

Abstract
During investigations of cyclization reactions between chiral allylsilanes and N-acyliminium ions, it was discovered that a suitably positioned benzyloxy group on the allylsilane component caused a reversal in the diastereoselectivity of these reactions relative to that normally observed with alkyl-substituted allylsilanes. This effect was subsequently observed in two other reaction types. Investigations into this effect led to the proposal of product formation through thermodynamic control facilitated by neighboring group interactions with a transient cationic species. This hypothesis was experimentally supported by the isolation of an intermediate in the proposed mechanistic pathway.
The reactions of allylsilanes with electrophilic species, in particular iminium ions, have become standard means for the formation of carbon–carbon bonds.1-4 This strategy has been enriched by the introduction of chiral allylsilane derivatives, which can afford products with a high degree of stereochemical control.5-18 Still this strategy has considerable untapped promise, especially when combined with non-standard methods of generating the electrophilic component.
Previous work in this laboratory has shown that reactive iminium ions can be prepared through the intramolecular reactions of an aryl imine bearing an NHBoc substituent in the ortho position and that this reaction affords a route to interesting ring systems.19,20 As an extension of this methodology, we wished to examine chiral silanes in this reaction, with the overall goal being the synthesis of enantiomerically pure products. In this work, a most unexpected outcome was observed: the diastereoselectivity of the reactions can be reversed by the placement of a benzyloxy group γ to the silicon moiety. We have ascertained that this observation holds in at least two other reactions of allylsilanes. These observations will be discussed in the context of kinetic v. thermodynamic control of the stereochemistry of a cationic intermediate in the reaction through neighboring group interactions.
Results
Preparation of Chiral Allylsilanes
Initially, chiral alkenyl silanes 1 and 2, each containing a terminal azido group were prepared in enantiomerically pure form. Thus, enantiomerically pure (S)-1,2,4-butanetriol derived from malic acid was converted into allylic alcohol 3 by a standard series of functional group transformations and protecting group manipulation steps (Scheme 1; see Supporting Information for details). Conversion of this allylic alcohol to alkenyl silane 5a was accomplished by the 1,3-transpositive bis-silylation protocol developed by Ito.21-23 This process has been shown to proceed with syn relative stereocontrol and with high retention of enantiomeric purity. Conversion of the product 5a to azide 1 was accomplished through straightforward transformations.
Scheme 1.

We synthesized chiral allylsilanes containing more highly functionalized side chains as shown in Scheme 2. The oxygenated allylsilane 2, was originally envisioned as an intermediate en route to martinelline.20 Allylic alcohol 8 was furnished through Wipf's zirconium-mediated coupling24 between a suitably substituted alkyne and aldehyde 7. A Sharpless asymmetric kinetic resolution was used to prepare chiral allylic alcohol (R)-8, which was then subjected to the Ito protocol.25 Although exemplified for the O-benzyl derivative 2, this route was also adapted to the synthesis of several related allylsilanes chosen in response to observations made on the alkylation chemistry of 1 v. 2 (see below).
Scheme 2.

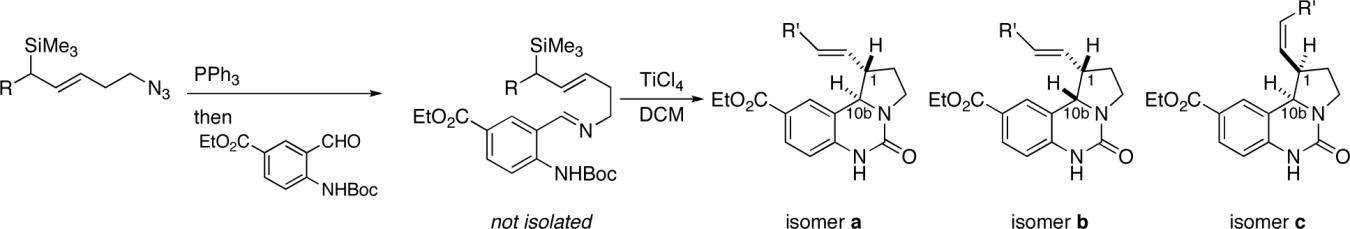
Once prepared, the azides (1 and 2) were separately ligated to ethyl 4-(N-tert-butoxycarbonylamino)-3-formylbenzoate20 to afford imines that were then treated with TiCl4 (Scheme 3). This generated an iminium ion by attack of the imine nitrogen on the ortho Boc group (possibly via the isocyanate intermediate shown). The electrophilic species thus generated smoothly reacted with the appended allylsilane species to afford a mixture of stereoisomeric products (Table 1). Interestingly, a benzylic allylsilane (i.e., 1 in which the terminal ethyl group is replaced by phenyl; not shown) did not undergo cyclization in several attempts. When the chiral allylsilanes 1 and 2 were incorporated into this scheme (through imines 10 and 11), the product ureas were isolated in good yields and as a mixture of inseparable stereoisomers. In the reaction of the terminal benzyloxy-substituted example 11, the benzyl group was lost during the course of cyclization, affording alcohols 13a–c. The removal of benzyl groups under Lewis acid conditions has been previously observed.26
Scheme 3.

Table 1.
TiCl4-promoted cyclizations of allylsilane-tethered aryl imines.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| isomer ratio (%)b |
||||||||
| entry | imine | product | yield (%)a | a | b | c | cis/trans ratioc | overall E/Z ratiod |
| 1 | (S)-10, R = CH2CH3 | 12, R′ = CH2CH3 | 85 | 15 | 66 | 19 | 5.7 : 1 | 81 : 19 |
| 2 | (S)-11, R = CH2CH2OBn | 13, R′ = CH2CH2OH | 80 | 87 | 10 | 3 | 1 : 6.7 | 97 : 3 |
| 3 | (±)-14, R = CH2(CH2)3OBne | 15, R′ = CH2(CH2)3OH | 73 | 20 | 68 | 12 | 4.0 : 1 | 88 : 12 |
| 4 | (±)-16, R = CH2CH2OTBDPSe | 13, R′ = CH2CH2OH | 64 | 53 | 42 | 5 | 1 : 1.1 | 95 : 5 |
| 5f | 17, R = H | 18, R′ = H | 80g | 15 | 85 | — | 5.7 : 1 | — |
Overall yield of isolated product from starting azide (2 steps).
Determined by 1H NMR integration and normalized to 100%.
Refers to the relative stereochemistry of H1 and H10b; ratio of (b+c)/a.
Double bond isomers; ratio of (a+b)/c.
Prepared in analogy to compound 2 (see Supporting Information).
Reference 20, included for comparison.
Yield based on purified imine.
The stereoselectivity of each reaction was determined by 1H NMR spectroscopy of the crude reaction mixture. In each of the reactions, three of the four possible diastereomers were readily apparent in the NMR spectrum and could be assigned by coupling constant analysis to isomers a, b, and c as shown in Table 1. Details of all structural assignments are provided in the Supporting Information. We cannot rule out the formation of small amounts of the Z-olefin analog of isomer a, but it was not detected spectroscopically. Absolute configurations were determined for both cases by preparing derivatives of 12b and 13a, and subjecting the derivatives to anomalous dispersion X-ray analysis (Scheme 4). Derivatization of 12b was effected through an oxidative cleavage/reduction sequence of the product mixture 12a–c followed by chromatographic separation of the resulting diastereomeric mixture of alcohols. Subsequent esterification of the major diastereomer 19 (derived from 12b,c) gave the corresponding p-bromobenzoyl ester. Preparation of the p-bromobenzoyl derivative of 13a (isolated from the product mixture containing 13a–c by crystallization) required only esterification of the terminal alcohol (Scheme 4(b)). Chiral chromatography of product 12 derivatives and of purified 13a verified that the trans isomers were obtained in high enantiomeric purity (12a in 97:3 er; 13a in ≥99:1 er). However, the corresponding enantiomeric ratios for the cis isomers were not determined due to incomplete resolution.
Scheme 4.

The most striking observation in these experiments concerned the diastereoselectivity of the reactions, specifically the cis/trans ratio with respect to H1 and H10b. Thus, the ethyl-substituted silane, 1, reacted (via imine 10) to afford products 12, in which the alkenyl side chain emerged cis to the bulky aryl substituent in an overall ratio of 5.7:1 (counting both E and Z double bond isomers together). This ratio is similar to that observed in the unsubstituted case previously reported (shown in Table 1, entry 5, for comparison).20 In contrast, imine 11, containing a benzyloxy group at the terminus, reacted to afford analogs 13 in which the trans isomer predominated (1:6.7 cis/trans). This remarkable stereochemical dependence on a remote substituent merited additional attention.
We hypothesized a change in mechanism involving the interaction of the ether oxygen with an intermediate along the reaction pathway and synthesized additional examples to address this point. Thus, compound 14, in which the ether was moved two methylene units further from the allylsilane moiety, was synthesized in racemic form and brought into the reaction manifold (Table 1, entry 3). This compound gave results similar to those obtained using the nonfunctionalized examples, giving a 4:1 ratio favoring the cis isomer of products 15. To determine whether the Lewis basicity of the oxygen in benzyloxy example 2 was a factor, the analogue (±)-16 containing a bulky trialkylsilyl group, known to limit the coordinating ability of the oxygen atom,27,28 was tested (entry 4). In this case, a nearly 1:1 mixture of isomers was obtained, strongly suggesting an involvement of the oxygen atom lone pairs in this phenomenon. Finally, a comparison of the overall product E/Z olefin ratios shows that the highest E-olefin selectivity occurred with the terminally benzyloxy-substituted allylsilane (entry 2), while the lowest E-selectivity occurred in the absence of a terminal ether substituent (entry 1).
Extension to Other Cyclization Types
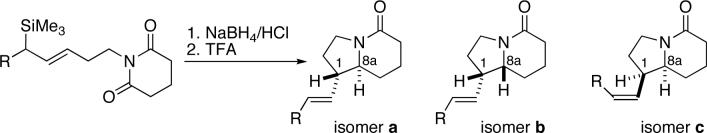
In an effort to explore the generality of the influence of a terminal benzyloxy substituent on the diastereoselectivity of intramolecular allylsilane reactions, we investigated two additional reaction types. Thus, a series of Speckamp/Royer acyliminium cyclization substrates was prepared from the corresponding alcohols by a Mitsunobu/reduction sequence (Supporting Information).29-31 Reaction under protic acid promotion gave good yields of the cyclization products, again isolated as isomeric mixtures (Table 2).
Table 2.
TFA-promoted cyclizations of hydroxy lactams.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| isomer ratio (%)b |
||||||||
| entry | imide | product | yield (%)a | a | b | c | cis/trans ratioc | overall E/Z ratiod |
| 1 | (S)-21, R = CH2CH3 | 22 | 92 | 8 | 28 | 64 | 12 : 1 | 36 : 64 |
| 2 | (S)-23, R = CH2CH2OBn | 24 | 85 | 83 | 5 | 12 | 1 : 5 | 88 : 12 |
| 3 | (±)-25, R = CH2(CH2)3OBn | 26 | 98 | 19 | 25 | 56 | 4.2 : 1 | 44 : 56 |
| 4 | (±)-27, R = CH2CH2OTBDPS | 28 | 74 | 33 | 19 | 48 | 2.0 : 1 | 52 : 48 |
| 5 | 29, R = H | 30 | 71 | 10 | 90 | — | 9 : 1 | — |
Overall isolated yield.
Determined by 1H NMR integration and normalized to 100%.
Refers to the relative stereochemistry of H1 and H8a; ratio of (b + c)/a.
Double bond isomers; ratio of (a+b)/c.
We again observed three of the four possible isomers in the 1H NMR of the product mixtures, which were assigned by 2D NMR experiments and coupling constant analysis as isomers a, b, and c (cis/trans descriptors refer to the relative stereochemistry of H1 and H8a). The diastereomeric trends observed in these reactions paralleled those of the o-NHBoc arylimine cyclizations (cf. Table 1). Most notably, reversal of diastereoselectivity was again observed with the benzyloxy-substituted allylsilane, which afforded a 1:5 cis/trans diastereomeric ratio (entry 2); all other examples provided cis products as major isomers. The highest product E-olefin selectivity was also observed in the presence of a γ-benzyloxy substituent (entry 2). Absolute configurations of 22c and 24a were determined by anomalous dispersion X-ray analysis of the corresponding p-bromobenzoyl derivatives. In general, p-bromobenzoyl derivatives were prepared through an oxidative cleavage/reduction sequence, followed by esterification of the resulting alcohol (Scheme 5 and Supporting Information). Single isomers for X-ray analysis were obtained by crystallization of the alcohol mixture (affording 32) or after esterification (affording 31c). Alcohol 32 was converted to 32a to obtain heavy atom-containing crystals; oddly, the double addition product shown was obtained in this step (probably during an acidic workup). Chiral chromatography of derivatized purified product mixtures (22 and 24) again showed that the trans products were formed in high enantiomeric purity (22b in 93:7 er; 24a in 97:3 er). The corresponding determinations for the cis isomers could not be made due to incomplete resolution.
Scheme 5.

Allylsilanes also react with tethered oxenium ions (via initial acetal formation between an alcohol moiety on the allylsilane and an aldehyde) to afford substituted tetrahydrofurans.17,32 Accordingly, we reacted racemic versions of 1 and 2 with benzaldehyde under TMSOTf promotion at low temperature (Scheme 6). As before, remarkably different diastereomeric outcomes were realized, with compound 33 being obtained with completely cis stereoselectivity at C-2/C-3 and 34 arising from a strongly 2,3-trans-biased reaction. The stereochemical assignments for the cyclized products 33 and 34 were made on the basis of NOE difference spectra.
Scheme 6.

Discussion
The reaction shown in Scheme 7 is for tricyclic urea formation under TiCl4 promotion, but analogous analyses may be extended to the other reaction types. If one assumes kinetic control, anti addition relative to the C–Si bond, and coordinated C–C bond formation with loss of SiMe3, product types a–c could be rationalized as shown. The predominant isomers for unfunctionalized side chains (i.e., where X = alkyl or H) have cis stereochemistry across the pyrrolidine or tetrahydrofuran ring in all three series (major compounds 12b, 18b, 22c, 30b, and 33). These products are consistent with the chairlike transitions states shown. Differences in double bond geometry are known to be coupled with the face selectivity of the allylsilane component (cf. products a and c),5 which was confirmed in the four cases where absolute stereochemistry was determined. The E/Z isomer ratios in a given series (i.e., in the competition between the two chairs leading to isomers b and c) also depend on the minimization of non-bonded interactions in the specific reaction being studied. Differences observed in the E/Z ratios for the examples shown in Tables 1 vs. 2 likely depend on the interplay between the steric demands of the substrates and the timing of the transition state (early or late). The only observed trans isomer (relative to the five-membered ring being formed) requires a boat-like transition state as shown for series a.
Scheme 7.

The observation that a particularly situated basic oxygen could reverse the stereoselectivity of these reactions was unexpected. Other researchers have shown that β-silyl cation intermediates in allylsilane reactions can be intercepted and lead to alternative reaction product types, as occurs in formal [2+2] and [3+2] cycloadditions with aldehydes, imines, or isocyanates.33-48 In this case, we propose that the presence of a suitably positioned basic ether oxygen either changes the mechanism of the reaction or otherwise participates in the transition state to effect a change in the diastereoselectivity. In considering these options, we noted that the presence of the γ ether turned the reaction in favor of the thermodynamically more favored isomer in all three reaction types; i.e., the heterocycles containing an exo side chain in compounds 13 and 24 and the trans isomer of tetrahydrofuran 34. One way that this could happen is suggested in Scheme 8.
Scheme 8.

In reactions with the γ-benzyloxy-substituted allylsilane, the potential for interception of the emerging β-silyl cation by the alkoxy group, forming a five-membered oxonium ion arises. The participation of ether groups with cations49 has been recently invoked by Angle and coworkers in their annulation chemistry37,38 and by Molander in explaining long-range stereoselectivity.50 In the present case, such participation could lead to a kinetic distribution of products giving rise to the same major product as observed in the non-functionalized allylsilane reactions. However, the fact that trans-fused, rather than cis-fused, products predominate for reactions with a γ-benzyloxy-substituted allylsilane suggests that an additional element may be operative in this process. A likely explanation is that the oxonium species shown in Scheme 8 (and the analogous intermediates that would be formed in the other reaction types) are sufficiently stable to allow equilibrium between the cis- and trans-fused oxonium ions and probably the analogous carbocations, and that the thermodynamically favored trans products therefore result. This additionally assumes that SiR3 elimination from the initial addition step (as depicted in Scheme 7) or from the oxonium-containing intermediates (in which the SiR3 group cannot readily achieve overlap with the ether oxygen) is slow enough to allow for equilibrium of the cation pool.51,52 Moreover, bond rotation would more likely occur before elimination of the silyl group, which would explain the higher amounts of E olefin isomers in two of the three reaction types investigated.
On the basis of the proposed mechanistic rationale, the cis products predominated, or reactions were poorly selective, in cases in which the allylsilane contained structural elements inconsistent with the formation of the cyclic oxonium intermediate. Thus, the presence of the bulky OTBDPS group attenuated the ability of these substituents to engage in neighboring group participation relative to the OBn substituent, resulting in a nearly 1:1 mixture of cis and trans isomers in one case (Table 1, entry 4), and the predominant formation of the cis isomer in another (Table 2, entry 4). When the benzyloxy substituent is moved two additional methylene units away from the silyl group (ε-OBn), the cis products predominate as a result of the decreased ability of the more distal ether oxygen to engage in the formation of the cyclic oxonium species through interaction with the emerging β carbocation, as this would require the formation of a less favorable 7-membered ring (cf. entry 3 in Table 1 and Table 2).
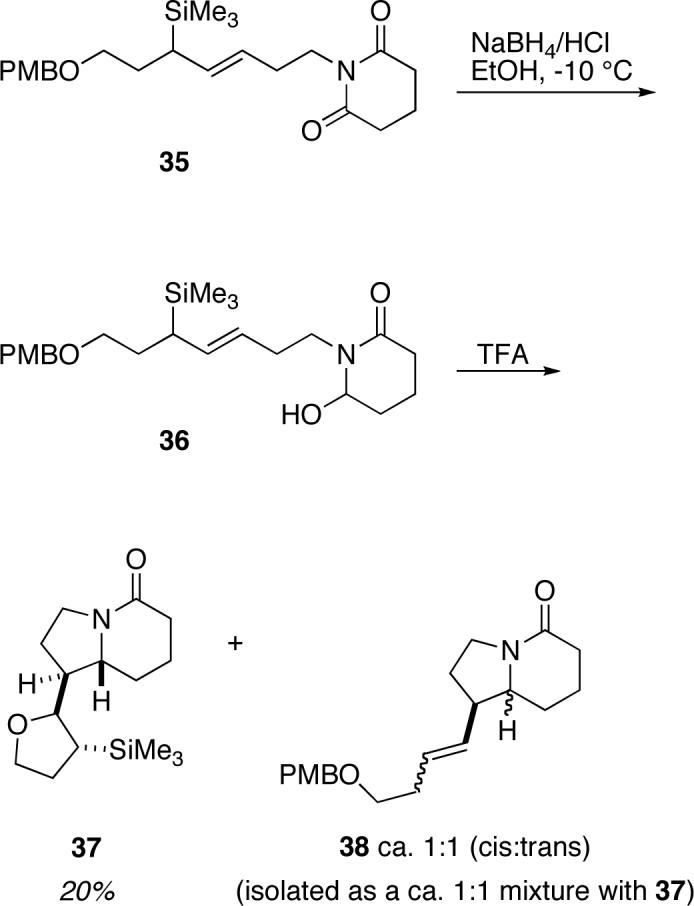
With the aim of providing experimental support for the proposed mechanism of diastereoselection through ether oxygen participation, we considered substrates that would allow for the isolation of a silyl-substituted tetrahydrofuran-containing product (formed via the oxonium intermediate proposed in Scheme 8). We reasoned that this might be accomplished by using an acid-labile p-methoxy benzyl (PMB) ether-substituted allylsilane in the N-acyliminium cyclization. Thus, incorporation of a PMB ether-substituted allylsilane into the cyclization substrate 35 followed by acid-promoted cyclization gave a product mixture of the cyclic ether 37 and the lactam products 38, from which cyclic ether-containing product 37 could be isolated in 20% yield (along with a mixture of 37 contaminated with additional 38; Scheme 9). Unfortunately, it was not possible to unambiguously determine the relative stereochemistry of 37 by NMR. The stereostructure shown is hypothesized on the basis of the proposal shown in Scheme 8. The fact that compound 37 was observed in this reaction lends support for the proposed intermediacy of the cyclic oxonium species arising from neighboring group participation.
Scheme 9.
Summary
These studies show that a reversal in the diastereoselectivity in intramolecular reactions of chiral allylsilanes can be effected by a γ-benzyloxy substituent. The postulated mechanism involves interaction of the terminal ether oxygen with the incipient β-silyl carbocation, ultimately forming the thermodynamically favored trans products. Experiments to determine the generality of this phenomenon are ongoing.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM-49093). The authors thank the referees for their careful review and useful suggestions as well.
Footnotes
Supporting Information.\ Details of starting material preparations, experimental details, determination of isomer ratios and enantiomeric purities, and copies of 1H and 13C spectra for new compounds. This material is available free of charge via the Internet at pubs.acs.org.
References
- 1.Chabaud L, James P, Landais Y. Eur. J. Org. Chem. 2004:3173–3199. [Google Scholar]
- 2.Langkopf E, Schinzer D. Chem. Rev. 1995;95:1375–1408. [Google Scholar]
- 3.Masse CE, Panek JS. Chem. Rev. 1995;95:1293–1316. [Google Scholar]
- 4.Fleming I. Org. React. 1989;37:57–575. [Google Scholar]
- 5.Fleming I, Barbero A, Walter D. Chem. Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi T, Konishi M, Kumada M. J. Am. Chem. Soc. 1982;104:4962–4963. [Google Scholar]
- 7.Fleming I, Kindon ND, Sarkar AK. Tetrahedron Lett. 1987;28:5921–5924. [Google Scholar]
- 8.Coppi L, Asessandro M, Mauizio T. Tetrahedron Lett. 1987;28:969–972. [Google Scholar]
- 9.Ward RA, Procter G. Tetrahedron Lett. 1992;33:3363–3366. [Google Scholar]
- 10.Panek JS, Yang M, Xu F. J. Org. Chem. 1992;57:5790–5792. [Google Scholar]
- 11.Dias LC, Meira PRR. Synlett. 2000:37–40. [Google Scholar]
- 12.Yamaguchi R, Tanaka M, Matsuda T, Okano T, Nagura T, Fujita K. Tetrahedron Lett. 2002;43:8871–8874. [Google Scholar]
- 13.Mikami K, Maeda T, Kishi N, Nakai T. Tetrahedron Lett. 1984;25:5151–5154. [Google Scholar]
- 14.Hong CY, Kado N, Overman LE. J. Am. Chem. Soc. 1993;115:11028–11029. [Google Scholar]
- 15.Denmark SE, Almstead NG. J. Org. Chem. 1994;59:5130–5132. [Google Scholar]
- 16.Masse CE, Dakin LA, Knight BS, Panek JS. J. Org. Chem. 1997;62:9335–9338. [Google Scholar]
- 17.Suginome M, Iwanami T, Ito Y. J. Org. Chem. 1998;63:6096–6097. doi: 10.1021/jo981173y. [DOI] [PubMed] [Google Scholar]
- 18.Huang H, Spande TF, Panek JS. J. Am. Chem. Soc. 2003;125:626–627. doi: 10.1021/ja028937i. [DOI] [PubMed] [Google Scholar]
- 19.Frank KE, Aubé J. Tetrahedron Lett. 1998;39:7239–7242. [Google Scholar]
- 20.Frank KE, Aubé J. J. Org. Chem. 2000;65:655–666. doi: 10.1021/jo990843c. [DOI] [PubMed] [Google Scholar]
- 21.Murakami M, Andersson P, Suginome M, Ito Y. J. Am. Chem. Soc. 1991;113:3987–3988. [Google Scholar]
- 22.Suginome M, Matsumoto A, Nagata K, Ito Y. J. Organomet. Chem. 1995;499:C1–C3. [Google Scholar]
- 23.Suginome M, Matsumoto A, Ito Y. J. Am. Chem. Soc. 1996;118:3061–3062. [Google Scholar]
- 24.Wipf P, Xu W. Org. Synth. 1997;74:205–211. [Google Scholar]
- 25.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765–5780. [Google Scholar]
- 26.Hori H, Nishida Y, Ohrui H, Meguro H. J. Org. Chem. 1989;54:1346–1353. [Google Scholar]
- 27.Frye SV, Eliel EL. Tetrahedron Lett. 1986;27:3223–3226. [Google Scholar]
- 28.Chen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1992;114:1778–1784. [Google Scholar]
- 29.Hiemstra H, Sno MHAM, Vijn RJ, Speckamp WN. J. Org. Chem. 1985;50:4014–4020. [Google Scholar]
- 30.Hubert JC, Steege W, Speckamp WN. Synth. Commun. 1971;1:103–109. [Google Scholar]
- 31.Hubert JC, Wijnberg JBPA, Speckamp WN. Tetrahedron. 1975;31:1437–1441. [Google Scholar]
- 32.Mohr P. Tetrahedron Lett. 1993;34:6251–6254. [Google Scholar]
- 33.Peng Z-H, Woerpel KA. Org. Lett. 2000;2:1379–1381. doi: 10.1021/ol005676d. [DOI] [PubMed] [Google Scholar]
- 34.Akiyama T, Yamanaka M. Synlett. 1996:1095–1096. [Google Scholar]
- 35.Schinzer D, Panke G. J. Org. Chem. 1996;61:4496–4497. doi: 10.1021/jo952207u. [DOI] [PubMed] [Google Scholar]
- 36.Angle SR, El-Said NA. J. Am. Chem. Soc. 2002;124:3608–3613. doi: 10.1021/ja012193b. [DOI] [PubMed] [Google Scholar]
- 37.Angle SR, Belanger NA, El-Said NA. J. Org. Chem. 2002;67:7699–7705. doi: 10.1021/jo0203342. [DOI] [PubMed] [Google Scholar]
- 38.Osumi H, Sugimura H. Tetrahedron Lett. 1995;36:5789–5792. [Google Scholar]
- 39.Banuls V, Escudier J-M. Tetrahedron Lett. 1999;55:5831–5838. [Google Scholar]
- 40.Micalizio GC, Roush WR. Org. Lett. 2000;2:461–464. doi: 10.1021/ol9913082. [DOI] [PubMed] [Google Scholar]
- 41.Nair V, Rajesh C, Dhanya R, Rath NP. Tetrahedron Lett. 2002;43:5349–5351. [Google Scholar]
- 42.Panek JS, Hu T. J. Org. Chem. 1997;62:4914–4915. [Google Scholar]
- 43.Peng Z-H, Woerpel KA. Org. Lett. 2001;3:675–678. doi: 10.1021/ol0069960. [DOI] [PubMed] [Google Scholar]
- 44.Sugita Y, Kumura Y, Yokoe I. Tetrahedron Lett. 1999;40:5877–5880. [Google Scholar]
- 45.Panek JS, Yang M. J. Am. Chem. Soc. 1991;113:9868–9870. [Google Scholar]
- 46.Panek JS, Su Q. J. Am. Chem. Soc. 2004;126:2425–2430. doi: 10.1021/ja037957x. [DOI] [PubMed] [Google Scholar]
- 47.Isaka M, Willard PG, Nakamura E. Bull. Chem. Soc. Jpn. 1999;72:2115–2116. [Google Scholar]
- 48.Roberson CW, Woerpel KA. J. Am. Chem. Soc. 2002;124:11342–11348. doi: 10.1021/ja012152f. [DOI] [PubMed] [Google Scholar]
- 49.Winstein S, Allred E, Heck R, Glick R. Tetrahedron. 1958;3:1–13. [Google Scholar]
- 50.Molander GA, Haar JP., Jr. J. Am. Chem. Soc. 1993;115:40–9. [Google Scholar]
- 51.Yoshida J, Suga S, Suzuki S, Kinomura N, Yamamoto A, Fujiwara K. J. Am. Chem. Soc. 1999;121:9546–9549. [Google Scholar]
- 52.Yoshida J, Suga S. Chem. Eur. J. 2002;8:2650–2658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.