and VPg domains of 3AB are cytosolic. A similar non-TM arrangement of a potentially-TM sequence has seen in other proteins such as cytochrome b5 and caveloin, under at least some conditions (57, 58). The non-TM configuration is not a surprise, as formation of a TM configuration would have required either the large hydrophilic domain of 3A or that of 3B to translocate across the membrane. However, although translocation of a large hydrophilic domain would be very unusual, certain bacterial toxin proteins do catalyze self-translocation of hydrophilic domains, so this could not be ruled out a priori (59, 60).
In contrast to the behavior in the 3AB protein, the hydrophobic domain in 3A adopts both a TM and a non-TM structure. In order for a TM state to form, the sequence flanking the C-terminal tail of the hydrophobic domains (KLFAGHQ) must cross the bilayer, despite the presence of several hydrophilic residues. It is possible that the residual non-TM configuration is an artifact of the use of model membrane liposomes that do not adequately mimic natural membranes. The difference in hydrophobic anchor configuration in 3A and 3AB proteins is interesting because it illustrates one physical mechanism by which precursor (3AB) and mature (3A) proteins can have different structures that might lead to different functions. In this context it is also interesting to note that based on biochemical studies, Towner et al. (10) have suggested that the primary membrane anchor domain of 3A in the context of 3AB contains only the C-terminal 13 amino acids of the hydrophobic domain, which is too short to form an α–helical structure that spans the lipid bilayer.
It should also be noted that when we incorporated the anchor peptides or the intact 3A or 3AB proteins into vesicles composed of 70mol% DOPC/30mol% DOPG we obtained Trp λmax and quenching data that was very similar to that obtained in DOPC, indicating that the presence of the anionic DOPG did not affect the topography of the hydrophobic sequence (data not shown).
It is plausible that a fraction of the 3A hydrophobic anchors form TM helices once 3A is cleaved off from 3AB by 3Cpro or 3CDpro. It has been previously reported that the hydrophobic domain of the RNA polymerase (NS5B) of HCV forms a TM helix (61), and we have recently shown that this hydrophobic domain of NS5B can be replaced with that of poliovirus 3A without abrogating replication of an HCV replicon (62). This result can be easily rationalized if the hydrophobic anchor of 3A also has the ability to form a TM helix.
As discussed above, our results are in agreement with those of Towner et al. (48), in terms of suggesting that the hydrophobic domains of 2B and 3A interact with each other during poliovirus replication. Because of the precursor/mature protein relationships multiple interacting partners are possible: 3A/2B, 3A/2BC, 3AB/2BC, and 3AB/2B. In addition, if one of the interacting partners is 3A then two forms of the membrane bound protein (TM and non-TM) are available for these interactions. Whether one or more of these interactions are required for creating the correct environment for RNA replication is not known. It should be noted that any such effects could be exerted at different stages in the replication process. Previous studies with the 2B(C) protein of enteroviruses indicated that its membrane binding ability affects RNA replication at two different stages (2, 63). One of these involves the induction of vesicles, which are required for RNA replication (64). The second step involves RNA replication more directly and requires an interaction with one or more viral or cellular proteins (63).
Our studies using peptides containing the hydrophobic domain of 3A and liposomes with different acyl chain lengths indicated that the hydrophobic domain of 3A behaves as a short transmembrane helix consisting of approximately 16 amino acids. Based on the putative hydrophobic domain sequence, Phe at position 69 was originally believed to be near the center of the TM segment. However, λmax and Q-ratio of F69W peptide (anchor peptide-1) was higher compared to the V72W peptide (anchor peptide-2), indicating that the Val at position 72 is closer to the center of the transmembrane helix than the Phe at position 69. Therefore, the TM segment is most likely to be formed by residues 65–80, rather than by the entire hydrophobic sequence (residues 59–80), with Q64 forming the N-terminal boundary of the buried sequence (Fig. 1B; shaded area of the hydrophobic sequence). This is further supported by our findings that Gln has a strong tendency to locate at the membrane surface rather than being buried in the bilayer (S. Krishnakumar and E. London, unpublished data).
Effect of 3AB Membrane Binding Upon 3AB Function
Having determined that in our synthetic vesicles 3AB forms a membrane-bound structure, albeit non-TM, we were interested to find out whether 3A(B) insertion affects some of the biochemical activities of the 3A(B) protein. We have previously shown that in the presence of detergent, purified 3AB, but not 3A, stimulates the activity of 3Dpol about 50-fold in the elongation of a dT15 primer on a poly(A) template (14, 16). In the present study, we found that the membrane bound forms of the 3A(B) proteins behave the same way. The lack of stimulation by 3A is not surprising since 3AB and 3Dpol interact primarily via the VPg domain of 3AB (9). We did not observe any inhibition or stimulation of the elongating activity of 3Dpol in the presence of different concentrations of empty liposomes (data not shown), an observation indicating that the membrane itself has no effect on polymerase activity. The mechanism by which the elongating activity of 3Dpol is stimulated by 3AB is not fully understood. However, it has been suggested that the role of 3AB is to make the primer-template-3Dpol complex more stable (18).
Using an in vitro reconstituted system, we also showed that VPg derived from membrane bound 3AB by proteolytic cleavage with 3Cpro/3CDpro can be uridylylated by 3Dpol on a cre(2C) template. When we used 3CDpro with an active-site mutation or used 3AB in the presence of detergent, we could not detect any 3AB-specific signal. The fact that the active-site mutation in 3CDpro abolished uridylylation suggests that 3AB itself is not uridylylated. However, once 3AB is cleaved, VPg becomes a substrate for uridylylation. When membrane-bound 3AB was used in the uridylylation reaction, the yield of VPg-pU was poor presumably because of inefficient processing by 3Cpro/3CDpro. Processing in vitro may be inefficient because other proteins that normally enhance digestion are not present, because the normal substrate is a precursor to 3AB, or because the artificial membranes do not exactly mimic natural membranes due to a difference in lipid composition.
The functionality of VPg derived from 3AB is in agreement with the results of Cao and Wimmer, who have previously shown that a lethal mutation in VPg (Y3F/T4A) could be complemented, albeit very inefficiently, by 3AB, which was expressed from a different cistron in the same genome (65). In that study, heterologous VPg nucleotide sequences were used in the complementing 3AB to eliminate the possibility of genetic recombination. Viable virus was recovered, but the progeny was “quasi-infectious” suggesting that complementation by 3AB was inefficient. We interpret the ability to rescue replication to mean that the complementing 3AB protein was cleaved to 3A and VPg, which was subsequently uridylylated and used for RNA synthesis. In this context it is interesting to note that even if 3AB is the direct precursor of VPg in vivo, experiments in vitro suggest that it has to be delivered to the replication complex in the form of a large precursor such as P3. In the in vitro translation/RNA replication system the 3A F69H mutations in the PV template RNA could be complemented only by P3 and not by 3AB (56). It is also possible that in vivo the VPg used for uridylylation is derived primarily from other precursor proteins such 3BC, 3BCD, or 3ABC or alternatively, that these precursors themselves are the true substrates for uridylylation. This latter possibility is supported by the observation that in vitro 3BC and 3BCD are not only as good substrates for 3Dpol as VPg but they work at about 50 fold lower concentration than VPg (Pathak and Cameron, unpublished observations; Paul and Wimmer, unpublished observations).
Proposed model for the association of 3A/3AB with membranes and the initiation of PV RNA synthesis
The major cleavage products originating from the P3 region are 3AB and 3CDpro, which are slowly processed to the final mature proteins (Fig. 7). The 3AB protein associates with intracellular membranes via the hydrophobic domain of 3A and adopts a non-TM structure. Its soluble, cytosolic VPg domain then interacts with 3Dpol and recruits it to the membranes. Once 3AB is cleaved into 3A and VPg by 3Cpro/3CDpro the hydrophobic domain of 3A can adopt both a TM and a non-TM structure. Whether the TM and/or non-TM forms of 3A derived from membrane-bound 3AB, have a subsequent function in replication remains to be determined. The 3Dpol/VPg complex then binds to 3CDpro by means of an interaction between 3Dpol and the 3Cpro domain of 3CDpro (42, 66). Using the RNA binding activity of 3CDpro the complex is attached to the cre(2C) RNA where the uridylylation of VPg takes place (33, 67). It should be noted that the role of the 2B(C)/3A(B) interaction(s) in RNA replication also remains to be determined.
Figure 7.

Proposed model for the structures of 3AB and 3A on membranes and initiation of poliovirus replication. The P3 precursor is processed by proteinase 3CDpro to yield 3AB and 3CDpro. 3AB interacts with membranes forming a non-TM configuration and its VPg domain binds 3Dpol to the membranes. Protein 3CDpro cleaves off VPg releasing the 3A protein, which then assumes both a TM and a non-TM form. 3Dpol, complexed with VPg, then interacts with the 3Cpro domain of 3CDpro. Through its RNA binding domain 3CDpro binds the entire complex to the cre(2C) RNA structure where the uridylylation of VPg takes place yielding VPgpUpU. The 3A(B) interaction with 2B(C) is not indicated on the model because we do not yet know whether this interaction is required during the initiation of RNA replication.
Since cleavage of 3AB by 3CDpro does not take place in detergent (16), and the uridylylation reaction in HeLa cell extracts is abolished in the presence of detergent (36), reconstitution of proteoliposomes containing 3AB is a useful tool for studying the role of 3AB in poliovirus replication. In the future, it might be possible to reconstitute the entire poliovirus replication complex in vitro using proteoliposomes. Such a system might enhance our understanding of the protein network within the poliovirus replication complex and also the difference in the mechanism of negative- and positive-strand RNA synthesis.
Acknowledgments
We thank H. Pathak and C. Cameron for providing us with the cleavage site mutant of 3CDpro.
Footnotes
This work was supported by NIH grants AI015122 to E.W. and GM31986 to E.L.
References
- 1.Paul AV. Possible unifying mechanism of picornavirus genome replication. In: Semler BL, Wimmer E, editors. In molecular Biology of picornaviruses. ASM Press; Washington, DC: 2002. pp. 227–246. [Google Scholar]
- 2.Bienz K, Egger D, Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology. 1987;160:220–226. doi: 10.1016/0042-6822(87)90063-8. [DOI] [PubMed] [Google Scholar]
- 3.Egger D, Gosert R, Bienz K. Role of cellular structures in viral RNA replication. In: Semler BL, Wimmer E, editors. In Molecular Biology of Piocrnaviruses. ASM Press; Washington DC: 2002. pp. 20036–22904. [Google Scholar]
- 4.Schlegel A, Giddings TH, Jr, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS biology. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bienz K, Egger D, Pfister T, Troxler M. Structural and functional characterization of the poliovirus replication complex. J Virol. 1992;66:2740–2747. doi: 10.1128/jvi.66.5.2740-2747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrasco L, Guinea R, Irurzun A, Barco A. Effects of viral replication on cellular membrane metabolism and function. In: Semler BL, Wimmer E, editors. In molecular Biology of picornaviruses. ASM Press; Washington, DC: 2002. pp. 20036–22904. [Google Scholar]
- 8.Lyle JM, Clewell A, Richmond K, Richards OC, Hope DA, Schultz SC, Kirkegaard K. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. J Biol Chem. 2002;277:16324–16331. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- 9.Xiang W, Cuconati A, Hope D, Kirkegaard K, Wimmer E. Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J Virol. 1998;72:6732–6741. doi: 10.1128/jvi.72.8.6732-6741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Towner JS, Ho TV, Semler BL. Determinants of membrane association for poliovirus protein 3AB. J Biol Chem. 1996;271:26810–26818. doi: 10.1074/jbc.271.43.26810. [DOI] [PubMed] [Google Scholar]
- 11.Hope DA, Diamond SE, Kirkegaard K. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J Virol. 1997;71:9490–9498. doi: 10.1128/jvi.71.12.9490-9498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strauss DM, Glustrom LW, Wuttke DS. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. J Mol Biol. 2003;330:225–234. doi: 10.1016/s0022-2836(03)00577-1. [DOI] [PubMed] [Google Scholar]
- 13.Harris KS, Xiang W, Alexander L, Lane WS, Paul AV, Wimmer E. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J Biol Chem. 1994;269:27004–27014. [PubMed] [Google Scholar]
- 14.Paul AV, Cao X, Harris KS, Lama J, Wimmer E. Studies with poliovirus polymerase 3Dpol. Stimulation of poly(U) synthesis in vitro by purified poliovirus protein 3AB. J Biol Chem. 1994;269:29173–29181. [PubMed] [Google Scholar]
- 15.Xiang W, Harris KS, Alexander L, Wimmer E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J Virol. 1995;69:3658–3667. doi: 10.1128/jvi.69.6.3658-3667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lama J, Paul AV, Harris KS, Wimmer E. Properties of purified recombinant poliovirus protein 3aB as substrate for viral proteinases and as co-factor for RNA polymerase 3Dpol. J Biol Chem. 1994;269:66–70. [PubMed] [Google Scholar]
- 17.Plotch SJ, Palant O. Poliovirus protein 3AB forms a complex with and stimulates the activity of the viral RNA polymerase, 3Dpol. J Virol. 1995;69:7169–7179. doi: 10.1128/jvi.69.11.7169-7179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards OC, Ehrenfeld E. Effects of poliovirus 3AB protein on 3D polymerase-catalyzed reaction. J Biol Chem. 1998;273:12832–12840. doi: 10.1074/jbc.273.21.12832. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Wells V, Plotch SJ, DeStefano JJ. Primer-dependent synthesis by poliovirus RNA-dependent RNA polymerase (3D(pol)) Nucleic Acids Res. 2001;29:2715–2724. doi: 10.1093/nar/29.13.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molla A, Harris KS, Paul AV, Shin SH, Mugavero J, Wimmer E. Stimulation of poliovirus proteinase 3Cpro-related proteolysis by the genome-linked protein VPg and its precursor 3AB. J Biol Chem. 1994;269:27015–27020. [PubMed] [Google Scholar]
- 21.Datta U, Dasgupta A. Expression and subcellular localization of poliovirus VPg-precursor protein 3AB in eukaryotic cells: evidence for glycosylation in vitro. J Virol. 1994;68:4468–4477. doi: 10.1128/jvi.68.7.4468-4477.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choe SS, Kirkegaard K. Intracellular topology and epitope shielding of poliovirus 3A protein. J Virol. 2004;78:5973–5982. doi: 10.1128/JVI.78.11.5973-5982.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giachetti C, Hwang SS, Semler BL. cis-acting lesions targeted to the hydrophobic domain of a poliovirus membrane protein involved in RNA replication. J Virol. 1992;66:6045–6057. doi: 10.1128/jvi.66.10.6045-6057.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz BA, Vance LM. Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J Virol. 1996;70:4854–4857. doi: 10.1128/jvi.70.7.4854-4857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belov GA, Fogg MH, Ehrenfeld E. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J Virol. 2005;79:7207–7216. doi: 10.1128/JVI.79.11.7207-7216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodd DA, Giddings TH, Jr, Kirkegaard K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J Virol. 2001;75:8158–8165. doi: 10.1128/JVI.75.17.8158-8165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doedens JR, Giddings TH, Jr, Kirkegaard K. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J Virol. 1997;71:9054–9064. doi: 10.1128/jvi.71.12.9054-9064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. Embo J. 1995;14:894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teterina NL, Levenson E, Rinaudo MS, Egger D, Bienz K, Gorbalenya AE, Ehrenfeld E. Evidence for functional protein interactions required for poliovirus RNA replication. J Virol. 2006;80:5327–5337. doi: 10.1128/JVI.02684-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teterina NL, Rinaudo MS, Ehrenfeld E. Strand-specific RNA synthesis defects in a poliovirus with a mutation in protein 3A. J Virol. 2003;77:12679–12691. doi: 10.1128/JVI.77.23.12679-12691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul AV, van Boom JH, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 33.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray KE, Barton DJ. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol. 2003;77:4739–4750. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Ooij MJ, Vogt DA, Paul A, Castro C, Kuijpers J, van Kuppeveld FJ, Cameron CE, Wimmer E, Andino R, Melchers WJ. Structural and functional characterization of the coxsackievirus B3 CRE(2C): role of CRE(2C) in negative- and positive-strand RNA synthesis. J Gen Virol. 2006;87:103–113. doi: 10.1099/vir.0.81297-0. [DOI] [PubMed] [Google Scholar]
- 36.Fogg MH, Teterina NL, Ehrenfeld E. Membrane requirements for uridylylation of the poliovirus VPg protein and viral RNA synthesis in vitro. J Virol. 2003;77:11408–11416. doi: 10.1128/JVI.77.21.11408-11416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda N, Kuhn RJ, Yang CF, Takegami T, Wimmer E. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J Virol. 1986;60:43–53. doi: 10.1128/jvi.60.1.43-53.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Werf S, Bradley J, Wimmer E, Studier FW, Dunn JJ. Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc Natl Acad Sci U S A. 1986;83:2330–2334. doi: 10.1073/pnas.83.8.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herold J, Andino R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J Virol. 2000;74:6394–6400. doi: 10.1128/jvi.74.14.6394-6400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molla A, Paul AV, Wimmer E. Cell-free, de novo synthesis of poliovirus. Science. 1991;254:1647–1651. doi: 10.1126/science.1661029. [DOI] [PubMed] [Google Scholar]
- 41.Pata JD, Schultz SC, Kirkegaard K. Functional oligomerization of poliovirus RNA-dependent RNA polymerase. Rna. 1995;1:466–477. [PMC free article] [PubMed] [Google Scholar]
- 42.Pathak HB, Ghosh SK, Roberts AW, Sharma SD, Yoder JD, Arnold JJ, Gohara DW, Barton DJ, Paul AV, Cameron CE. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J Biol Chem. 2002;277:31551–31562. doi: 10.1074/jbc.M204408200. [DOI] [PubMed] [Google Scholar]
- 43.Ren J, Lew S, Wang J, London E. Control of the transmembrane orientation and interhelical interactions within membranes by hydrophobic helix length. Biochemistry. 1999;38:5905–5912. doi: 10.1021/bi982942a. [DOI] [PubMed] [Google Scholar]
- 44.Ren J, Lew S, Wang Z, London E. Transmembrane orientation of hydrophobic alpha-helices is regulated both by the relationship of helix length to bilayer thickness and by the cholesterol concentration. Biochemistry. 1997;36:10213–10220. doi: 10.1021/bi9709295. [DOI] [PubMed] [Google Scholar]
- 45.Caputo GA, London E. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic alpha-helices. Biochemistry. 2003;42:3275–3285. doi: 10.1021/bi026697d. [DOI] [PubMed] [Google Scholar]
- 46.Lew S, Ren J, London E. The effects of polar and/or ionizable residues in the core and flanking regions of hydrophobic helices on transmembrane conformation and oligomerization. Biochemistry. 2000;39:9632–9640. doi: 10.1021/bi000694o. [DOI] [PubMed] [Google Scholar]
- 47.Caputo GA, London E. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic alpha-helix locations within membranes. Biochemistry. 2003;42:3265–3274. doi: 10.1021/bi026696l. [DOI] [PubMed] [Google Scholar]
- 48.Towner JS, Brown DM, Nguyen JH, Semler BL. Functional conservation of the hydrophobic domain of polypeptide 3AB between human rhinovirus and poliovirus. Virology. 2003;314:432–442. doi: 10.1016/s0042-6822(03)00448-3. [DOI] [PubMed] [Google Scholar]
- 49.Caputo GA, London E. Position and ionization state of Asp in the core of membrane-inserted alpha helices control both the equilibrium between transmembrane and nontransmembrane helix topography and transmembrane helix positioning. Biochemistry. 2004;43:8794–8806. doi: 10.1021/bi049696p. [DOI] [PubMed] [Google Scholar]
- 50.Wimmer E, Hellen CU, Cao X. Genetics of poliovirus. Annu Rev Genet. 1993;27:353–436. doi: 10.1146/annurev.ge.27.120193.002033. [DOI] [PubMed] [Google Scholar]
- 51.Lewis BA, Engelman DM. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J Mol Biol. 1983;166:211–217. doi: 10.1016/s0022-2836(83)80007-2. [DOI] [PubMed] [Google Scholar]
- 52.Kuhn RJaWE. The Replication of Picornaviruses. In: Rowlands DJ, Mahy BWJ, Mayo M, editors. In The Molecular Biology of Positive Strand RNA Viruses. Academic Press; London: 1987. pp. 17–51. [Google Scholar]
- 53.Richards OC, Spagnolo JF, Lyle JM, Vleck SE, Kuchta RD, Kirkegaard K. Intramolecular and intermolecular uridylylation by poliovirus RNA-dependent RNA polymerase. J Virol. 2006;80:7405–7415. doi: 10.1128/JVI.02533-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt-Mende J, Bieck E, Hugle T, Penin F, Rice CM, Blum HE, Moradpour D. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J Biol Chem. 2001;276:44052–44063. doi: 10.1074/jbc.M103358200. [DOI] [PubMed] [Google Scholar]
- 55.Lama J, Sanz MA, Carrasco L. Genetic analysis of poliovirus protein 3A: characterization of a non-cytopathic mutant virus defective in killing Vero cells. J Gen Virol. 1998;79(Pt 8):1911–1921. doi: 10.1099/0022-1317-79-8-1911. [DOI] [PubMed] [Google Scholar]
- 56.Towner JS, Mazanet MM, Semler BL. Rescue of defective poliovirus RNA replication by 3AB-containing precursor polyproteins. J Virol. 1998;72:7191–7200. doi: 10.1128/jvi.72.9.7191-7200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dailey HA, Strittmatter P. Orientation of the carboxyl and NH2 termini of the membrane-binding segment of cytochrome b5 on the same side of phospholipid bilayers. J Biol Chem. 1981;256:3951–3955. [PubMed] [Google Scholar]
- 58.Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- 59.London E. Diphtheria toxin: membrane interaction and membrane translocation. Biochim Biophys Acta. 1992;1113:25–51. doi: 10.1016/0304-4157(92)90033-7. [DOI] [PubMed] [Google Scholar]
- 60.Zhang S, Finkelstein A, Collier RJ. Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc Natl Acad Sci U S A. 2004;101:16756–16761. doi: 10.1073/pnas.0405754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ivashkina N, Wolk B, Lohmann V, Bartenschlager R, Blum HE, Penin F, Moradpour D. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J Virol. 2002;76:13088–13093. doi: 10.1128/JVI.76.24.13088-13093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee H, Liu Y, Mejia E, Paul AV, Wimmer E. The C-terminal hydrophobic domain of hepatitis C virus RNA polymerase NS5B can be replaced with a heterologous domain of poliovirus protein 3A. J Virol. 2006;80:11343–11354. doi: 10.1128/JVI.02072-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Jong AS, Melchers WJ, Glaudemans DH, Willems PH, van Kuppeveld FJ. Mutational analysis of different regions in the coxsackievirus 2B protein: requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J Biol Chem. 2004;279:19924–19935. doi: 10.1074/jbc.M314094200. [DOI] [PubMed] [Google Scholar]
- 64.Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao X, Wimmer E. Intragenomic complementation of a 3AB mutant in dicistronic polioviruses. Virology. 1995;209:315–326. doi: 10.1006/viro.1995.1263. [DOI] [PubMed] [Google Scholar]
- 66.Boerner JE, Lyle JM, Daijogo S, Semler BL, Schultz SC, Kirkegaard K, Richards OC. Allosteric effects of ligands and mutations on poliovirus RNA-dependent RNA polymerase. J Virol. 2005;79:7803–7811. doi: 10.1128/JVI.79.12.7803-7811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yin J, Paul AV, Wimmer E, Rieder E. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J Virol. 2003;77:5152–5166. doi: 10.1128/JVI.77.9.5152-5166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]