

Abstract
The present work reveals that four proteins, MceCDIJ, encoded by the MccE492 gene cluster are responsible for the remarkable post-translational tailoring of Microcin E492 (MccE492), an 84-residue protein toxin secreted by Klebsiella pneumonaie RYC492 that targets neighboring gram-negative species. This modification results in attachment of a linearized and monoglycosylated derivative of enterobactin, a nonribosomal peptide and iron scavenger (siderophore), to the MccE492m C-terminus. MceC and MceD derivatize enterobactin by C-glycosylation at the C5 position of a N-(2,3-dihydroxybenzoyl) serine (DHB-Ser) moiety and regiospecific hydrolysis of an ester linkage in the trilactone scaffold, respectively. MceI and MceJ form a protein complex that attaches C-glycosylated enterobactins to the C-terminal serine residue of both aC10 model peptide and full-length MccE492. In the enzymatic product, the terminal serine residue is covalently attached to the C4′ oxygen of the glucose moiety. Non-enzymatic and base-catalyzed migration of the peptide to the C6′ position affords the C6′ glycosyl ester linkage observed in the mature toxin, MccE492m, isolated from bacterial cultures.
Introduction
Microcins are low molecular weight (< 10 kDa) ribosomal peptide toxins that enterobacteria produce to inhibit the growth of competing bacteria and aid in host colonization.1 Many microcin peptides exhibit unusual post-translational modifications that contribute to their bactericidal effects.2 The microcin B17 (MccB17) precursor peptide undergoes tandem heterocyclization, which generates thiazole and oxazole rings in the peptide backbone and confers activity against DNA gyrase.3,4 Installation of adenosine-5′-monophosphate via a phosphoramidate bond at the C-terminus of the microcin C7 (MccC7) precursor forms an inhibitor of aspartyl-tRNA-synthethase.5 The post-translational modification of microcin E492 (MccE492) creates a ribosomal peptide -nonribosomal peptide conjugate with a glucose bridge.6 Together, these two scaffolds afford a toxin targeted to certain species of gram-negative bacteria. The biosynthetic logic and remarkable post-translational maturation of MccE492m (where m is for modified) are subjects of this work.
MccE492m, secreted by Klebsiella pneumonaie RYC492 and active against several species of Enterobacteriaceae, is an 84-residue protein toxin exhibiting a striking C-terminal post-translational modification comprised of a C-glycosylated and linearized enterobactin moiety (Figure 1). Enterobactin (Ent, Figure 1), a cyclic trimer of N-2,3-dihydroxybenzoyl serine (DHB-Ser), is a prototypic siderophore produced by gram-negative enterobacteria for iron acquisition in the host environment.7 It is also the metabolic precursor to the recently discovered salmochelins,8,9 C-glycosylated and linearized enterobactin congeners that may confer more efficient iron acquisition in vivo because of their increased hydrophilicity10 and ability to evade the mammalian protein lipocalin 2,11,12 a component of the innate immune response.13–15 Post-translational modification of MccE492m enables it to target virulent bacteria that express siderophore uptake pumps (FepA, Cir, Fiu) on the cell surface/outer membrane,16,17 making it a Trojan horse toxin. Following recognition, pore formation, membrane depolarization, and cell death occur.18–20 The mechanistic details of these processes are unclear, but recent work indicates that the inner membrane protein components of the mannose permease are required for MccE492m toxicity.21
Figure 1.

Structures of enterobactin (Ent), linearized enterobactin (lin-Ent), monoglycosylated enterobactin (MGE), linearized MGE (lin-MGE) and a truncated depiction of microcin E492m (MccE492m) showing the last eleven amino acids of the microcin peptide. Diglycosylated (DGE) and triglycosylated enterobactin (TGE) have glucose moieties attached to the C5 positions of either two or three DHB-Ser units, respectively.
The MccE492 gene duster (Figure 2) encodes ten genes (mceABCDEFGJIH) that are necessary and sufficient for MccE492m production in a strain that produces enterobactin.22–25 mceA is the structural gene for a 99- or 103-residue precursor protein (MceA) of ribosomal origin. The precursor protein gets cleaved at amino acid 15 or 19, which yields the active 84-residue MccE492 peptide.23 Microcin gene clusters often produce immunity proteins and the mceB gene product is a 95-residue protein that confers resistance to MccE492(m) by an unknown mechanism.23 Knock-out studies have implicated mceCDIJ in the maturation of MccE492m.24 The mceG and mceH genes encode an ABC transporter and accessory protein, respectively, that are necessary for microcin export. The functions of mceE and mceF are as yet undefined, but the mceF gene product may also be involved in MccE492(m) export.24
Figure 2.
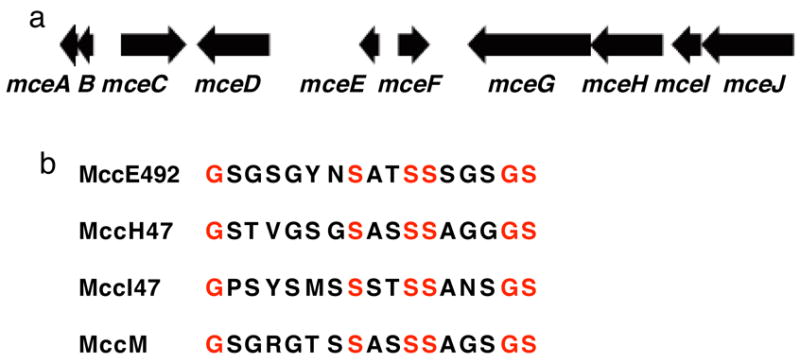
(a) The MccE492 gene cluster: mceA, 312 bp; mceB, 288 bp; mceC, 1113 bp; mceD, 1245 bp; mceE, 345 bp; mceF, 540 bp; mceG, 2097 bp; mceH, 1242 bp; mceI, 492 bp; mceJ, 1575 bp (ref. 1). (b) Amino acid sequences for the serine-rich C-termini of MccE492, MccH47, MccI47 and MccM. The sequence of the C-terminus of MccE492 was determined experimentally. MccH47, MccI47 and MccM have not been isolated and their C-terminal sequences were deduced from the respective genes.
In this work, we examine several facets of MccE492m biosynthesis. We focus on four proteins, MceCDIJ, and demonstrate that they catalyze the series of post-translational modifications required for MccE492m formation. We were led to MceC and MceD because the MccE492 gene cluster shares some commonality with the iroA locus,26–28 which encodes proteins required for salmochelin synthesis (IroB, IroD, IroE) and transport (IroC, IroN). MceC and MceD are IroB (C-glycosyItransferase)29 and IroD/IroE (esterase)30 homologs, respectively. MceI is a homolog of HlyC, the acyltransferase required for hemolysin toxin activation,31 and is essential for MccE492m formation. We therefore presumed that it is required for linking the siderophore to the MccE492 C-terminus. Genetic studies have also shown that MceJ is necessary for MccE492m maturation.24,25 The function of MceJ was difficult to predict because it has no known homologs other than its counterpart in the microcin H47 gene cluster (MchC),32 but we speculated that it is also necessary for siderophore attachment. Herein we report the over-production and purification of MceCDIJ, establish that MceC and MceD are a C-glycosyltransferase and esterase, respectively, and show that MceIJ are sufficient to derivatize both the C-terminal decapeptide of MccE492 and native MccE492 with monoglycosylated enterobactins in vitro. We also demonstrate that the MceIJ product exhibits the decapeptide attached to monoglycosylated enterobactin at the C4′ oxygen via an ester linkage. This ester undergoes non-enzymatic and base-catalyzed rearrangement to the C6′ glycosyl ester, which is the observed connectivity in MccE492m isolated from bacterial cultures.
Experimental
Materials and Methods
Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA.). All chemicals were purchased from Sigma-Aldrich. Analytical and preparative HPLC were performed on a Beckman System Gold (Beckman Coulter) instrument. A Vydac (Hesperia, CA) small pore C18 (4.6 × 50 mm) column at 4 mL/min was generally employed for analytical HPLC assays. A Vydac small pore C18 (4.6 × 250 mm) column at 1 mL/min was employed for the MceIJ/lin-MGE assays and some MceIJ/MGE assays. A Vydac C18 (22 × 250 mm) column at 10 mL/min was used for preparative HPLC. A Shimadzu LCMS-QP8000a outfitted with a Higgins Analytical (Mountain View, CA) Sprite Targa C18 column (2.1 × 20 mm) at 0.5 mL/min was employed for LCMS. All calculated and found m/z ratios are the monoisotopic values. Absorption at 220 and 316 nm was monitored for all liquid chromatography and 316 nm absorption traces are presented in all Figures unless otherwise noted. MALDI-TOF spectra were collected on an Applied Biosystems Voyager mass spectrometer. With the exception of the NOESY spectra that were acquired as described below in the “Structural Elucidation of MceIJ Products” section, NMR spectra were collected on a Varian 600 MHz instrument and referenced to internal solvent peaks. A Cary 50 Bio scanning spectrometer was used to record absorption spectra. The concentrations of the siderophore stock solutions were verified using the reported33 extinction coefficients for enterobactin (316 nm, 9500 M−1cm−1), linear enterobactin (315 nm, 9700 M−1cm−1) and [Fe(enterobactin)]3” (338 nm, 15100 M−1cm−1) with the assumption that the glucose moieties would have no effect on siderophore absorption. Siderophore stock solutions were generally prepared in DMSO (~3 to ~10 mM), divided into aliquots and stored at −20 °C. The Bradford assay was employed to determine protein concentration.34 The decapeptide, SATSSSGSGS, was prepared by standard Fmoc solid phase peptide synthesis by staff at the Biopolymers Facility at Harvard Medical School. Its purity and identity were verified by analytical HPLC and LCMS. Concentrations of peptide stock solutions were verified by quantitative amino acid analysis (Dana Farber Cancer Institute). Details for substrate preparations are included in the Supporting Information section.
Cloning, Overexpression and Purification of MceD
The mceD gene was PCR amplified from pJEMIS using the forward primer 5′-ggaattccatatgccatttatgaggaatcatccatc-3′ (NdeI restriction site underlined) and reverse primers 5′-gatcctcgagttacacatctgataatccatcgataattg-3′ (pET-28b, XhoI restriction site underlined) and 5′-gatcctcgagcacatctgataatccatcgataattg-3′ (pET-29a, XhoI restriction site underlined). PCR reactions were performed with Pfu Turbo DNA polymerase (Stratagene) and the amplified gene sequences were digested with NdeI and XhoI (New England Biolabs), ligated into pET-28b and pET-29a expression vectors using T4 DNA ligase (New England Biolabs) and transformed into E. coli TOP10 cells (Stratagene). The identities of the resulting pET-28b (N-His) and pET-29a (C-His) constructs were confirmed by DNA sequencing. A discrepancy (G to A) at position 5378 with the published sequence was observed for the pET28b-mceD construct. Expression constructs were transformed into E. coli BL21(DE3) cells, grown to saturation in LB medium supplemented with 50 μg/mL kanamycin at 37 °C and diluted 1:100 into LB medium containing 30 μg/mL kanamycin. The cultures were incubated at 37 °C with shaking, induced with 400 μM IPTG at OD600 = 0.5–0.6, and then incubated at 15 °C for ~20 h. Cells from 4 L of culture were pelleted by centrifugation (6,000 rpm × 10 min), resuspended in 60 mL of buffer A (20 mM Tris-HCl pH 8, 500 mM NaCl and 10 mM MgCl2) and homogenized. The homogenate was passed through a cell disrupter (Avestin EmulsiFlex-C5) twice at 5,000 – 10,000 psi and the lysate was clarified by ultracentrifugation (35,000 rpm × 35 min). The supernatant was incubated with ~1.5 mL of Ni-NTA resin (Qiagen) for 2 h at 4 °C and the mixture was centrifuged (3,000 rpm × δ min). The unbound fraction was discarded and the Ni-NTA resin resuspended in 10 mL of buffer B (20 mM Tris-HCl pH 8, 500 mM NaCl and 10 mM MgCl2, δ mM imidazole) and loaded onto a column and washed with 10 mL buffer B. MceD was eluted from the column using a stepwise imidazole gradient (25 to 200 mM). SDS-Page analysis (4–15% Tris-HCl gel, BioRad) was employed to ascertain the presence and purity of MceD in each fraction. Fractions containing pure MceD were combined and dialyzed twice against buffer C (25 mM Tris-HCl pH 8, 50 mM NaCl, 1 mM DTT, 10% glycerol). This procedure afforded yields of >15 mg/L for both N- and C-terminally tagged MceD. Gel filtration (Supradex 200) indicated that the protein is monomeric. The protein was frozen in liquid N2 and stored at −80 °C. Initial activity assays indicated that both N-terminally and C-terminally His6 tagged MceD were active. All subsequent assays were conducted with the N-terminally tagged enzyme.
Cloning, Overexpression and Purification of MceC
The mceC gene was subcloned from pJEM15 using the forward primer 5′-ggaattccatatgcgtattctctttattggccctcc-3′ (NdeI) and reverse primer 5′-gatcctcgagttgccagatggttttcagtttcgc-3′ (XhoI). Pfu Turbo DNA polymerase was employed for PCR reactions and the amplified genes were digested with NdeI and XhoI, ligated into the pET-29a expression vector using T4 DNA ligase and transformed into E. coli TOP10 cells. The identity of the construct was confirmed by DNA sequencing. E. coli BL21(DE3) cells were transformed with the construct and grown to saturation in LB media containing 50 μg/mL kanamycin. MceC was subsequently overexpressed, purified and stored as described for MceD with one modification. During initial purifications, MceC precipitated within minutes of elution from the Ni-NTA column. Immediate addition of enterobactin to fractions containing MceC remedied this problem. For a typical Ent addition, a 700-μL aliquot of Ent (10 mM in DMSO) was added to 12 mL of eluted MceC. This mixture was immediately transferred to a dialysis cassette (10 kDa cut-off) and dialyzed twice against buffer C. This approach afforded MceC in yields of ~4 mg/L.
Cloning, Expression and Purification of MceIJ
The fragment encoding mceIJ was subcloned from pJEM15 as a bicistronic operon using the forward primer 5′-ggaattccatatggtaacggagaaatacagtcaaatg-3′ (NdeI) and the reverse primers 5′-gatcctcgagtcaaagttctttctgtgtctccggg-3′ (XhoI, pET-28b) and 5′-gatcctcgagaagttctttctgtgtctccggggcc-3′ (XhoI, pET-22b). The genes were amplified and ligated into E. coli expression vectors as described for MceD above. The pET-28b-mceIJ vector encodes MceJ as a N-terminal His6 fusion and untagged MceI. The pET-22-mceIJ vector encodes MceI as a C-terminal His6 fusion and untagged MceJ. The proteins were over-expressed and purified as described for MceD. SDS-Page analysis indicated that both MceI and MceJ co-elute from the Ni-NTA column regardless of which component bears a His6 tag. All subsequent work was conducted with the MceIJ complex in which the His6 tag is fused to MceJ, which was generally obtained in yields of >3 mg/L. Gel filtration analysis (Supradex 200) of MceIJ showed a retention volume of ~150 mL, which is consistent with a molecular weight range of 150–200 kDa and suggests a tetrameric A2B2 state. Several variations in the MceIJ purification protocol were tested and had negligible effect on activity. In particular, the presence of DTT has no effect on MceIJ activity.
Large-Scale Enzymatic Preparation of MceC Glycosylation Products
A 48 mL aqueous solution containing 75 mM Tris-HCl pH 8, δ mM MgCl2 2.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 100 μM enterobactin, and 600 μM uridine diphosphoglucose (UDP-Glc) was prepared and divided into 48 aliquots of 900 μL. To each aliquot was added 100 μL of 10 μM MceC in 75 mM Tris-HCl pH 8 buffer. The solutions were mixed with a pipette, incubated at room temperature for 90 min, and each quenched with 500 μL of 2.5 N HCl in MeOH. The reactions were centrifuged (13,000 rpm × 10 min), combined and concentrated to ~16 mL on a lyophilizer. The concentrated material was centrifuged (13,000 rpm × 10 min) again. MGE (1.3 mg) and DGE (0.9 mg) were obtained as white powders following preparative HPLC using a solvent gradient of 0 to 40% B over 40 min (solvent A, 0.2% TFA/H2O; solvent B, 0.2% TFA/MeCN) and lyophilization. 1H NMR (CD3OD, 600 MHz) δ for MGE: 7.38 (1H, s), 7.16 (2H, dd), 7.00 (1H, d), 6.94 (2H, dq), 6.72 (2H, td), 5.03 (3H, m), 4.67 (3H, m), 4.59 (3H, m), 4.01 (1H, d), 3.86 (1H, d), 3.69 (1H, m), 3.42 (2H, m), 3.36 (2H, m); δ for DGE: 7.39 (1H, dd), 7.27 (1H, dd), 7.06 (2H, s), 6.96 (1H, dd), 6.74 (1H, t), 5.05 (3H, m), 4.70 (3H, m), 4.59 (3H, m), 4.02 (2H, m), 3.86 (2H, dd), 3.71 (2H, m), 3.27–3.47 (8H, m). MS (ESI): [M+H]+ m/z calc 832.2 (MGE), 994.3 (DGE); found, 832.1 (MGE), 994.3 (DGE).
Large-Scale Enzymatic Preparation of MceD Hydrolysis Products
For MGE: A portion (15.6 mg, 18.6 μmol) of MGE was dissolved in 200 μL of DMSO and diluted to a final volume of 15 mL with 75 mM Hepes pH 7.5. A 135 μL aliquot of MceD (71 μM in buffer C) was added and the reaction was mixed thoroughly and incubated for 20 min at room temperature. The reaction was quenched with addition of 2.5 N HCl in MeOH (7 mL), vortexed and filtered through a 0.2 μm membrane. The products were separated and purified by preparative HPLC using a solvent gradient of 0 to 40% B in 40 min (solvent A, 0.2% TFA/H2O; solvent B, MeCN), which afforded linear MGE (lin-MGE, 2.5 mg) and the glycosylated DHB-Ser dimer (Glc-Dimer, 1.8 mg) as white powders following lyophilization. Product purity was verified by analytical HPLC. 1H NMR (CD3OD, 600 MHz) δ for lin-MGE: 7.45 (1H, d), 7.27 (2H, qd), 7.04 (1H, d), 6.93 (2H, dq), 6.70 (2H, td), 5.05 (1H, q), 5.00 (1H, q), 4.81 (1H, d), 4.76–4.81 (2H, m), 4.69 (1H, t), 4.60 (1H, m), 4.51 (1H, m), 4.05 (1H, d), 3.96 (1H, m), 3.87 (2H, m), 3.71 (1H, m), 3.44 (2H, m), 3.39 (2H, m); for Glc-Dimer: 7.44 (1H, d), 7.32 (1H, dd), 7.04 (1H, d), 6.94 (1H, dd), 6.73 (1H, t), 5.04 (1H, m), 4.89 (2H, m), 4.78 (1H, t), 4.52 (1H, m), 4.04 (2H, m), 3.95 (1H, d), 3.93 (1H, d), 3.87 (1H, d), 3.85 (1H, d), 3.72 (2H, m), 3.46 (2H, m), 3.36 (2H, m). MS (ESI) [M+H]+ m/z calc 850.2 (lin-MGE), 627.2 (Glc-dimer); found, 850.2, 627.4. For DGE: As described for MGE except that DMSO was not employed because DGE (16 mg, 16 μmol) exhibited sufficient water solubility. Linear DGE (lin-DGE) and the glycosylated DHB-Ser dimer were obtained as white powders in 6.5 and 4.0 mg quantities, respectively. 1H NMR (CD3OD, 600 MHz) δ lin-DGE: 7.45 (1H, s), 7.40 (1H, s), 7.30 (1H, d), 7.03 (2H, m), 6.94 (1H, d), 6.72 (1H, t), 5.08 (1H, m), 5.00 (1H, t), 4.68 (1H, t), 4.56 (1H, m), 4.51 (1H, m), 4.04 (2H, t), 3.96 (1H, m), 3.86 (3H, m), 3.70 (2H, m), 3.35–3.49 (8H, m). MS (ESI) [M+H]+m/z calc 1012.3 (lin-DGE), 628.2 (Glc-Dimer); found, 1012.2, 627.3.
Large-Scale Preparation of MceIJ Reaction Products
A 102 mL solution containing 75 mM Tris-HCl pH 8, 5 mM MgCl2, 2.5 mM TCEP, 5 mM 100 μM MGE, 550 μM C10 (C-terminal decapeptide, SATSSSGSGS) and was divided into 202 aliquots of 460 μL. A 40 μL portion of ~20 μM MceIJ in buffer C was added to each aliquot and the reactions were incubated at room temperature for 7.25 h. Each reaction was quenched with 100 μL of 0.3% TFA/H2O and centrifuged (13,000 rpm × 10 min). The supernatants were combined and concentrated on a lyophilizer. The two products from the MceIJ-catalyzed reactions were separated by preparative HPLC using a solvent gradient of 0 to 40% B over 40 min (solvent A, 0.2% TFA/H2O; solvent B, 0.2% TFA/MeCN). The collected fractions were lyophilized to dryness, which afforded white powders (1.9 mg, product 1; 3.2 mg, product 2). The purity of each product was verified by analytical HPLC. 2-dimensional (2-D) NMR was employed to determine product identity as described below. MS (ESI) [M+H]+ ra/z calc, 1640.5; found, 1640.2 for both products.
Structural Elucidation of MceIJ Products
The products from the large-scale MceIJ reaction were structurally characterized using 2-D NMR spectroscopy. Data acquisition was performed on a Bruker Avance 500 spectrometer with a 1H-Lamor frequency of 500.13 MHz equipped with a triple resonance z-gradient cryogenic probe. The sample temperature was 298 K. The samples were dissolved in 500 μL of 9:1 H2O/D2O (pH ~ 4) for final concentrations of 1.9 (peak 1) and 3.8 mM (peak 2). For spectral analysis and resonance assignment, a sect of two 2D-1H homonuclear NOESY spectra with correlation via dipolar coupling was recorded for each compound. A water gate W5 pulse sequence was applied for water suppression.35 For each set of experiments, the mixing time was varied from 80 to 250 ms to distinguish between short- and long-range interactions and to support the resonance-specific assignments. All spectra were processed with XWINNMR 3.1 and analyzed with SPARKY 3.111.36 For spectra processing, a square sine bell apodization function was used for Fourier Transformation. For both compounds, all proton resonances were assigned with the exceptions of the hydroxyl protons from the hexose, the serine sidechains and the benzoic acid residues of the 2,3-DHB units, which could not be observed in aqueous solution. Starting with the unambiguous low field amide proton resonances from the serine residues of the three DHB-Ser units, the aromatic moieties of the DHB units were assigned by intra- and inter-residual NOEs. NOE correlations of one DHB unit into the hexose residue and further to the attached peptide supported the sequential assignment of the attached peptide. The peptide was completely sequentially assigned using the amide-to-amide proton correlations.
Initial Activity Assays with MceC
Initial activity assays with MceC were conducted in buffer D (75 mM Tris-HCl pH 8, δ mM MgCl2 and 2.5 mM TCEP). For a typical assay, a 500 μL solution containing 100 μM siderophore, 600 μM UDP-Glc and 1 μM MceC was prepared, mixed and incubated at room temperature. A solution of 2.5 N HCl in MeOH (75 μL) was used to quench reaction aliquots (75 μL) at various time points. The quenched solutions were centrifuged (13,000 rpm × 10 min) and analyzed by HPLC. A solvent gradient of 0 to 40% B in 8 min (solvent A, 0.1% TFA/H2O; solvent B, MeCN) was employed. Product identity was confirmed by co-elution with hydrolysis products of IroB-catalyzed reactions, LCMS and NMR.
Kinetic Investigations of MceC
Kinetic investigations of MceC-catalyzed glycosylation of Ent, linear Ent (lin-Ent) and MGE were conducted in buffer D. To determine kcat and KM for the donor substrate, the UDP-Glc concentration was varied (0 – 256 μM) in the presence of 100 μM Ent and 200 nM MceC, and the reactions (100 μL volume) were quenched at t = 20 min with 75 μL of 2.5 N HCl in MeOH. To determine the kinetic parameters for the acceptor substrates, the UDP-Glc concentration was held constant at 500 μM and the siderophore concentrations were generally varied from 0 – 256 μM with a MceC concentration of 100 (Ent) or 200 (MGE, lin-Ent) nM. The 100-μL reactions were quenched at t = 1 (Ent), 7 (MGE) or 8 (lin-Ent) min with 75 μL of 2.5 N HCl in MeOH. The quenched solutions were centrifuged (13,000 rpm × 10 min), stored on ice and analyzed by HPLC using a solvent gradient of 0 to 40% B in 8 min (solvent A, 0.1% TFA/H2O; solvent B, MeCN). Product quantification was based on the area of the 316 nm absorption peaks. All kinetic runs were repeated in triplicate and the data fit to the Michaelis-Mention equation. The Ent present as a result of the MceC preparation was taken into account in total [Ent] used in the analysis of MceC glycoslyation of Ent.
Kinetic Investigations of IroB
Kinetic studies of IroB-catalyzed glycosylation of Ent, lin-Ent, MGE, lin-MGE and DGE were conducted as described for MceC. The UDP-Glc concentration was 500 μM and the siderophore concentrations were generally varied from 0 – 256 μM in the presence of 100 nM (Ent, MGE, DGE), 200 (lin-Ent) nM or 1 μM (lin-MGE) IroB. The 100 μL reactions were quenched at t = 2.5 (Ent), 4 (MGE), 20 (lin-Ent, lin-MGE) or 30 (DGE) min with 75 μL of 2.5 N HCl in MeOH and analyzed as described for MceC.
Initial Activity Assays with MceD
Initial activity assays with MceD were conducted in 75 mM Hepes buffer adjusted to pH 7.5. In general, a 500 μL solution containing 32 μM siderophore and 20 nM MceD was employed. Reaction aliquots (100 μL) were quenched at t = 0, 2, 8, 25 min with 2.5 N HCl in MeOH (100 μL), centrifuged (13,000 rpm × 10 min) and stored on ice. In all cases, the reaction progress was monitored by analytical HPLC using a solvent gradient of 0 to 40% B in 8 min (solvent A, 0.1% TFA/H2O; solvent B, MeCN). Product identity was confirmed by co-elution with hydrolysis products of IroD catalyzed reactions and by LCMS.
Kinetic Investigations of MceD
Kinetic studies of MceD-catalyzed hydrolysis of apo Ent, MGE, DGE, lin-MGE and lin-DGE and [Fe(Ent)]3−, [Fe(MGE)]3− and [Fe(DGE)]3− were conducted at room temperature in 75 mM Hepes buffer adjusted to pH 7.5. The enzyme concentration was 5 nM and the substrate concentration was generally varied from 0 – 256 μM for apo and from 0 – 32 μM for ferric substrates. Each 100-μL reaction was quenched with 50 μL of 2.5 N HCl in MeOH at t = 30 sec, centrifuged (13,000 rpm × 10 min) stored on ice and analyzed as described above. Negative control reactions with no enzyme were conducted to determine if background hydrolysis needed to be taken into account. This treatment was necessary for Ent and lin-Ent.30 Kinetic studies of the MceD-catalyzed hydrolysis of C10-C6′-MGE and C10-C4′-MGE were conducted in a similar manner except that the reactions were quenched at t = 1 min. Some (<10%) migration of the C10 peptide from the C4′ to the C6′ position was observed when C10-C4′-MGE was employed as a substrate and the area of both peaks were considered in the data analysis.
Activity Assays with McelJ
Activity assays with MceIJ were generally conducted at pH 8 (75 mM Tris-HCl) and in the presence of 2.5 mM TCEP and 5 mM ATP with 100 μM MGE/lin-MGE, 550 μM C10 peptide and 2 μM MceIJ. The reactions (300 μL) were incubated at room temperature, quenched at various time points with an equal volume of 0.6% TFA/H2O, vortexed and centrifuged (13,000 rpm × 10 min). The reaction progress was monitored by analytical HPLC using a solvent gradient of 0 to 40% B (MGE) or 15 to 35% B (lin-MGE) where solvent A is 0.2% TFA/H2O and solvent B is 0.2% TFA/MeCN. Product identities were determined by LCMS and NMR as described above.
Results and Discussion
MccE492m exhibits a number of remarkable structural features (Figure 1). The antibiotic illustrates an intersection of ribosomal and non-ribosomal peptide synthesis. It bears an unusual post-translational modification at the C-terminus comprised of an ester linkage between the ribosomally-derived MccE492 peptide and the C6′ oxygen of the non-ribosomally synthesized monoglycosylated and linearized enterobactin (lin-MGE). The glucose moiety in MccE492m is a bridging element that connects its two functional units, the toxic microcin peptide and the siderophore recognition element. The serine-rich C-terminus of MccE492 is another striking feature shared by several other microcins including MccH47, MccI47 and MccM (Figure 2).37–42 This commonality may point to similar post-translational modifications at the C-termini and a general strategy for destroying competing bacterial species that express siderophore receptors.43,42 The biosynthesis of the siderophore moiety itself requires multiple layers of secondary metabolism. Its central scaffold, enterobactin, is the product of the enterobactin synthetase and requires both C-glycosylation and linearization for MccE492m maturation. Unveiling the biosynthetic tailoring of MccE492m provides a foundation for future studies of structurally related microcins, including MccH47, MccI47 and MccM, and a platform for the design of new Trojan horse antibiotics.
Expression and Purification of MceC, MceD and MceIJ
We chose first to investigate MceC and MceD based on their respective amino acid sequence homology to IroB (C-glycosyItransferase) and IroD/IroE (esterases) from the iroA gene cluster. IroB carries out regiospecific C-glycosylation of enterobactin at the C5 positions of the DHB rings and IroD/E catalyze hydrolysis of the ester linkages in the enterobactin macrolactone to afford linearized congeners.29,30 IroD and IroE both accept glycosylated enterobactins as substrates and IroD processes these forms regioselectively.
The mceC and mceD genes were subcloned from pJEM15 into E. coli expression vectors as either N- and/or C-terminal His6 fusions. MceD (45.8 kDa, 414 aa) was over-expressed in E. coli BL21(DE3) cells as N- and C-terminally His6 fusions and purified by Ni-NTA affinity chromatography with yields of >15 mg/L (Figure 3). MceC (39.9 kDa, 370 aa) was over-expressed as a C-terminal His6 fusion and purified in a similar manner (Figure 3), but exhibited very poor solubility following elution from the Ni-NTA column. We reasoned that addition of enterobactin, a putative substrate of MceC, might stabilize the enzyme and afford solubility. We subsequently added Ent to several MceC-containing fractions and immediately dialyzed, which conferred solubility.
Figure 3.
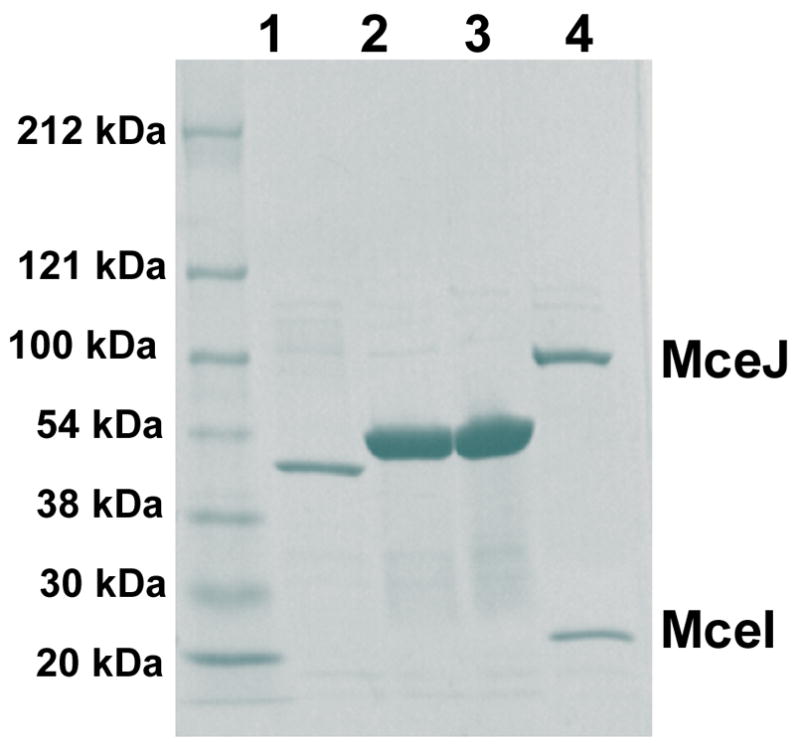
SDS-Page gel (4–15% Tris-HCl) of purified His6 fusions of MceC (1, C-terminal His6 fusion), MceD (2, C-terminal His6 fusion; 3, N-terminal His6 fusion) and MceIJ (4, MceJ bears a N-terminal His6 tag).
Genetic studies revealed that mcel and mcej are required for MccE492m production.24,25 In related work, initial attempts to overexpress and purify MceIJ homologs from the microcin H47 gene cluster, MchD and MchC, did not yield appreciable quantities of soluble protein.43 Co-expression of MchC and MchD afforded both proteins as a soluble complex. We therefore subcloned the bicistronic operon encoding mceIJ with a His6 tag fused either to the C-terminus of MceI (19.8 kDa, 163 aa) or the N-terminus of MceJ (59.1 kDa, 524 aa). The proteins were over-expressed in E. coli BL21(DE3) cells and purified using Ni-NTA column chromatography. SDS-PAGE analysis showed that MceI-C-His6 pulled down MceJ and that MceJ-N-His6 pulled down Mcel, indicating formation of a Mcelj complex (Figure 3). Gel filtration supported complex formation and the retention volume suggested an A2B2 tetramer, although further work is required to verify this notion.
MceC is a C-Glycosyltransferase that Modifies Apo Siderophores
Based on the MccE492m structure and our previous studies of homolog IroB,29 we anticipated that MceC would catalyze C-glycosidic bond formation between C1′ of the glucose and C5 of the DHB ring to form monoglycosylated enterobactin (MGE). Because the Ent derivative in MccE492m has only one glucose moiety, we hypothesized that MceC might differ from IroB in its ability to successively glycosylate the Ent scaffold. Incubation of IroB with Ent and UDP-Glc resulted in the distributive formation of three products, mono-, di- and triglycosylated enterobactin. MceC, in contrast, might prefer to glycosylate Ent only once, generating a product that would be on-pathway to MccE492m.
Preliminary activity assays revealed that incubation of MceC with Ent and UDP-Glc for t < 30 min resulted in formation of a new peak in the analytical HPLC trace with a retention time and mass consistent with MGE ([M+H]+ ra/z calc, 832.2; found, 832.1) (Figure 4). Following accumulation of MGE, a second peak appeared at t > 30 min with a retention time and mass consistent with DGE ([M+H]+ m/z calc, 994.3; found, 994.3). MceC did not generate TGE (Figure SI, Supporting Information), a novel enterobactin derivative isolated from IroB-catalyzed reactions in vitro,29 even in the presence of 1 mM UDP-Glc (t = 180 min). 1H (MGE, DGE) and HMBC NMR spectra (DGE) analysis of the products obtained from a large-scale MceC-catalyzed glycosylation of Ent confirmed the C-glycosidic linkage between the C1′ of the glucose moiety and C5 of the DHB unit (Supporting Information). The spectroscopic data for DGE are in agreement with those of DGE isolated from an IroB-catalyzed reaction29 and the structure of salmochelin S4, which was recently isolated from Salmonella enterica.9
Figure 4.

MceC C-glycosylates Ent. (a) Representation of the MceC-catalyzed converstion of Ent to MGE and DGE. (b) HPLC analysis of the MceC-catalyzed glycosylation of Ent. (c) Kinetic traces for MceC-catalyzed glycosylation of Ent, MGE and lin-Ent. The corresponding kcat and Km values are listed in Table 1.
Because the enterobactin scaffold in MccE492m is linearized, we investigated whether MceC would also accept linearized enterobactin (lin-Ent) as a substrate. Incubation of MceC with lin-Ent and UDP-Glc resulted in the formation of a new peak in the analytical HPLC trace with a mass consistent with linear MGE (calc [M+H]+ m/z calc, 850.2; found, 850.5) and retention time equivalent to that of lin-MGE isolated from MceD-catalyzed hydrolysis of MGE (vide infra). We also surveyed lin-MGE and linearized DGE (lin-DGE), prepared by large-scale MceD-catalyzed reactions, and found that these siderophores are not good substrates for MceC. No glycosylation of lin-MGE or lin-DGE was observed.
Enterobactin is an iron-chelator produced by bacteria existing in iron-limiting conditions and could therefore be a substrate for MceC in its apo or holo form. We therefore questioned if MceC would also recognize [Fe(Ent)]3−, [Fe(MGE)]3−, and [Fe(DGE)]3−. Initial activity assays indicated that the ferric siderophores are not viable substrates for MceC. Incubation of MceC with a ferric siderophore and UDP-Glc resulted in either negligible or no glycosylation of the DHB moieties (t = 60 min). This behavior is analogous to that of IroB, which does not accept [Fe(Ent)]3− or its glycosylated congeners.44
To gain further insight into the substrate specificity of MceC and its homolog IroB, kinetic studies were undertaken with the apo siderophores and the resulting data are listed in Table 1 (Figures 4 and S1). The kinetic parameters for the acceptor substrates reveal that the catalytic efficiency of MceC decreases with either glycosylation or linearization of the enterobactin scaffold with Ent > MGE > lin-Ent. A comparison of data for enterobactin and MGE indicates a ~40-fold reduction in kcat/Km for the second glycosylation event. Likewise, linearization of the Ent scaffold results in an ~4.4-fold decrease in kcat and ~19-fold increase in Km, affording an overall ~97-fold reduction in kcat/Kmfor lin-Ent relative to enterobactin. Substrate inhibition was also observed with [lin-Ent] > 64 μM. In total, these data indicate that enterobactin is the preferred substrate for MceC and that MceC acts prior to MceD in MccE492m maturation.
Table 1.
Kinetic Data for C-Glycosyltransferases MceC and IroBa
| Ent | MGE | DGE | lin-Ent | lin-MGE | UDP-Glce | ||
|---|---|---|---|---|---|---|---|
| MceC | kcat (min−1) | 2.7 ± 0.3 | 1.22 ± 0.05 | n.d.b | 0.61 ± 0.06c | n.d. | 1.19 ± 0.03 |
| Km(μM) | 0.94 ± 0.38 | 18.5 ± 2.4 | n.d.b | 17.8 ± 4.6c | n.d. | 87 ± 6 | |
| kcat/Km (min−1μM−1) | 2.9 | 0.07 | n.d. | 0.03 | n.d. | 0.014 | |
| IroB | kcat (min−1) | 1.8 ± 0.1 | 5.1 ± 0.2 | 3.0 ± 0.1 | 0.57± 0.002 | 0.44 ± 0.03d | n.d. |
| Km(μM) | 2.0 ± 0.3 | 48 ± 6 | 72 ± 5 | 36 ±4 | 111 ± 15d | n.d. | |
| kcat/Km (min−1μM−1) | 0.9 | 0.11 | 0.04 | 0.02 | 0.004 | n.d. |
Experiments were conducted at room temperature in buffer D (75 mM Tris-HCl pH 8, 5 mM MgCl2 and 2.5 mM TCEP) and in the presence of 500 μM UDP-Glc.
n.d. = not determined.
Substrate inhibition was observed at [lin-Ent] > 64 μM.
Substrate inhibition was observed at [lin-MGE] > 256 μM.
The UDP-Glc concentration was varied in the presence of 100 μM Ent.
The kinetic data for IroB show generally similar trends. IroB has a broader substrate scope than MceC and accepts DGE and lin-MGE, substrates with greater steric bulk. The catalytic efficiency decreases with glycosylation and linearization in the order Ent > MGE > DGE > lin-Ent > lin-MGE (Table 1, Figure S1).
MceD is an Esterase and Hydrolyzes Apo and Ferric Siderophores
MceD is a homolog of IroD/IroE, the enterobactin hydrolases encoded by the iroA gene cluster. Although IroD and IroE both accept apo and ferric enterobactin and their glycosylated congeners, IroD prefers to process [Fe(MGE)]3−/[Fe(DGE)]3− whereas IroE preferentially hydrolyzes the apo forms. IroD cuts MGE and DGE regioselectively and readily degrades the linear DHB-Ser trimer products to monomers and dimers. IroE, in contrast, hydrolyzes glycosylated Ent species with no regioselectivity and cuts only once to yield trimers.30 With the MccE492m structure in mind, we anticipated that MceD would exhibit the regioselectivity of IroD and hydrolyze the macrolactone ring once in an IroE-like manner.
Incubation of Ent with MceD in pH 7.5 buffer resulted in hydrolysis of the lactone scaffold (Figure 5). At t > 2 min, several Ent hydrolysis products were observed in the analytical HPLC trace, which correspond to lin-Ent and its dimeric and monomeric derivatives, arising from two and three hydrolytic cuts, by retention time and MS analysis (Table S1). This distribution indicates that the initial lin-Ent product, generated by Ent hydrolysis, is also a good substrate for MceD. This behavior is similar to that of IroD, which readily hydrolyzes the ester linkages in both cyclic and linear Ent trimers.30
Figure 5.

MceD hydrolyzes apo Ent, MGE and DGE. (a) HPLC analysis of MceD-catalyzed hydrolysis of Ent. Dimer and monomer refer to the DHB-Ser dimer and DHB-Ser monomer, respectively, (b) HPLC analysis of MceD-catalyzed hydrolysis of MGE. Glc-Dimer is the monoglycosylated DHB-Ser dimer and “Monomers” refer to the Glc-DHB-Ser (elution time ~ 2 min) and DHB-Ser (elution time ~2.5 min) monomers, the former of which results from hydrolysis of the Glc-DHB-Ser dimer. (c) Kinetic traces for the MceD-catalyzed hydrolysis of Ent, lin-Ent, MGE, lin-MGE, and DGE. Corresponding kinetic parameters are listed in Table 2. All reactions were carried out at room temperature (75 mM Hepes, pH 7.5). (d) Schematic of the MceD-catalyzed hydrolysis of MGE illustrating the regioselectivity of the cut as determined by 2-D NMR. HPLC traces for assays with DGE are given in Figure S2 and the m/z data for Ent, MGE and DGE hydrolysis products are listed in Table S1.
Initial assays with MGE and DGE under the same conditions indicated that the presence of glucose moieties alters the product distribution of MceD (Figures 5, S2). MGE and DGE are both hydrolyzed by MceD, but much more readily than lin-MGE and lin-DGE. As a result, build-up of lin-MGE and lin-DGE occurs followed by some formation of the dimeric and monomeric hydrolysis products. The kinetic data corroborate these observations (Table 2). Comparisons of kcat/Km for the apo substrates reveal that (i) MceD hydrolyzes Ent and lin-Ent with comparable efficiency, (ii) successive glycosylation of Ent results in increases in both kcat and Km, yielding an overall >2-fold decrease in catalytic efficiency and (iii) linearization of the MGE scaffold results in a ~50-fold decrease in MceD efficiency relative to the macrolactone because of a ~ 19-fold decrease in kcat and ~3-fold increase in Km. The ~50-fold preference of MceD for MGE over lin-MGE suggests that MceD is capable of producing a linearized trimer like the one in MccE492m. NMR spectra (1H and 13C 1-D, HMBC, 13C-HSQC) of the trimeric and dimeric products from MGE and DGE hydrolysis are in agreement with those obtained for the analogous species obtained from IroD-catalyzed reactions,30 which indicates that both enzymes cleave MGE and DGE with the same regioselectivity (Figure 5). This mode of cutting affords the lin-MGE isomer observed in MccE492m.6
Table 2.
Kinetic Data for Esterase MceDa
| Ent | MGE | DGE | lin-Ent | lin-MGE | |
|---|---|---|---|---|---|
| kcat(min−1) | 207 ± 10 | 564 ± 20 | 910 ± 33 | 139 ±4 | 30 ± 2.5 |
| Km(μM) | 2.5 ± 0.4 | 45 ±9 | 29 ± 3 | 1.7 ± 0.2 | 45 ±9 |
| kcat/Km(min−1μM−1) | 83 | 34 | 31 | 82 | 0.67 |
| [Fe(Ent)]3− | [Fe(MGE)]3− | [Fe(DGE)]3− | C10-C6′-MGE | C10-C4′-MGE | |
|
| |||||
| kcat(min−1) | 43 ± 1.3 | 39 ± 2 | 29 ± 1.5 | 606 ± 53 | 767 ± 93 |
| Km(μM) | 2.5 ± 0.3 | 7.7 ± 1.4 | 7.2 ± 1.1 | 15.5 ± 3.6 | 26 ± 7 |
| kcat/Km(min−1μM−1) | 17 | 5 | 5 | 39 | 29.5 |
All reactions were carried out at room temperature (75 mM Hepes, pH 7.5).
MceD also hydrolyzes the ferric siderophores with product distributions that mirror those observed for the apo congeners (Figure S3). Prior studies of the related esterase, Fes, came to contradictory conclusions depending on assay conditions and subsequent interpretation.30,45,46 Because siderophores are quickly exported for iron acquisition following biosynthesis, the intracellular concentrations are most likely lower than the Km value. As a result, the apparent second-order rate constant, kcat/Km, should be considered when quantifying the throughput of siderophore-tailoring enzymes. The relative catalytic efficiencies for apo and ferric Ent, MGE and DGE reveal that MceD prefers the apo congeners (Table 2). Although the Km values are generally lower for the ferric siderophores than their apo counterparts, the kcat values are substantially reduced and the net effect is a ≤5-fold decrease in kcat/Km. A similar trend was previously observed for IroE.30
MceIJ is Responsible for Post-Translational Modification of MccE492
The post-translational modification that gives rise to MccE492m is unusual because it involves modification of the peptide C-terminus. Other C-terminal modifications have been documented in the esterification of small GTPases, including Ras,47 and in the structure of MccC7.48–50 Some microcins, including MccH47, MccI47 and MccM, exhibit serine-rich C-terminal regions like that of MccE492 (Figure 2) and may exhibit post-translational modifications identical to that of MccE492m.42 A recent study of microcin MccH47/ColV chimeras points to a modular structural organization where N- (toxin) and C-terminal (recognition) domains can be exchanged readily to yield active bacteriocins.51 This modularity may have played a role in the evolution of this family of peptide toxins.
Previous genetic studies suggest that MceI and MceJ catalyze the post-translational modification that converts MccE492 to MccE492m.24 Removal of mceI from the MccE492 gene cluster prevents maturation of MccE492. Its protein product, MceI, is a 19.8 kDa enzyme with homology to HlyC, the acyltransferase responsible N-acylation of lysine residues in hemolysin toxin,31 and MchD, which is required for MccH47 maturation. The MccE492 gene cluster also encodes MceJ, a 59.1 kDa protein of unknown function and with no known homologs other than its counterpart in the microcin H47 gene cluster (MchC), which has also been implicated in MccE492m maturation.
We therefore subcloned the bicistronic operon encoding MceIJ and found that the two proteins form a complex. We also designed a model system for assaying MceIJ activity. We hypothesized that the serine-rich portion of the MccE492 C-terminus might be sufficient for recognition by MceIJ, and utilized a model decapeptide comprised of the last ten residues of MccE492, SATSSSGSGS (C10), as a peptide substrate analog. We employed MGE rather than lin-MGE as the siderophore substrate because (i) the preparation of MGE requires one less enzymatic reaction, (ii) MGE decomposes less readily than lin-MGE in buffered aqueous solution at room temperature, and (iii) we were uncertain as to the order of MceD and MceIJ action during MccE492m biosynthesis.
Initial activity assays with this model system revealed several important facets of MccE492m assembly: (i) MceIJ recognizes the C10 peptide substrate, (ii) MceIJ accepts MGE as a substrate, (iii) only ATP and MgCl2 are required as co-factors for attachment of MGE to the decapeptide, and (iv) two new peaks, 1 and 2, form in the analytical HPLC trace following incubation of MceIJ with C10, MGE, ATP and MgCl2 (Figure 6). Isolation and MS analysis of the two product peaks indicated that they have the same molecular weight equivalent to C10+MGE-H2O that is consistent with attachment of MGE to the C10 peptide through an ester linkage ([M+H]+ m/z calc, 1640.5; found, 1640.2 (1), 1640.2 (2)).
Figure 6.
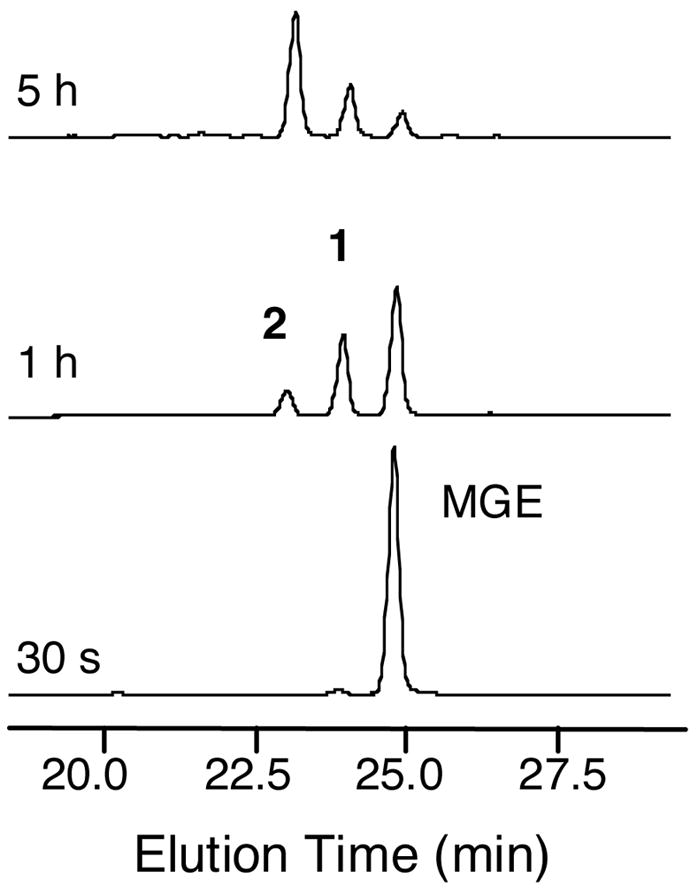
HPLC monitoring of a reaction containing 550 μM C10, 100 μM MGE and 2 μM MceIJ, 5 mM ATP and 5 mM MgCl2 (75 mM Tris-HCl pH 8, 2.5 mM TCEP). The two new peaks, 1 and 2, both have a mass equivalent to C10+MGE-H2O, which suggests attachment of MGE to the C10 peptide through an ester linkage.
To gain further insight into the origins of peaks 1 and 2, both products were obtained in multi-milligram quantities and in high purity from large-scale MceIJ-catalyzed reactions (Figure 7). A series of activity assays indicated that (i) formation of 1 is enzyme-dependent, (ii) formation of 2 from 1 does not require enzyme, (iii) conversion of 1 to 2 does not happen in water (pH ~ 5), but occurs in solutions buffered in the pH 7 to 9 range, (iv) the conversion rate of 1 to 2 is accelerated by base, and (v) incubation of peak 2 in the absence or presence of MceIJ does not result in significant formation of 1 in the pH range of 5.5 to 9 (Figure S4). In particular, analytical HPLC studies showed that a 50 μM solution of 1 is converted to ~20% (pH 7), ~50% (pH 8) or ~80% (pH 9) 2 following a 30 min incubation at room temperature. Incubation of 2 at room temperature for 30 min resulted in ~10% conversion to 1 in the pH 7 to 9 range.
Figure 7.

(a) Section of the 2D-1H homonuclear NOESY spectra for C10-C4′-MGE (peak 1). (b) Section of the 2D-1H homonuclear NOESY spectra for C10-C6′-MGE (peak 2). Both spectra were acquired using a W5 pulse sequence for water suppression and a 250 msec mixing time. The dotted lines indicate the positions of the sugar protons and the corresponding NOE correlations. The proton assignments are listed in Table S2. (c) Analytical HPLC (220 nm absorption) of purified C10-C6′-MGE. (d) Analytica HPLC trace (220 nm absorption) of purified C10-C4′-MGE. A gradient of 0 to 40% MeCN in 8 min was employed for both samples, (e) Numbering scheme for the glucose and DHB moieties.
To verify that MceIJ is indeed responsible for the MccE492m post-translational modification, an assay with MceIJ, ATP, MgCl2, MGE and native MccE492, isolated from E. coli VCS257 harboring pJAM434 according to a published protocol,6 was performed. A new peak in the HPLC trace exhibiting catecholate (316 nm) absorption was observed and MALDI-TOF analysis indicated a mass consistent with MccE492+MGE-H2O ([M+H]+ m/z calc, 8700.2; found, 8704.4; [M+H]+ for unmodified MccE492 m/z calc, 7887.5; found, 7892.3).
Structural Analysis of the MceIJ Reaction Products: Initial Reaction at the C4′ Hydroxyl of MGE
We reasoned that peaks 1 and 2 are two isomers of the C10-MGE structure with one exhibiting C10 linked to MGE through the C6′ oxygen on the glucose moiety as seen in the MccE492m structure and the other a novel isomer. We employed 2-D-1H-homonuclear NMR to determine the product identities. Table S2 lists the resonance assignments for all observed protons in 1 and 2 and sections of the corresponding NOESY spectra are presented in Figure 7. The published assignment of MccE492m(75–84)6 was largely reproduced for both compounds. The C10 peptide was assigned using amide-to-amide proton correlation and the sequence was corroborated by identification of the distinct side chain proton resonances of the Ala-2 2 Hβ methyl protons and the Thr-3 Hγ proton. The structures of the MGE moieties in 1 and 2 were confirmed as enterobactin linked to a hexose by a C-glycosidic bond between the C1′ atom of the sugar and the C5 position of a DHB-Ser unit. The C-C bond between C5 of DHBS1 and C1′ of the glucose was confirmed by the observed NOE correlations between the H4 and H6 protons of DHBS1 and the H1′ proton of the sugar. The absence of the H5 proton resonance of DHBS1 and the identifiable H6-H5 and H4-H5 correlations for DHBS2 and DHBS3 further verifies the nature of the C-glycosidic bond.
The proton resonances and NOE correlations for the sugar moieties in 1 and 2 reveal key differences. For 2, correlations between H1′-H3′, H3-′H5′ and H5′-H1′ were observed, in addition to a weak correlation between H2′-H4′. Together with the observed coupling constants, these NOE correlations indicate that H1′, H3′ and H5′ are in the axial positions and confirm the β-D-glucose arrangement. The averaged chemical shift for the H6′ protons in 2 are 4.45 and 4.24 ppm, considerably different from the expected chemical shift of free methylene protons at the C6′ position of β-D-glucose (3.25 ppm). Specific NOE correlations were observed between (i) HN of Ser-10 and H6′ and (ii) the Ser-10 Hα proton resonance and H6′ of the glucose. These correlations demonstrate that the C-terminal Ser residue of the C10 peptide is attached on the C6′ position of the glucose moiety in 2. This arrangement is in full accord with the assigned ester connectivity in MccE492m.
The chemical shifts for the H1′, H2′, H3′ and H5′ protons of the glucose in 1 are comparable to those in 2. In contrast, the proton resonances and NOE correlations for H4′ and H6′ differ in the NOESY and COSY spectra. Using the 1H-COSY spectra to assign the protons of the sugar moiety in 1, we assert that the chemical shift for H4′ is 4.96 ppm, whereas the chemical shift of H6′ is 3.98 ppm. With respect to 2, this is a flip-like change of the chemical shifts for H4′ and H6′. The J-coupling constants and observed intra-moiety NOEs support the configuration of the sugar in 1 as a β-D-glucose. Furthermore for 1, new NOE correlations were identified between H4′ and one of the Hβ protons of Ser-10 and between H4′ and HN of Gly-9 and H3′ and HN Ser-10. The inter-residual NOE contacts to H6′ observed in 2 could not be identified for 1. These data indicate that the C-terminal serine residue of the C10 peptide is attached to the C4′ position in the MceIJ-generated product.
The structural analysis and activity assays establish that MceIJ installs C10 onto the C4′ oxygen of MGE via an ester linkage from Ser-10 and subsequent migration of C10 to the C6′ oxygen moiety is enzyme-independent and occurs in the presence of a base to yield the connectivity observed in a sample of MccE492m that was isolated from bacterial cultures and structurally characterized (Scheme 1). The former observation was contrary to our expectation that MceIJ would link C10 to the C6′ position of the MGE moiety. We rationalize the migration of the decapeptide from C4′ to C6′ with a six-membered intermediate where the deprotonated C6′ hydroxyl attacks the carbonyl of the ester bond between Ser-10 and the C4′ hydroxyl (Scheme 1). This intermediate was previously suggested in the base-dependent migration of simple acyl groups from C4′ to C6′ of glucose52 and analogous transformations have been reported for acetylated pyranoses, including 1,2,3,4-tetra-O-acetyl-α-D-glucopyranose,53–56 and suggested in the isomerization of 1-O-indol-3-ylacetyl-β-D-glucose, an ester conjugate of a plant growth hormone.57 These observations raise the question of whether other natural products containing ester linkages at the C6′ position of glucose were first generated enzymatically with alternative connectivity.
Scheme 1.

(a) Attachment of MGE to the MccE492m C-terminal decapeptide (C10) by MceIJ through the C4′ hydroxyl and subsequent migration to the C6′ position, (b) Formation of the six-membered intermediate between the C4′ and C6′ positions.
Further Considerations Regarding MccE492m Maturation
Having established that MceIJ can transfer MGE to the terminal serine residues of C10 and MccE492, we questioned if lin-MGE is also a substrate. Incubation of MceIJ with C10, lin-MGE, ATP and MgCl2 at pH 8 resulted in the formation of three new peaks with catecholate absorption in the analytical HPLC trace (Figure S5). Isolation and mass spectral analysis indicated that each product has a molecular weight equivalent to C10+(lin-MGE)-H2O and thus connection of C10 to lin-MGE through an ester linkage ([M+H]+ m/z calc 1658.5; found 1658.1, 1658.4, 1658.0). We presume that peak 2 (Figure S5) corresponds to the C10-C4′-(lin-MGE) isomer and peaks 3 and 4 (Figure S5) to structural isomers, one of which is C10-C6′-(lin-MGE), but NMR structural analysis is required to validate this notion.
With knowledge that MceIJ accepts both lin-MGE and MGE, we also sought to ascertain if MceD recognizes the C10-MGE products from MceIJ-catalyzed reactions. Incubation of C10-C6′-MGE with MceD at pH 7.5 for 2 min resulted in formation of a new peak in the analytical HPLC trace with a mass consistent with hydrolysis of the MGE lactone ring and formation of C10-C6′-(lin-MGE) ([M+H] m/z calc, 1658.5; found, 1659.1) (Figure S6). The product distribution at longer time points resembles that of MceD-catalyzed MGE hydrolysis (Figure 5) with initial build-up of the linear product and then further degradation (Figure S6). MS analysis confirmed formation of the C10-linked monoglycosylated DHB-Ser dimer ([M+H] m/z calc, 1436.5; found, 1435.8) and monomer ([M+H] m/z calc, 1212.4; found, 1212.7). MceD is therefore selective for hydrolysis of the macrolactone over hydrolysis of the ester linkage between C10 and the glucose moiety. The C10-C4′-MGE enzymatic product is also recognized and processed similarly (data not shown). The HPLC assays suggest that some migration of the C10 peptide from C4′ to the C6′ position occurs in the MceD-generated product during the course of the reaction. Kinetic characterization of MceD-catalyzed hydrolysis of both C10-MGE isomers was therefore conducted (Table 2, Figure S6). A comparison of kcat and Km for MGE and its peptide-derivatized analogs reveals that the C10 peptide has negligible effect on MceD activity.
We depict the maturation of MccE492m in Scheme 2. This process commences with C-glycosylation of Ent by MceC. The resulting MGE is either attached to MceA, the peptide precursor of MccE492, by MceIJ or hydrolyzed by MceD to yield lin-MGE. In the former case, MceIJ attaches MceA to the C4′ hydroxyl of MGE and base-catalyzed migration of the glycosyl ester to the C6′ position affords the MccE492m connectivity. MceD linearizes the lactone scaffolds of these C10-MGE conjugates and cleavage of the MceA precursor peptide occurs during export,58 which together yield MccE492m. In the latter case, MceIJ attaches lin-MGE to the MceA C-terminus, presumably at the C4′ position. Migration of the glycosyl ester to the C6′ position and cleavage of MceA affords MccE492m, which is exported by MceGH. The versatility of MceIJ and MceD points to a mechanism of efficient toxin assembly and enhanced survival. Although not considered in Scheme 2, MccE492m analogs bearing the glycosylated DHB-Ser dimer and monomer have been indentified from bacterial cultures.6,58 These species may arise from the MceD-catalyzed hydrolysis of C10-(lin-MGE), as shown in this work. MceIJ-catalyzed attachment of these hydrolysis products to the peptide C-terminus is another possible route that merits consideration.
Scheme 2.

Maturation of MccE492m as defined by in vitro studies. MceA is the peptide precursor to MccE492. It undergoes cleavage at amino acid 15 or 19 during export to yield the 84-residue MccE492(m).
Summary and Perspectives
Bacteria elaborate peptide-based antibiotics during periods of stress to compete and survive in the host environment. Some of these peptides, including the beta lactams of the penicillin/cephalosporin classes and glycopeptides of the vancomycin class, are synthesized by nonribosomal peptide synthetases. Others, including the microcin family, are produced by the ribosome. Many microcin peptide scaffolds are subject to distinct post-translational modifications that are required for their biological activity. Enzymes co-expressed with the structural gene in the microcin cluster catalyze these modifications. The post-translational tailoring of MccE492 is noteworthy in several respects. Structurally, modification of the C-terminal residue of a protein with an ester linkage to a glucose moiety is novel. Because the glucose is also connected to enterobactin by a C-glycosidic bond, it bridges the ribosomal and nonribosomal peptide fragments of MccE492m and thereby joins two elements with distinct function. The lin-MGE added to MccE492 during maturation, formed by glycosylation of enterobactin, constitutes a second layer of secondary metabolism. Subsequent attachment of MccE492 to lin-Ent may be therefore viewed as a third layer of secondary metabolism and further exemplifies Nature’s biosynthetic ingenuity. Further study of the MceIJ-catalyzed attachment of MGE to the MccE492 C-terminus is particularly warranted for mechanism and selectivity.
From a functional standpoint, maturation of MccE492m creates a Trojan horse toxin directed to virulent bacteria that express catecholate siderophore receptors. Other toxic natural products exploit this type of recognition strategy. Albomycin, produced by Streptomyces, is comprised of a nucleoside antibiotic linked to a hydroxamate siderophore and it binds siderophore-specific porins,59–61 as does Colicin Ia.62 Siderophore uptake pumps also grant cell entry to larger cargo, such as bacteriophage H8,63 and have been considered as targets for combating Pseudomonas infections.64,65 This siderophore-based approach to cell penetration may be applicable in the design of new antibiotics that use iron-scavengers to target specific microbial populations, including gram-negative and multi-drug resistant strains.
Supplementary Material
Complete ref. 27, substrate preparations, Tables S1 and S2, Figures S1–S6 and representative NMR spectra. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH Grant AI47238 (C.T.W.). NMR instrumentation was supported by Grant GM47467 from the National Institute of General Medical Sciences to Prof. Gehard Wagner. We thank Prof. Rosalba Lagos, Claudia Estevez and Mario Tello for a gift of the pJEM15 plasmid and for insightful discussions. We also thank Carl Balibar and Jay Read for helpful suggestions. EMN acknowledges the NIH for a post-doctoral fellowship, MAP thanks the Hertz Foundation for a graduate fellowship and AK is a recipient of a long-term fellowship from the Human Frontier Science Program Organization.
References
- 1.Duquesne S, Destoumieux-Garzón D, Peduzzi J, Rebuffat S. Nat Prod Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- 2.Pons AM, Lanneluc I, Cottenceau G, Sable S. Biochimie. 2002;84:531–537. doi: 10.1016/s0300-9084(02)01416-5. [DOI] [PubMed] [Google Scholar]
- 3.Vizán J, Hernandex-Chico C, del Castillo I, Moreno F. EMBO J. 1991;10:467–476. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heddle JG, Blance SJ, Zamble DB, Hollfelder F, Miller DA, Wentzell LM, Walsh CT, Maxwell A. J Mol Biol. 2001;307:1223–1234. doi: 10.1006/jmbi.2001.4562. [DOI] [PubMed] [Google Scholar]
- 5.Metlitskaya A, Kazakov T, Kommer A, Pavlova O, Praetorius-Ibba M, Ibba M, Krasheninnikov I, Kolb V, Khmel I, Severinov K. J Biol Chem. 2006;281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- 6.Thomas X, Destoumieux-Garzón D, Peduzzi J, Afonso C, Blond A, Birlirakis N, Goulard C, Dubost L, Thai R, Tabet JC, Rebuffat S. J Biol Chem. 2004;279:28233–28242. doi: 10.1074/jbc.M400228200. [DOI] [PubMed] [Google Scholar]
- 7.Raymond KN, Dertz EA, Kim SS. Proc Natl Acad Sci USA. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hantke K, Nicholson G, Rabsch W, Winkelmann G. Proc Natl Acad Sci USA. 2003;100:3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bister B, Bischoff D, Nicholson GJ, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Süssmuth RD. BioMetals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- 10.Luo M, Lin H, Fischbach MA, Liu DR, Walsh CT, Groves JT. ACS Chem Biol. 2006;1:29–32. doi: 10.1021/cb0500034. [DOI] [PubMed] [Google Scholar]
- 11.Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. Proc Natl Acad Sci USA. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischbach MA, Lin H, Liu DR, Walsh CT. Nat Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 13.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 14.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 15.Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. Structure. 2005;13:29–41. doi: 10.1016/j.str.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Strahsburger E, Baeza M, Monasterio O, Lagos R. Antimicrob Agents Chemother. 2005;49:3083–3086. doi: 10.1128/AAC.49.7.3083-3086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Destoumieux-Garzón D, Peduzzi J, Thomas X, Djediat C, Rebuffat S. BioMetals. 2006;19:181–191. doi: 10.1007/s10534-005-4452-9. [DOI] [PubMed] [Google Scholar]
- 18.Lagos R, Wilkens M, Vergara C, Cecchi X, Monasterio O. FEBS Lett. 1993;321:145–148. doi: 10.1016/0014-5793(93)80096-d. [DOI] [PubMed] [Google Scholar]
- 19.Destoumieux-Garzón D, Thomas X, Santamaria M, Goulard C, Barthélémy M, Boscher B, Bessin Y, Molle G, Pons AM, Letellier L, Peduzzi J, Rebuffat S. Mol Microbiol. 2003;49:1031–1041. doi: 10.1046/j.1365-2958.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- 20.Hertz C, Bono MR, Barros LF, Lagos R. Proc Natl Acad Sci USA. 2002;99:2696–2701. doi: 10.1073/pnas.052709699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieler S, Silva F, Soto C, Belin D. J Bacteriol. 2006;188:7049–7061. doi: 10.1128/JB.00688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilkens M, Villanueva JE, Cofré J, Chnaiderman J, Lagos R. J Bacteriol. 1997;179:4789–4794. doi: 10.1128/jb.179.15.4789-4794.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagos R, Villanueva JE, Monasterio O. J Bacteriol. 1999;181:212–217. doi: 10.1128/jb.181.1.212-217.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagos R, Baeza M, Corsini G, Hetz C, Strahsburger E, Castillo JA, Vergara C, Monasterio O. Mol Microbiol. 2001;42:229–243. doi: 10.1046/j.1365-2958.2001.02630.x. [DOI] [PubMed] [Google Scholar]
- 25.Corsini G, Baeza M, Monasterio O, Lagos R. Biochimie. 2002;84:539–544. doi: 10.1016/s0300-9084(02)01415-3. [DOI] [PubMed] [Google Scholar]
- 26.Bäumler AJ, Tsolis RM, van der Velden AWM, Stojijkovic I, Anic S, Heffron F. Gene. 1996;183:207–213. doi: 10.1016/s0378-1119(96)00560-4. [DOI] [PubMed] [Google Scholar]
- 27.Welch RA, et al. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu WS, Hsieh PC, Huang TM, Chang YF, Chang CF. DNA Seq. 2002;13:333–341. doi: 10.1080/1042517021000039221. [DOI] [PubMed] [Google Scholar]
- 29.Fischbach MA, Lin H, Liu DR, Walsh CT. Proc Natl Acad Sci USA. 2005;102:571–576. doi: 10.1073/pnas.0408463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin H, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2005;127:11075–11084. doi: 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trent MS, Worsham LMS, Ernst-Fonberg ML. Biochemistry. 1999;38:3433–3439. doi: 10.1021/bi982491u. [DOI] [PubMed] [Google Scholar]
- 32.Gaggero C, Moreno F, Laviña M. J Bacteriol. 1993;175:5420–5427. doi: 10.1128/jb.175.17.5420-5427.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scarrow RC, Ecker DJ, Ng C, Liu S, Raymond KN. Inorg Chem. 1991;30:900–906. [Google Scholar]
- 34.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 35.Lui M, Mao X, He C, Huang H, Nicholson JK, Lindon JC. J Magn Reson. 1998;132:125–129. [Google Scholar]
- 36.Goddard TD, Kneller DG. SPARKY-3. University of California; San Francisco: 2006. [Google Scholar]
- 37.Laviña M, Gaggero C, Moreno F. J Bacteriol. 1990;172:6586–6588. doi: 10.1128/jb.172.11.6585-6588.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patzer SI, Baquero MR, Bravo D, Moreno F, Hantke K. Microbiology. 2003;149:2557–2570. doi: 10.1099/mic.0.26396-0. [DOI] [PubMed] [Google Scholar]
- 39.Rodriquez E, Laviña M. Can J Microbiol. 1998:44. doi: 10.1139/cjm-44-7-692. [DOI] [PubMed] [Google Scholar]
- 40.Rodriquez E, Gaggero C, Laviña M. Antimicrob Agents Chemother. 1999;43:2176–2182. doi: 10.1128/aac.43.9.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azpiroz MF, Laviña M. Antimicrob Agents Chemother. 2004;48:1235–1241. doi: 10.1128/AAC.48.4.1235-1241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poey ME, Azpiroz MF, Laviña M. Antimicrob Agents Chemother. 2006;50:1411–1418. doi: 10.1128/AAC.50.4.1411-1418.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischbach MA, Walsh CT. Unpublished results. [Google Scholar]
- 44.Lin H, Fischbach MA, Gatto GJ, Liu DR, Walsh CT. J Am Chem Soc. 2006;128:9324–9325. doi: 10.1021/ja063236x. [DOI] [PubMed] [Google Scholar]
- 45.Brickman TJ, McIntosh MA. J Biol Chem. 1992;267:12350–12355. [PubMed] [Google Scholar]
- 46.Langman L, Young IG, Frost GE, Rosenberg H, Gibson F. J Bacteriol. 1972;112:1142–1149. doi: 10.1128/jb.112.3.1142-1149.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujiyama A, Tsunasawa S, Tamanoi F, Sakiyama F. J Biol Chem. 1991;266:17926–17931. [PubMed] [Google Scholar]
- 48.González-Pastor J, San Millán JL, Moreno F. Nature. 1994;369:281. doi: 10.1038/369281a0. [DOI] [PubMed] [Google Scholar]
- 49.Guijarro JI, González-Pastor JE, Baleux F, San Millán JL, Castilla MÃ, Rico M, Moreno F, Delepierre M. J Biol Chem. 1995;270:23520–23532. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- 50.González-Pastor JE, San Millán JL, Castilla MÃ, Moreno F. J Bacteriol. 1995;177:7131–7140. doi: 10.1128/jb.177.24.7131-7140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azpiroz MF, Laviña M. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.01606-06. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonner WA. J Am Chem Soc. 1958;80:3697–3700. [Google Scholar]
- 53.Bastida A, Fernández-Lafuente R, Fernández-Lafuente G, Guisán JM. Bioorg Med Chem Let. 1999;9:633–636. doi: 10.1016/s0960-894x(99)00053-0. [DOI] [PubMed] [Google Scholar]
- 54.Terreni M, Salvetti R, Linati L, Fernández-Lafuente R, Fernández-Lorente G, Bastida A, Guisan JM. Carbohydrate Res. 2002;337:1615–1521. doi: 10.1016/s0008-6215(02)00113-1. [DOI] [PubMed] [Google Scholar]
- 55.Rodrguez-Pérez T, Lavandera I, Fernández S, Sanghvi YS, Ferrero M, Gotor V. Eur J Org Chem. 2007:2769–2778. [Google Scholar]
- 56.Fernández-Lafuente G, Palomo JM, Cocca J, Mateo C, Moro P, Terreni M, Fernández-Lafuente R, Guisan JM. Tetrahedron. 2003;59:5705–5711. [Google Scholar]
- 57.Kowalczyk S, Bandurski RS. Plant Physiol. 1990;94:4–12. doi: 10.1104/pp.94.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vassiliadis G, Peduzzi H, Zirah S, Thomas X, Rebuffat S, Destoumieux-Garzón D. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.00261-07. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pramanik A, Braun V. J Bacteriol. 2006;188:3878–3886. doi: 10.1128/JB.00205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Braun V. Drug Resist Update. 1999;2:363–369. doi: 10.1054/drup.1999.0107. [DOI] [PubMed] [Google Scholar]
- 61.Ferguson AD, Braun V, Fielder HP, Coulton JW, Diederichs K, Welte W. Protein Sci. 2000;9:956–963. doi: 10.1110/ps.9.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao Z, Klebba PE. Biochimie. 2002;84:399–412. doi: 10.1016/s0300-9084(02)01455-4. [DOI] [PubMed] [Google Scholar]
- 63.Rabsch W, Ma L, Wiley G, Najar FZ, Kaserer W, Schuerch DW, Klebba JE, Roe BA, Laverde Gomez JA, Schallmey M, Newton SMC, Klebba PE. J Bacteriol. 2007 doi: 10.1128/JB.00437-07. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banin E, Vasil ML, Greenberg EP. Proc Natl Acad Sci USA. 2005;102:11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Budzikiewicz H. Current Topics Med Chem. 2001;1:73–82. doi: 10.2174/1568026013395524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete ref. 27, substrate preparations, Tables S1 and S2, Figures S1–S6 and representative NMR spectra. This information is available free of charge via the Internet at http://pubs.acs.org.