

Graphical abstract
.
Keywords: Chemo-enzymatic synthesis; Sialic acid; Acetylation; CMP-Neu5,9Ac2; Immunogens
Abstract
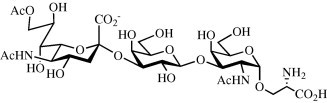
A chemo-enzymatic synthesis of [(5-acetamido-9-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-(β-d-galactopyranosyl)-(1→3)-O-(2-acetamido-2-deoxy-α-d-galactopyranosyl)]-l-serine acetate (1) has been accomplished by a regioselective chemical acetylation of Neu5Ac (2) to give 9-O-acetylated sialic acid 3, which was enzymatically converted into CMP-Neu5,9Ac2 (4) employing a recombinant CMP-sialic acid synthetase from Neisseria meningitis [EC 2.7.7.43]. The resulting compound was then employed for the enzymatic glycosylation of the C-3′ hydroxyl of chemically prepared glycosylated amino acid 10 using recombinant rat α-(2→3)-O-sialyltransferase expressed in Spodooptera frugiperda [EC 2.4.99.4] to give, after deprotection of the Nα-benzyloxycarbonyl (CBz)-protecting group of serine, target compound 1. The Nα-CBz-protected intermediate 11 can be employed for the synthesis of glycolipopeptides for immunization purposes.
1. Introduction
Sialic acids are a family of approximately 50 naturally occurring 2-keto-3-deoxy-nononic acids that are involved in a wide range of biological processes.1, 2, 3 The C-5-amino derivative represents the long-known neuraminic acid and its amino function can either be acetylated (Neu5Ac) or glycolylated (Neu5Gc). As terminal components of glycoproteins and glycolipids, neuraminic acids are α-(2→3)- or α-(2→6)-linked to galactosides or α-(2→6)-linked to 2-acetamido-galactosides.4, 5, 6
Enzymatic acetylation, lactylation, sulfation, methylation and phosphorylation of free hydroxyls give a large structural diversity of sialic acids.3, 7 O-Acetylation of sialic acids takes place in the Golgi apparatus by membrane bound O-acetyltransferases. Interestingly, different cell types show different substrate selectivity for acetylation of sialic acid indicating that there are different classes of acetyltransfereases.2, 3, 8 Alternatively, it may be possible that adaptor proteins control the substrate specificity. Initially, acetyl transferases introduce an acetyl ester at C-7 or C-9 of sialic acid and under physiological conditions, the C-7 acetyl ester can migrate to C-8 or C-9 making C-9 acetylation (Neu5,9Ac2) the most prevalent sialic acid modification of cell surface glycoconjugates.2, 7 Acetylation of the C-4 hydroxyl of sialosides has also been observed.9
It has been shown that 9-O-acetylation of sialic acids can dramatically influence biological properties of glycoconjugates.2, 7, 10 For example, O-acetylation of sialosides on the surface of thymocytes results in loss of binding to Siglec-2 (CD22β) on B-cells.11 Abrogation of 9-O-acetylated deleteriously affects development of transgenic mice. This modification is also involved in cell specific recognition by influenza C and corona viruses.12, 13 It has also been demonstrated that sialoglycoproteins having 9-O-acetylation are expressed on lymphoblasts of children with acute lymphoblastic leukemia.14
Glycoproteins and gangliosides from rat, mouse and chicken erythrocytes express Neu5,9Ac2-α-(2→3)-Gal-β-(1→3)-GalNAc, which serves as targets for the binding of naturally occurring antibodies.12, 15, 16 In microorganisms, such as the trypanosoma parasite Crithidia fasciculata and the pathogenic fungus Cryptococcus neoformans, Neu5,9Ac2 can be glycosidically linked to Gal-β-(1→3)-GalNAc.17, 18 The potential impact of 9-O-acetylation of the GD1a ganglioside on the overall conformation and recognition by a natural human antibody has been investigated by molecular dynamics simulations and NMR. It was found that acetylation did not affect the conformational properties of the oligosaccharide. However, it was shown that an antibody specific for a 9-O-acetylated ganglioside can make direct interactions with the acetyl group rationalizing the selective recognition of acetylated derivatives.16, 19
As part of a program to develop monoclonal antibodies against sialo-oligosaccharides bearing 9-O-acetyl esters, we report here an efficient chemo-enzymatic synthesis of Neu5,9Ac2-α-(2→3)-Gal-β-(1→3)-GalNAc-O-Ser (1). It is to be expected that this compound can be employed for the preparation of fully synthetic immunogens for the development of monoclonal antibodies.20, 21, 22
2. Results and discussion
While relatively efficient methods have been developed for the introduction of sialosides, the preparation of acetylated analogues is less well developed. It was anticipated that compound 1 could be prepared by a regioselective chemical acetylation23 of Neu5Ac (2) to give 9-O-acetylated sialic acid 3, which can then be enzymatically converted into CMP-Neu5,9Ac2 (4) employing the recombinant CMP-sialic acid synthetase from Neisseria meningitis. This enzyme possesses a flexible substrate specificity and has been employed for the synthesis of CMP-sialic acid analogues such as CMP-Neu5,9Ac2.24, 25, 30 The resulting compound can then be used for the enzymatic glycosylation26 of the C-3′ hydroxyl of chemically prepared glycopeptide 10 using the recombinant rat α-(2→3)-sialyltransferase [EC 2.4.99.4] expressed in Spodooptera frugiperda to give, after removal of the N α-CBz protecting group of serine, target compound 1.
Sialyltransferases exhibit some flexibility with regard to the structure of their acceptors and are able to transfer modified sialic acids from corresponding CMP-derivatives.24, 25, 27 For example, a rat liver α-(2→3)-sialyltransferase has been used to transfer Neu5,9Ac2 from the corresponding CMP-donor to various glycoproteins,28 and a porcine liver α(2→6)-sialyltransferase has been employed for the chemo-enzymatic synthesis of Neu5,9Ac2-α-(2→6)-Gal-β-(1→4)-GlcNAc-α-OCH3.24 Recently, a library of (2→6)-linked modified sialosides of Gal-β-(1→4)-GlcNAc-α-O-(CH2)3N3, which includes four 9-O-acetylated sialoside derivatives, has been prepared using the flexible recombinant Photobacterium damsela [EC 2.4.99.1] α-(2→6)-sialyltransferase by a one-pot three-enzyme system.29 The enzymatic synthesis of the Neu5,9Ac2-α-(2→3)-Gal-O-p-nitrophenyl glycoside has also been reported.30
It was anticipated that C-9 derivatives of sialic acid (Neu5Ac) would be readily available from Neu5Ac by a chemical regioselective acetylation.23 Thus, treatment of Neu5Ac (2) with trimethylorthoacetate in DMSO in the presence of a catalytic amount of p-TsOH·H2O gave, after purification by an ion exchange column chromatography (Dowex 50W-X2, form, eluent: a mixture of H2O and formic acid) followed by a size exclusion chromatography (Biogel P-2; eluent: H2O) Neu5,9Ac2 (3) in an excellent yield of 92% (Scheme 1 ). Condensation of Neu5,9Ac2 (3) with CTP in the presence of the commercial recombinant CMP-sialic acid synthetase from N. meningitis [EC 2.7.7.43] and the inorganic pyrophosphatase from Thermus thermophilus [EC 3.1.3.1] afforded CMP-Neu5,9Ac2 (4). The reported purification strategy25 gave a compound 4 contaminated with phosphorylated nucleotides (CTP, CDP and CMP) and a small amount of CMP-Neu5Ac and Neu5Ac. However, it was found that highly pure 4 could be obtained by removal of inorganic salts by selective precipitation31 in an ethanol–H2O mixture (9:1, v:v, twice) followed by size exclusion chromatography over extra fine Biogel P-2 using ammonium bicarbonate buffer as eluent at 4 °C.
Scheme 1.
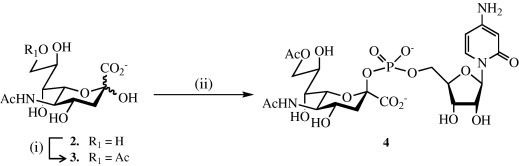
Reagents and conditions: (i) CH3(OCH3)3, DMSO, p-TsOH·H2O, 92%; (ii) CTP, 3, Tris buffer (0.1 M, pH 7.5), CMP-sialic acid synthetase, inorganic pyrophosphatase.
Glycosylation of thioglycoside 5 32 with properly protected serine derivative 6 26 in the presence of Ph2SO–Tf2O and DTBMP provided the α-linked glycopeptide 7 in 83% yield with excellent α-anomeric selectivity (Scheme 2 ). Treatment of 7 with aqueous acetic acid removed the benzylidene acetal and acetylation of the resulting free hydroxyls gave compound 8 in excellent overall yield. Next, the azido moiety of 8 was converted into an acetamido derivative by reaction with thioacetic acid in pyridine33, 34, 35 and the resulting compound 9 was treated with a mixture of TFA and water to remove the t-butyl ester, and then with NaOMe in methanol to cleave the acetyl esters to give glycopeptide 10.36 At this stage of the synthesis, the Cbz group was kept intact because protection of the amino group of serine is required for glycopeptide synthesis.
Scheme 2.
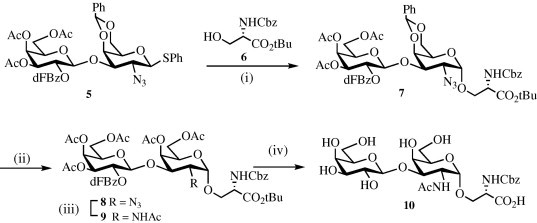
Reagents and conditions: (i) Ph2SO, DTBMP, CH2Cl2, −70 °C, Tf2O then 6, 83%; (ii) (a) CH3CO2H–H2O, 80 °C, 1 h; (b) Ac2O, Py, DMAP, 92%; (iii) CH3COSH, Py, 16 h, 67%; (iv) (a) TFA, H2O, 1 h; (b) CH3ONa, CH3OH, pH < 10, 86%.
Next, attention was focused on the enzymatic sialylation of 10 using a CMP-Neu5,9Ac2 donor (4) (Scheme 3 ). Thus, the recombinant rat α-(2→3)-sialyltransferase [EC 2.4.99.4] expressed in S. frugiperda and a calf intestine alkaline phosphatase [EC 3.6.1.1] were added to a mixture of 4 and 10 dissolved in cacodylate buffer (50 mM; pH 6.2) containing Triton X-100 and BSA.37 After incubation at 37 °C for 2 h, another portion of alkaline phosphatase and CMP-Neu5,9Ac2 (4) was added and after occasional stirring for an additional 3.5 h, TLC analysis showed complete consumption of the starting material 4 and the formation of a new product. Purification of the crude reaction mixture by a reverse-phase C18 column chromatography followed by an ion exchange chromatography using Biorad AG 50W-X8 (100–200 mesh, Na+ form) at 4 °C, gave a pure trisaccharide 11 in a yield of 69%. In addition, a small amount (<5%) of 9-O-deacetylated product was also isolated.
Scheme 3.
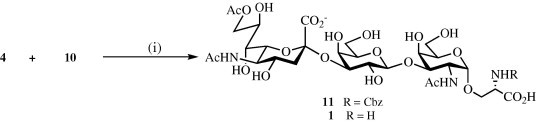
Reagents and condition: (i): α-(2→3)-sialyltransferase, cacodylate buffer (50 mM, pH 6.2), alkaline phosphatase; Pd–C, CH3OH–H2O–AcOH.
Compound 11 is appropriately protected for glycopeptide synthesis. However, fully unprotected 1 could be obtained by catalytic hydrogenation over Pd–C to remove the N α-Cbz group followed by purification by a Biogel P-2 size exclusion column chromatography. The chemical shift of the C-9 protons clearly demonstrated the presence of the C-9 acetyl ester: 4.37 (dd, 1H, Hz, Hz, H-9″a) and 4.19 (dd, 1H, Hz, Hz, H-9″b).
In conclusion, we have developed an efficient chemo-enzymatic synthesis of the trisaccharide 11, which is appropriately protected for glycopeptide synthesis. It has been shown that the rat recombinant α-(2→3)-sialyltransferase expressed in S. frugiperda can accept CMP-Neu5,9Ac2 as a donor. The improved purification protocol for CMP-Neu5,9Ac2 was essential for the successful transfer of CMP-Neu5,9Ac2 to glycosyl acceptor Gal-β-(1→3)-GalNAc-O-Ser.
3. Experimental
3.1. General methods
The recombinant N. meningitis CMP-sialic acid synthetase expressed in Escherichia coli [EC 2.7.7.43], the recombinant rat α-(2→3)-sialyltransferase [EC 2.4.99.4] expressed in S. frugiperda and calf intestine alkaline phosphatase [EC 3.6.1.1] were purchased from Calbiochem. The inorganic pyrophosphatase from T. thermophilus [EC 3.1.3.1], bovine serum albumin (BSA), cytidine 5′-monophosphate (CMP), cytidine 5′-triphosphate (CTP), 1,4-dithioerythritol (DET), manganese(II) chloride (MnCl2) and Triton X-100 were purchased from Sigma. All other chemicals were purchased from Aldrich. Solvents were dried using standard procedures, and all chemical reactions were performed under an atmosphere of Argon. Column chromatography was performed on F60 Silica Gel (60–200 μm) from Silicycle. Bio-Gel P-2 Gel fine (200–3000 Da; 45–90 μm), Bio-Gel P-2 extra fine (200–3000 Da, <45 μm) and AG 50W-X8 (100–200 mesh) ion exchange resin were obtained from Bio-Rad; preparative C18 silica gel (125 Å, 55–105 μm) was obtained from Waters and Iatrobeads 6RS-8060 (60 μm) were purchased from Bioscan. Thin layer chromatography (TLC) was performed using Kieselgel 60 F254 (Merck) plates with detection by UV and/or by charring with 5% sulfuric acid in ethanol, cerium ammonium nitrate or p-anisaldehyde reagents. Solvent evaporations were done in vacuo with a bath temperature <25 °C. Optical rotations were measured with a Jasco P-1020 polarimeter. Both positive and negative ion matrix-assisted laser desorption ionization time of flight (MALDI-TOF) mass spectra were recorded using an Applied Biosystems 4700 instrument with gentisic acid as matrix. 1D and 2D 1H NMR and 13C NMR spectra were recorded at 25 °C on Varian Inova300, Varian Inova500, Varian Inova600 or Varian Inova900 spectrometers. Assignments were obtained from COSY and HSQC spectra. For 1H spectra recorded in CDCl3, D2O and CH3OD and 13C spectra recorded in CDCl3, chemical shifts are given in ppm relative to solvents peaks (CDCl3, 1H 7.24; 13C, 77.00; D2O, 1H, 4.76; CH3OD, 1H, 3.31, 4.78). 13C Spectra recorded in D2O were indirectly referenced.
3.2. N-(Benzyloxycarbonyl) [2-azido-4,6-O-benzylidene-2-deoxy-3-O-(3,4,6-tri-O-acetyl-2-O-(2,5-difluorobenzoyl)-β-d-galactopyranosyl)-(1→3)-α-d-galactopyranosyl]-l-serine t-butyl ester (7)
Ph2SO (83 mg, 0.411 mmol), di-t-butyl-3-methyl pyridine (90.4 mg, 0.441 mmol) and crushed activated molecular sieves 4 Å (500 mg) were added to a cooled solution (−70 °C) of disaccharide 5, (120 mg, 0.147 mmol) in CH2Cl2 (6 mL) prior to the addition of trifluoromethanesulfonic anhydride (58 mg, 36 μL, 0.205 mmol). After stirring the mixture at −70 °C for 5 min, a solution of 6 (85.5 mg, 0.29 mmol) in CH2Cl2 (0.5 mL) was added dropwise by syringe. The resulting reaction mixture was stirred at −60 °C for 1 h and then allowed to warmed to room temperature over a period of 3 h. TLC analysis (hexanes–acetone 70:30, v:v) indicated completion of the reaction, which was quenched by the addition of solid NaHCO3. After addition of CH2Cl2, the mixture was filtered, washed with H2O, aqueous NaHCO3 and brine, dried (MgSO4), filtered and concentration in vacuo. The residue was purified by silica gel column chromatography (hexanes–EtOAc 65:35, v:v) to provide 7 (122 mg, 83%); +22 (c 0.2; CHCl3); 1H NMR (CDCl3): δ 7.30–7.00 (m, 13H, aromatics), 5.66 (d, 1H, J NH,Hα = 8.0 Hz, NH), 5.51 (m, 2H, H-2′, CH, benzylidene), 5.43 (d, 1H, Hz, H-4′), 5.16 (dd, Hz, Hz, H-3′), 5.10 (s, 2H, PhCH 2), 4.91 (d, 1H, J 1,2 = 3.0 Hz, H-1), 4.86 (d, 1H, Hz, H-1′), 4.40–3.66 (m, 12H), 2.16, 2.03, 1.93 (3s, 3H each, 3 × CH3, OAc), 1.44 (s, 9H, CH3 t-Bu); 13C NMR: δ 170.5, 170.3, 137.7, 129.1 128.8, 128.6, 128.4, 128.3, 126.3, 118.4, 118.1, 102.4 (C-1), 100.8 (PhCh), 99.9 (C-1), 83.1, 75.8, 75.7, 71.3, 71.1, 70.0, 69.9, 69.2, 67.3, 63.7, 61.6, 58.9, 55.1, 28.1, 20.9, 20.7; HRMALDI-ToF-MS: calcd for C47H52F2N4O18 [M+Na]+: m/z: 1021.3231; found m/z: 1021.3224.
3.3. N-(Benzyloxycarbonyl) [4,6-di-O-acetyl-2-azido-2-deoxy-3-O-(3,4,6-tri-O-acetyl-2-O-(2,5-difluorobenzoyl)-β-d-galactopyranosyl)-(1→3)-α-d-galactopyranosyl]-l-serine t-butyl ester (8)
A solution of disaccharide 7 (143 mg, 0.142 mmol) in a mixture of CH3CO2H (4 mL) and H2O (1 mL) was kept at 80 °C for 1 h after which TLC analysis (hexanes–EtOAc 1:1, v:v) indicated completion of the reaction. The mixture was co-evaporated with toluene and the residue dried in vacuo. The recovered material was acetylated with pyridine (2 mL) and Ac2O (0.2 mL) in the presence of a catalytic amount of dimethylaminopyridine (DMAP) for 12 h. After the addition of CH3OH, the mixture was co-evaporated with toluene, the residue dissolved in CH2Cl2 and washed with aqueous NaHCO3, H2O and brine. The organic solvents were dried (MgSO4), filtered and the filtrate concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes–acetone 70:30, v:v) to afford 8 (129 mg, 92%); +52.8 (c 0.2; CHCl3); 1H NMR (CDCl3): δ 7.3–7.0 (m, 8H, aromatics), 5.65 (d, 1H, J NH,Hα = 8.0 Hz, NH), 5.45–5.35 (m, 3H, H-4, H-2′, H-4′), 5.16–5.08 (m, 3H, H-3′, PhCH 2), 4.85 (d, J 1,2 = 3,3 Hz, H-1), 4.77 (d, 1H, Hz, H-1′), 4.40 (m, 1H, HαSer), 4.18–3.84 (m, 10H), 3.58 (dd, 1H, J 1,2 = 3.3 Hz, J 2,3 = 10.7 Hz, H-2), 2.16, 2.10, 2.03, 2.02, 1.92 (5s, 15H, 5 × CH3, OAc), 1.42 (s, 9H, CH3, t-Bu); 13C NMR: δ 170.7, 170.6, 170.4, 170.2, 169.8 168.6, 136.3, 129.2, 128.8, 128.6, 128.5, 128.4, 101.5, 98.9, 83.1, 74.6, 71.2, 70.8, 70.1, 69.6, 69.4, 68.1, 67.3, 67.0, 62.8, 61.2, 59.6, 55.0, 28.0, 20.9, 20.8, 20.7; HRMALDI-ToF-MS: calcd for C44H52F2N4O20 [M+Na]+: m/z: 1017.3124; found m/z: 1017.3141.
3.4. N-(Benzyloxycarbonyl) [-2-acetamido-4,6-di-O-acetyl-2-deoxy-3-O-(3,4,6-tri-O-acetyl-2-O-(2,5-difluorobenzoyl)-β-d-galactopyranosyl)-(1→3)-α-d-galacto-pyranosyl]-l-serine t-butyl ester (9)
Freshly distilled thioacetic acid (0.6 mL) was added by syringe to a solution of disaccharide 8 (128 mg, 0.128 mmol) in pyridine. The resulting reaction mixture was stirred under an atmosphere of argon for 18 h after which TLC (hexanes–EtOAc 1:1, v:v) indicated completion of the reaction. The mixture was diluted with CH2Cl2, successively washed with H2O, aqueous NaHCO3 and brine, dried (MgSO4), filtered and the filtrate concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes–EtOAc 55:45, v:v) to give 9 (86 mg, 67%); +66 (c 0.1; CHCl3); 1H NMR (CDCl3): δ 7.3–7.0 (m, 8H, aromatics), 5.55 (d, 1H, J NH,Hα = 8.0 Hz, NH), 5.45–5.35 (m, 3H, H-4, H-2′, H-4′), 5.16–5.30 (m, 3H, H-3′, PhCH 2), 4.78 (d, J 1,2 = 3,3 Hz, H-1), 4.68 (d, 1H, Hz, H-1′), 4.50 (m, 1H, H-2), 3.38 (m, 1H, HαSer), 4.20–3.80 (m, 8H), 3.70 (dd, 1H, J Hα,Hβ = 3.0 Hz, Hz, HβSer), 2.14, 2.10, 2.03, 2.02, 1.92 (6s, 18H, 5 × CH3, OAc and NHAc), 1.39 (s, 9H, CH3, t-Bu); 13C NMR: δ 170.5, 170.4, 170.3, 170.1, 170.0, 169.5, 168.9, 135.9, 128.6, 128.5, 128.3, 118.4, 118.0, 100.5 (C-1′), 98.2 (C-1), 82.8, 74.8, 70.9, 70.7, 70.2, 68.6, 68.2, 67.8, 67.2, 66.7, 62.8, 60.8, 54.5, 48.2, 22.6, 20.7, 20.6, 20.5; HRMALDI-ToF-MS: calcd for C46H56F2N2O4 [M+Na]+: m/z: 1033.3362; found m/z: 1033.3341.
3.5. N-(Benzyloxycarbonyl)-[-2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-α-d-galactopyranosyl]-l-serine (10)
H2O (50 μL) was added to a solution of the disaccharide 9 (83 mg, 0.082 mmol) in trifluoroacetic acid (0.95 mL). The mixture was stirred at 22 °C for 1 h until TLC (hexanes–acetone 1:1, v:v) indicated completion of the reaction. The mixture was co-evaporated with toluene and the residue dried in vacuo. The crude material was dissolved in dry CH3OH (3 mL), and a solution of NaOCH3 in CH3OH (0.5 M) was carefully added while keeping the pH < 10 and the reaction monitored by TLC (CHCl3–CH3OH–H2O 65:35:5, v:v:v). The reaction was neutralized by the addition of Dowex 50W-X2 (H+ form). After filtration, the product was dry-loaded on Iatrobeads (200 mg) and purified by chromatography on a Iatrobeads column (1.8 g) (CHCl3–CH3OH–H2O 65:35:5, v:v:v) to afford disaccharide 10 (43 mg, 83%); +85 (c 0.09; H2O); 1H NMR (CD3OD, 500 MHz): δ 7.30 (m, 5H, aromatics), 5.04 (m, 2H, PhCH 2), 4.73 (J 1,2 = 3.2 Hz, H-1), 4.33–4.25 (m, 3H, H-1′, H-2, HαSer), 4.06 (br d, J 3,4 = 2.6 Hz, H-4), 3.87 (dd, 1H, J Hα,Hβ = 3.3 Hz, Hz, HβSer), 3.83–3.75 (m 3H, H-3, H-5, Hβ′Ser), 3.72 (d, 1H, Hz, H-4′), 3.68–3.57 (m, 4H, H6a, H6b, H6′a, H6′b), 3.47–3.38 (m, 2H, H-2′, H-5′), 3.34 (dd, 1H, Hz, Hz, H-3′), 1.86 (s, 3H, CH3, NHAc); 13C NMR (D2O): δ 174.8, 174.7, 136.4, 129.9, 128.9, 128.6, 127.9, 104.7, 98.2, 77.0, 75.1, 72.7, 71.0, 70.8, 68.7, 68.2, 67.3, 61.2, 61.1, 55.3, 48.7, 22.16; HRMALDI-ToF-MS: calcd for C25H36N2O15 [M+Na+]: m/z: 627.2147; found m/z: 627.2156.
3.6. 5-Acetamido-9-O-acetyl-3,5-dideoxy-d-glycero-β-d-galacto-2-nonulopyranosylonic acid (3)
Trimethyl orthoacetate23 (2.0 mL, 16.0 mmol) and p-TsOH·H2O (20 mg) were added to a solution of Neu5Ac 2 (0.5 g, 1.6 mmol) in DMSO (5.0 mL) under an atmosphere of argon. The reaction mixture was stirred for 1.5 h and then purified by anion exchange resin column chromatography using Dowex 50W-X2 ( form, 40 × 1 cm). Elution with H2O (110 mL), followed by formic acid (1 N, 75 mL) provided appropriate fractions (detected by TLC), which were combined and first evaporated under vacuum (T < 25 °C) and then lyophilized to afford crude Neu5,9Ac2 3 (0.54 g, 1.54 mmol, 96%). The resulting material was dissolved in H2O (1 mL) and then loaded on a Biogel P-2 fine column (75 × 2.5 cm, flow rate 20.5 mL/h, H2O) to give pure 3 (0.52 g, 92.5%); [α]D −8.6 (c 1; H2O); 1H NMR (D2O, 300 MHz): δ 4.36 (dd, 1H, J 8,9a = 2.4 Hz, J 9a,9b = 11.6 Hz, H-9a), 4.17 (dd, 1H, J 8,9b = 5.5 Hz, J 9a,9b = 11.6 Hz, H-9b), 4.10–3.80 (m, 4H), 3.60 (d, 1H, J 7,8 = 9.4 Hz, H-7), 2.30 (dd, 1H, J 3eq,4 = 4.9 Hz, J 3eq,3ax = 13.0 Hz, H-3eq), 2.10 and 2.04 (2s, 6H, 2 × CH3, OAc, NHAc), 1.86 (dd, J 3ax,4 = 12.2 Hz, J 3eq,3ax = 13.0 Hz, H-3ax); HRMALDI-ToF-MS: calcd for C13H21NO10 [M+Na]+: m/z: 374.1118; found m/z: 374.1134.
3.7. Cytidine 5′-(5-acetamido-9-O-acetyl-3,5-dideoxy-β-d-glycero-d-galacto-2-nonulopyranosylonic acid monophosphate) ammonium salt (4)
CTP (126 mg, 0.239 mmol) was added to a solution of Neu5,9Ac2 3 (50 mg, 0.142 mmol) in a Tris–HCl buffer (0.1 M, 9 mL, pH 7.5) containing DET (4.75 mM) and MnCl2 (13.80 mM). Addition of CTP resulted in a change of pH from 7.5 to 6.5, which was re-adjusted to pH 7.5 using aqueous NaOH (0.3 M). The recombinant CMP-sialic acid synthetase from N. meningitis (9.2 U, 27.0 μL) and the inorganic pyrophosphatase from T. thermophilus (4.6 U, 60.0 μL) were added, and the reaction mixture was incubated at 37 °C for 1 h with occasional shaking. Another portion of CMP-sialic acid synthetase and inorganic pyrophosphatase (1.5 U, 19.5 μL) was added after 2.5 h. The formation of a white precipitate (presumably manganese–ammonium phosphate) was observed after 1.5 h. The progress of the reaction was monitored by TLC (EtOH–1 M NH4HCO3, 4:1, v:v), which after 5 h indicated completion of the reaction. Ethanol (80 mL) was added and the mixture was kept on ice for 2 h prior to centrifugation. The supernatant was decanted and the pellet (mostly inorganic salts) was re-suspended in EtOH (30 mL), cooled on ice for 1 h and centrifuged. The combined ethanol extracts were concentrated in vacuo providing crude material (230 mg). Ethanol (1.8 mL) was slowly added to the material dissolved in H2O (0.2 mL) and precipitation occurred immediately. The mixture was kept on ice for 2 h, and the supernatant was removed after centrifugation. After drying, NMR (D2O) of the first pellet (114 mg) indicated the presence of the expected product, some Tris, ethanol and a small amount of Neu5,9Ac2. The recovered material was again dissolved in H2O (0.2 mL) and re-precipitated by the addition of ethanol (1.80 mL) as described above providing 75 mg of material, which was loaded on a column of extra-fine Biogel P-2 (75 × 1.5 cm) and eluted with 0.1 M NH4HCO3 at 4 °C. The appropriate fractions were detected by UV and TLC (as above), collected, concentrated in vacuo (bath temperature <25 °C) and lyophilized from H2O (three times to completely remove NH4HCO3) to afford 3 (60 mg, 61%, ammonium salt). −4 (c 1.5; H2O); 1H NMR (D2O, 500 MHz): δ 7.94 (d, 1H, J 5,6 = 7.4 Hz, H-5, cyt.), 6.12 (d, 1H, J 5,6 = 7.4 Hz, H-5, cyt.), 6.00 (d, 1H, J 1,2 = 4.9 Hz, H-1 rib.), 4.40–4.10 (m, 8H), 4.03 (ddd, 1H, J 8,9b = 5.1 Hz, J 7,8 = 9.8 Hz, J 8,9a = 11.0 Hz, H-8), 3.95 (t, 1H, J 4,5 = J 5,6 = 10.4 Hz, H-5), 3.49 (d, 1H, J 7,8 = 9.8 Hz, H-7), 2.47 (dd, 1H, J 3eq,4 = 4.9 Hz, J 3eq,3ax = 13.4 Hz, H-3eq), 2.08 and 2.04 (2s, 6H, 2 × CH3, OAc, NHAc), 1.64 (ddd, 1H, J 3ax,P = 5.8 Hz, J 3ax,3eq = 13.4 Hz, J 3ax,4 = 12.5 Hz, H-3ax) in agreement with reported data;24 HRESIMS: calcd for C23H24N3O17P [M−H]−: m/z: 655.1626; found 654.1469.
3.8. Sodium N-(benzyloxycarbonyl)-[(5-acetamido-9-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-(β-d-galactopyranosyl)-(1→3)-O-(2-acetamido-2-deoxy-α-d-galactopyranosyl)]-l-serine sodium salt (11)
Recombinant rat α-(2→3)-sialyltransferase from S. frugiperda [EC 2.4.99.4] (10 mU, 7 μL) and calf intestine alkaline phosphatase (15 U, 15 μL) were added to a mixture of disaccharide 10 (4.6 mg, 7.1 μmol) in cacodylate buffer (50 mM, pH 6.2; 600 μL) containing 0.1% Triton X-100, 0.1% BSA and CMP-Neu5,9Ac2 4 (6.7 mg, 9.5 μmol) in an Eppendorf tube, and five similar reactions were run in parallel. The tubes were incubated at 37 °C, and progress of the reactions were monitored by TLC (CHCl3–CH3OH–H2O–AcOH 60:40:10:0.5, v:v:v:v). After 2 h, an additional amount of α(2→3)-sialyltransferase (10 mU), calf intestine alkaline phosphatase and CMP-Neu5,9Ac2 (7.0 mg) was added to each of the five tubes. After 5.5 h, TLC indicated the disappearance of disaccharide 10. Each reaction was quenched by the addition of CH3OH (10 μL), combined and freeze dried to provide a crude material (200 mg), which was applied on a Biogel fine P-2 column (75 × 1.5 cm, using H2O as eluent at 8 mL/h) at 4 °C. The product was detected by TLC, and appropriate fractions were combined and freeze dried to provide 11 (37 mg). This material was passed through a short column of BioRad AG 50W-X8 (100–200 mesh, Na+) resin at 4 °C, and the appropriate fractions freeze dried. The recovered material was purified by C-18 silica gel (5 g) column chromatography using a gradient of CH3CN in H2O (95:5→90:10, v:v, at 4 °C), which removed the 9-O-deacetylated trisaccharide (1.0 mg) from the product 11 (22.5 mg, 69%); +12 (c 0.05; H2O); 1H NMR (600 MHz): δ 7.40 (m, 5H, aromatics), 5.18–5.08 (m, 2H, PhCH 2), 4.85 (d, 1H, J 1,2 = 3.3 Hz, H-1), 4.50 (d, 1H, Hz, H-1′), 4.35 (br d, 1H, Hz, H-9″a), 4.28 (dd, 1H, J 1,2 = 3.3 Hz, J 2,3 = 11.0 Hz, H-2), 4.20–4.15 (m, 3H, H-4, H-9″b, HαSer), 4.08–4.00 (m, 3H, H-3, H-3′, H-8″), 3.95–3.90 (m, 3H), 3.83 (t, 1H, Hz, H-5″), 3.78 (m, 1H, Hβ′Ser), 3.73–3.55 (m, 8H), 3.50 (dd, 1H, Hz, Hz, H-2′), 2.74 (dd, 1H, Hz, Hz, H-3″eq), 2.10 (s, 3H, OAc), 2.01, 1.95 (2s, 6H, NHAc), 1.77 (t, 1H, 12.5 Hz, H-3″ax). HRMALDI-ToF-MS: calcd for C38H55N3O24 [M+Na]+: m/z: 960.3342; m/z: found 960.3351.
3.9. [(5-Acetamido-9-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-O-(β-d-galactopyranosyl)-(1→3)-O-(2-acetamido-2-deoxy-α-d-galacto-pyranosyl)]-l-serine acetate (1)
Pd–C (10%, 18 mg) was added to trisaccharide 11 (20.0 mg, 0.021 mmol) in a mixture of H2O and CH3OH (1:1, 3.5 mL) containing CH3CO2H (0.14 mL). The mixture was hydrogenated at 1 atm for 30 min after which TLC (C2H5OH–1 M NH4CH3CO2 8:2, v:v) indicated completion of the reaction. The mixture was filtered through a 0.2 μM PTFE syringe filter, the solvents were evaporated and the residue was freeze dried from H2O to provide trisaccharide 1 (18.1 mg, 100%). +8.4 (c 0.1; H2O); 1H NMR (900 MHz): δ 4.91 (d, 1H, J 1,2 = 3.0 Hz, H-1), 4.52 (d, 1H, Hz, H-1′), 4.37 (dd, 1H, Hz, Hz, H-9″a), 4.35 (dd, 1H, J 1,2 = 3.2 Hz, J 2,3 = 11.0 Hz, H-2). 4.23 (d, 1H, J 3,4 = 2.5 Hz, H-4), 4.19 (dd, 1H, Hz, Hz, H-9″b), 4.11 (dd, 1H, J Hα,Hβ = 2.8 Hz, Hz, HαSer), 4.09–4.03 (m, 3H, H-3, H-3′, H-8″), 3.97 (m, 2H, Hβ-Ser, H-5), 3.92 (d, 1H, Hz H-4′), 3.88 (dd, 1H, J Hα,Hβ = 5.2 Hz, Hβ-Ser), 3.85 (t, Hz, H-5″), 3.80–3.64 (m, 4H, H6a, H6b, H6′a, H6′b), 3.61 (m, 2H, H-5′, H-7″), 3.52 (dd, 1H, Hz, Hz, H-2′), 2.74 (dd, 1H, Hz, Hz, H-3″eq), 2.12 (s, 3H, OAc), 2.02, 1.98 (2s, 6H, NHAc), 1.97 (s, CH 3CO2H), 1.78 (t, 1H, Hz, H-3″ax); HRMALDI-ToF-MS: calcd for C30H49N3O22 [M+Na]+: m/z: 826.3025; found: m/z: 826.3033.
Acknowledgement
This research was supported by the National Cancer Institute of the National Institute of Health (Grant No. RO1 CA88986).
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.carres.2008.05.002.
Supplementary data
NMR spectra of compounds.
References
- 1.Schauer R. Zoology (Jena) 2004;107:49–64. doi: 10.1016/j.zool.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Traving C., Schauer R. Cell. Mol. Life Sci. 1998;54:1330–1349. doi: 10.1007/s000180050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angata T., Varki A. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura K., Kojima H., Suzuki M., Suzuki A., Tamai Y. Eur. J. Biochem. 2000;267:5198–5208. doi: 10.1046/j.1432-1327.2000.01590.x. [DOI] [PubMed] [Google Scholar]
- 5.Vankar Y.D., Schmidt R.R. Chem. Soc. Rev. 2000;29:201–216. [Google Scholar]
- 6.Brocke C., Kunz H. Bioorg. Med. Chem. 2002;10:3085–3112. doi: 10.1016/s0968-0896(02)00135-9. [DOI] [PubMed] [Google Scholar]
- 7.Schauer R. Glycoconjugate J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butor C., Diaz S., Varki A. J. Biol. Chem. 1993;268:10197–10206. [PubMed] [Google Scholar]
- 9.Tiralongo J., Schmid H., Thun R., Iwersen M., Schauer R. Glycoconjugate J. 2000;17:849–858. doi: 10.1023/a:1010965128335. [DOI] [PubMed] [Google Scholar]
- 10.Lewis A.L., Cao H., Patel S.K., Diaz S., Ryan W., Carlin A.F., Thon V., Lewis W.G., Varki A., Chen X., Nizet V. J. Biol. Chem. 2007;282:27562–27571. doi: 10.1074/jbc.M700340200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sjoberg E.R., Powell L.D., Klein A., Varki A. J. Cell Biol. 1994;126:549–562. doi: 10.1083/jcb.126.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers G.N., Herrler G., Paulson J.C., Klenk H.D. J. Biol. Chem. 1986;261:5947–5951. [PubMed] [Google Scholar]
- 13.Kunkel F., Herrler G. Virology. 1993;195:195–202. doi: 10.1006/viro.1993.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandal C., Chatterjee M., Sinha D. Br. J. Haematol. 2000;110:801–812. doi: 10.1046/j.1365-2141.2000.02105.x. [DOI] [PubMed] [Google Scholar]
- 15.Gowda D.C., Reuter G., Shukla A.K., Schauer R. Hoppe-Seylers Z. Physiol. Chem. 1984;365:1247–1253. doi: 10.1515/bchm2.1984.365.2.1247. [DOI] [PubMed] [Google Scholar]
- 16.Siebert H.C., von der Lieth C.W., Dong X., Reuter G., Schauer R., Gabius H.J., Vliegenthart J.F. Glycobiology. 1996;6:561–572. doi: 10.1093/glycob/6.6.561-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matta M.A.D., Alviano D.S., Couceiro J.N.D.S., Nazareth M., Meirelles L., Alviano C.S., Angluster J. Parasitol. Res. 1999;85:293–299. doi: 10.1007/s004360050551. [DOI] [PubMed] [Google Scholar]
- 18.Rodrigues M.L., Rozental S., Couceiro J.N.S.S., Angluster J., Alviano C.S., Travassos L.R. Infect. Immun. 1997;65:4937–4942. doi: 10.1128/iai.65.12.4937-4942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal S., Chatterjee M., Bhattacharya D.K., Bandhyopadhyay S., Mandal C. Glycobiology. 2000;10:539–549. doi: 10.1093/glycob/10.6.539. [DOI] [PubMed] [Google Scholar]
- 20.Buskas T., Ingale S., Boons G.J. Angew. Chem., Int. Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]
- 21.Ingale S., Wolfert M.A., Gaekwad J., Buskas T., Boons G.J. Nat. Chem. Biol. 2007;3:663–667. doi: 10.1038/nchembio.2007.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bundle D.R. Nat. Chem. Biol. 2007;3:604–606. doi: 10.1038/nchembio1007-605. [DOI] [PubMed] [Google Scholar]
- 23.Ogura H., Furuhata K., Sato S., Anazawa K., Itoh M., Shitori Y. Carbohydr. Res. 1987;167:77–86. doi: 10.1016/0008-6215(87)80269-0. [DOI] [PubMed] [Google Scholar]
- 24.Auge C., Fernandez-Fernandez R., Gautheron C. Carbohydr. Res. 1990;200:257–268. doi: 10.1016/0008-6215(90)84196-2. [DOI] [PubMed] [Google Scholar]
- 25.Higa H.H., Paulson J.C. J. Biol. Chem. 1985;260:8838–8849. [PubMed] [Google Scholar]
- 26.Schultz M., Kunz H. Tetrahedron: Asymmetry. 1993;4:1205–1220. [Google Scholar]
- 27.Blixt O., Paulson J.C. Adv. Synth. Catal. 2003;345:687–690. [Google Scholar]
- 28.Gross H.J., Rose U., Krause J.M., Paulson J.C., Schmid K., Feeney R.E., Brossmer R. Biochemistry. 1989;28:7386–7392. doi: 10.1021/bi00444a036. [DOI] [PubMed] [Google Scholar]
- 29.Yu H., Huang S.S., Chokhawala H., Sun M.C., Zheng H.J., Chen X. Angew. Chem., Int. Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chokhawala H.A., Yu H., Chen X. Chembiochem. 2007;8:194–201. doi: 10.1002/cbic.200600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon E.S., Bednarski M.D., Whitesides G.M. J. Am. Chem. Soc. 1988;110:7159–7163. [Google Scholar]
- 32.Cato D., Buskas T., Boons G.J. J. Carbohydr. Chem. 2005;24:503–516. [Google Scholar]
- 33.Rosen T., Lico I.M., Chu D.T.W. J. Org. Chem. 1988;53:1580–1582. [Google Scholar]
- 34.Klich G., Paulsen H., Meyer B., Meldal M., Bock K. Carbohydr. Res. 1997;299:33–48. doi: 10.1016/s0008-6215(96)00337-0. [DOI] [PubMed] [Google Scholar]
- 35.Shangguan N., Katukojvala S., Greenberg R., Williams L.J. J. Am. Chem. Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 36.Bezay N., Dudziak G., Liese A., Kunz H. Angew. Chem., Int. Ed. 2001;40:2292–2295. doi: 10.1002/1521-3773(20010618)40:12<2292::AID-ANIE2292>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Ajisaka K., Miyasato M. Biosci. Biotech. Biochem. 2000;64:1743–1746. doi: 10.1271/bbb.64.1743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NMR spectra of compounds.