

Abstract
Uracil DNA glycosylase (UNG) is an important DNA repair enzyme that recognizes and excises uracil bases in DNA using an extrahelical recognition mechanism. It is emerging as a desirable target for small molecule inhibitors given its key role in a wide range of biological processes including the generation of antibody diversity, DNA replication in a number of viruses, and the formation of DNA strand breaks during anticancer drug therapy. To accelerate the discovery of inhibitors of UNG we have developed a uracil-directed ligand tethering strategy. In this efficient approach, a uracil-aldehyde ligand is tethered via alkyloxyamine linker chemistry to a diverse array of aldehyde binding elements. Thus, the mechanism of extrahelical recognition of the uracil ligand is exploited to target the UNG active site, and alkyloxyamine linker tethering is used to randomly explore peripheral binding pockets. Since no compound purification is required, this approach rapidly identified the first small molecule inhibitors of human UNG with micromolar to submicromolar binding affinities. In a surprising result, these uracil-based ligands are found not only to bind to the active site, but also to a second noncompetitive site. The weaker noncompetitive site suggests the existence of a transient binding site for uracil during the multistep extrahelical recognition mechanism. This very general inhibitor design strategy can be easily adapted to target other enzymes that recognize nucleobases, including other DNA repair enzymes that recognize other types of extrahelical DNA bases.
Introduction
DNA repair pathways have been traditionally viewed as the cellular quality control machinery that preserves the coding potential of genomes1. However, there is emerging recognition that the repair mechanisms evolved to prevent accumulation of the RNA base uracil in DNA play a much broader role in a number of important areas of biomedicine that are divergent from genome preservation. Remarkable examples include the role of the uracil excision repair machinery in the process of generating genetic diversity during antibody maturation in B cells2–4, the importance of uracil incorporation and removal in the life cycles of herpes5, cytomegalo6, pox7, 8 and type 1 human immunodeficiency viruses (HIV-1)9, and the essential role of this pathway in generating pharmacologically active single and double strand DNA breaks during chemotherapy treatment with 5-flurouracil and methotrexate10, 11. The key enzyme player in all of these remarkably diverse processes is uracil DNA glycosylase (UNG), which cleaves the glycosidic bond between the uracil base and the deoxyribose sugar in DNA by flipping the uracil nucleotide from the DNA duplex into the enzyme active site (Figure 1A)12. Given that UNG is emerging as a very interesting pharmacologic target, we have sought out methods for the rapid and efficient identification of small molecule ligands that could inhibit its activity. Although potent nucleic acid-based and proteinaceous inhibitors are available that target UNG13–17, there are no small molecule inhibitors for this enzyme, and strategies for the discovery of such ligands are lacking.
Figure 1.
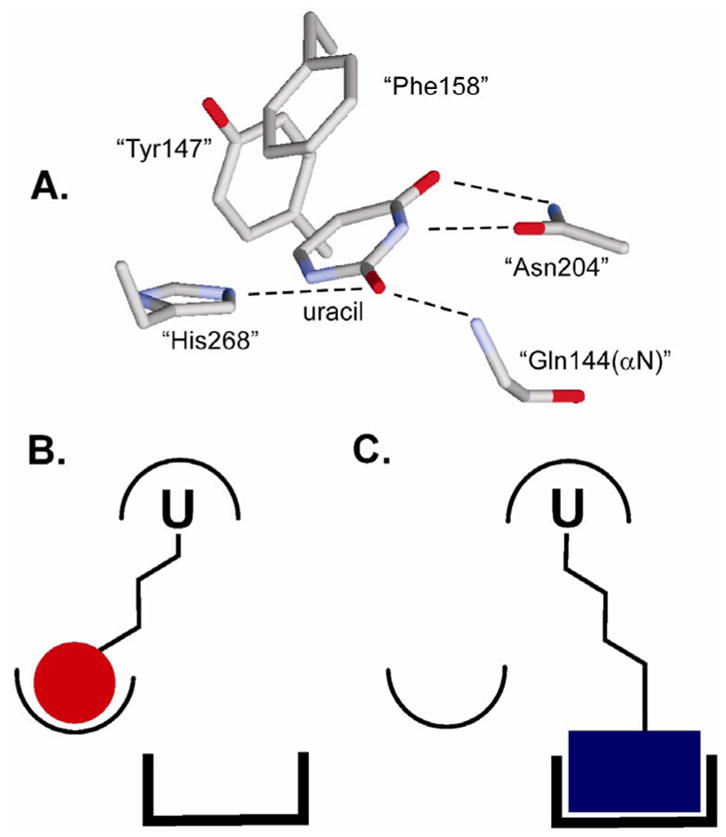
Extrahelical binding of uracil to the UNG active site and the general strategy for uracil-directed ligand tethering. (A) Structure of UNG bound to uracil (pdb code 2eug). The residue numbering is for the human enzyme. (B) and (C) The uracil ligand (U) that targets the UDG active site is covalently tethered to two different ligands that can interact with distinct binding surfaces near the active site.
One of the most exciting potential applications of small molecule human UNG inhibitors are as antiretroviral agents. Recent findings have established that HIV-1 specifically packages human UNG (hUNG) into virus particles via interaction with the virus encoded integrase protein (Int), or perhaps a ternary complex between UNG, Int and the viral Vpr protein5, 18–25. hUNG is required for infection of nondividing cells such as macrophages and resting T cells, and helps maintain a viral reservoir in the host that is crucial for virus spread to the lymphoid organs and T-helper lymphocytes, and ultimately, AIDS pathogenesis20, 26. UNG is apparently recruited to minimize uracil incorporation into the viral genome in these cells, which have naturally high levels of dUTP, a good substrate for the viral reverse transcriptase27. In the absence of UNG, the HIV-1 mutation rate is found to increase by 18-fold resulting in extremely inefficient virus replication in nondividing cells 20, and the virus particles produced from UNG depleted cells are incapable of infecting new target cells9, 28. Pharmacologic targeting of a human enzyme required for virus infectivity is extremely attractive because such a target would not be susceptible to the same high mutagenesis rate and resulting drug resistance as viral encoded proteins29. Targeting the human enzyme is a viable therapeutic strategy because it is not an essential enzyme. Thus, UNG knock-out mice display no remarkable phenotype, nor do UNG null yeast or human cell lines 30.
Herein, we report an integrated high-throughput (HTP) platform for discovering small molecule ligands that inhibit UNG. The strategy takes advantage of the extrahelical uracil recognition mechanism of UNG by using the specificity and binding energy of a uracil ligand to target the UNG active site14, 31, 32, and then covalent tethering of random functional groups for exploration of nearby binding pockets (Figure 1B). Library members can be rapidly screened using a robust HTP activity assay, and initial hits are quickly optimized using subsequent structure-activity studies. This tethering approach, which uses efficient oxime chemistry (Figure 2), is related to the “combinatorial target-guided ligand assembly” method of Ellman et al33, but differs in that the uracil ligand specifically targets the active site rather than irrelevant regions of the enzyme. Thus, the hit-rate and binding affinities of early hits are higher than the more random approach of Ellman and colleagues. This synthetic and screening strategy should be easily adaptable for the discovery of inhibitors of other enzymes that recognize extrahelical bases in DNA or free nucleosides.
Figure 2.

Synthesis of oxime libraries based on uracil and RCHO. (A) Synthesis of diaminoalkanediol tethers of variable length. (B) Construction of the uracil-oxime library based on the uracil aldehydes (1–3) and a series of aldehyde compounds (RCHO). The products consist of a 1:2:1 mixture of the heterodimer (U^R), and the two homodimers (U^U and R^R) connected via alkane linkers of lengths 2–6. One equivalent of total diaminoalkanediol is added to each reaction. Each linker length is present at one-fifth of the total concentration.
Results and Discussion
Synthesis of Uracil-Tethered Oxime Libraries and General Strategy
We sought an inhibitor development strategy that allowed rapid and economical synthesis of small molecule ligands that explore binding sites near the UNG active site, and which could be used directly in HTP screening applications without purification. One efficient synthesis strategy that meets these criteria is outlined in Figure 2. First, flexible diaminoalkanediol linkers of variable length are synthesized from the corresponding dibromoalkanes (Figure 2A). Then the linkers are used to tether uracil aldehyde binding elements (1–3) to a library of aldehyde binding elements (RCHO) via the formation of stable oxime linkages (Figure 2B). Each tethering reaction is carried out in one well of a 96-well microtiter plate that contains one equivalent uracil aldehyde, one equivalent RCHO library member, and a mixture of diaminoalkanediol linkers (n = 2–6). The reactions typically proceed to 85–99 % completion after overnight incubation (DMSO solvent, 37 °C), and produce a 1:2:1 statistical mixture of the homodimeric (U^U, R^R) and heterodimeric (U^R) oximes for each of the five linker lengths present (see Methods and Supplemental Figure S1). Although two geometric configurations are possible, oxime derivatives with bulky substituents are generally found to be ≥95 % in the trans configuration34. The unpurified oxime mixtures were directly screened for inhibition of UNG at ~100 μM total oxime concentration to ensure that each component in the mixture is present at a concentration in the range 5 to 10 μM. If significant inhibition is observed by any mixture, the linker length and RCHO binding element that gave rise to the inhibition can be identified by resynthesis of the individual oximes using a single linker length in each reaction (see below).
An important aspect of this approach is that the uracil homodimers present in some reaction mixtures are inhibitory even in the absence of any active heterodimer. For instance, the purified homodimers of various lengths that are based on 6-formyluracil (3) give rise to about 22 % inhibition in all the mixtures based on 3 under the screening conditions (not shown). In contrast, the homodimers of 1 and 2 show no detectable inhibition under the same conditions. Thus, the screening assay must be robust enough to detect any additional inhibition resulting from an active heterodimer in the mixture. Spectroscopic results for determining the purity and composition of representative reaction mixtures are available (see Supporting Information).
High-Throughput Screening of Uracil-Oxime Libraries
To test this directed library approach, we tethered the three uracil aldehydes (1, 2, 3) shown in Figure 2 to 14 aldehyde binding elements (RCHO) using the variable length diaminoalkanediol linkers (see Supporting Information Table S1 for RCHO structures). This library of uracil-linked binding elements was screened for inhibition of hUNG using a high-throughput molecular beacon activity assay (Figure 3) 35. In this assay, one DNA strand is labeled with a fluorescent 5′-FAM, and the complementary strand is modified with a 3′-dabsyl moiety that serves to efficiently quench the fluorescence of the FAM group through contact quenching. To increase stability, the two DNA strands are linked in a hairpin configuration using an 18 atom polyethylene glycol linker. When the substrate DNA is exposed to UDG, multiple uracils are removed, and eventually the two paired strands of the hairpin spontaneously separate, thus removing the dabsyl quencher from the proximity of the FAM group, and resulting in a 6-fold increase in the fluorescence of the system (Figure 3A). Under the assay conditions, the hairpin DNA substrate has a Km = 164 ± 10 nM and kcat = 0.33 ± 0.01 s−1 (Figure 3B). To enhance detection of competitive inhibitors during HTP screening we employed a molecular beacon substrate concentration equivalent to 1/3 Km (50 nM). Representative HTP screening results for several inactive and active oxime mixtures are shown in Figure 4 ([Total oxime] = 100 μM).
Figure 3.
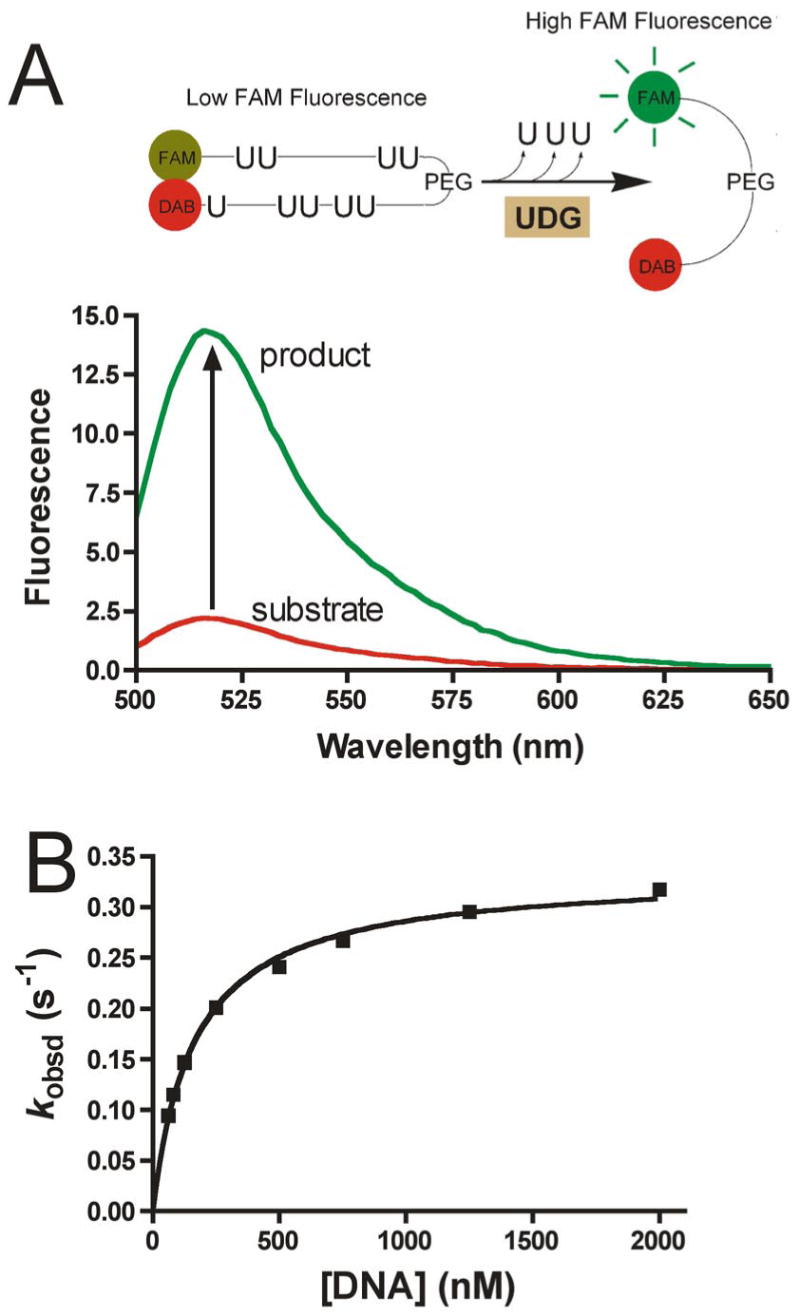
High-throughput (HTP) UDG kinetic assay. (A) The HTP assay relies on molecular beacon technology. Excision of multiple uracil bases by the enzyme destabilizes the hairpin structure thereby releasing the 5′ FAM fluorophore from the quenching effects of the 3′ dabsyl group. (B) Steady-state kinetic analysis of the hUDG reaction using the molecular beacon hairpin substrate.
Figure 4.
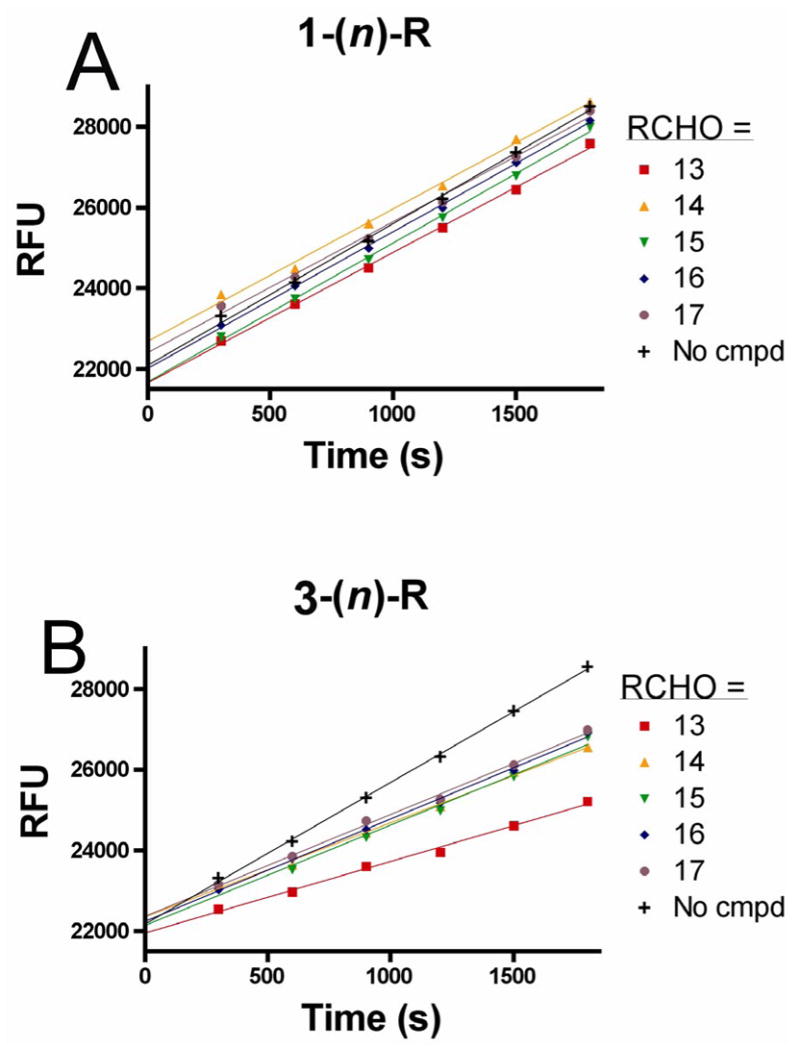
Representative HTP screening results using the molecular beacon substrate. (A) Screen of oxime dimer mixtures derived from uracil aldehyde 1 and aryl aldehydes 13–17. No inhibition was observed for any oxime derived from 1 regardless of linker length (n). (B) Screen of oxime dimer mixtures derived from uracil aldehyde 3 and aryl aldehydes 13–17. The mixed oxime derived from 3 and 13 shows significant inhibition and this derivative was further optimized. For 14–17, the observed inhibition represents that from the 3-3 homodimers that are present in the mixtures.
Several activity trends emerged immediately from the screening results shown in Figure 4. First, none of the mixtures derived from the uracil N1-acetaldehyde binding element (1) were inhibitory at the concentration used in the screen. In addition, none of the U^U homodimers derived from 2 were found to be inhibitory, nor were any of the R^R homodimers regardless of the linker length. (Inhibition by the homodimers is automatically assessed because these are present in multiple reaction mixtures.) In contrast, one oxime mixture derived from uracil aldehydes 2 and 3 and RCHO = 2, 4 dihydroxybenzaldehyde (13) showed inhibitory activity in the range 15 to 100 %, indicating that active heterodimers were present. The structures of the active heterodimers present in these two oxime mixtures are shown at the top of Table 1.
Table 1.
Structures of active heterodimers and dependence of inhibition on linker Lengtha
 | ||
|---|---|---|
| Mixture | Linker length (n) | % Inhibition |
| 2-(n)-13 | 2 | 50 |
| 3 | 40 | |
| 4 | 20 | |
| 5 | 20 | |
| 6 | 15 | |
| 3-(n)-13 | 2 | 57 |
| 3 | 100 | |
| 4 | 51 | |
| 5 | 48 | |
| 6 | 48 | |
Reactions were performed in the presence of 100 μM oxime mixture and 50 nM substrate concentration.
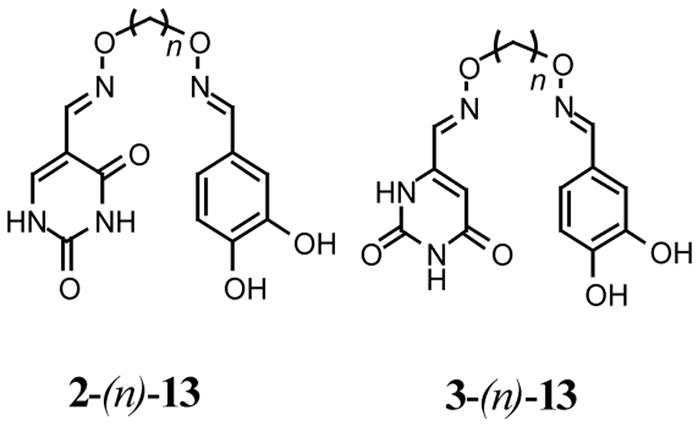
The two active mixtures were deconvoluted with respect to linker length by individually synthesizing each oxime dimer using a single diaminoalkanediol linker per reaction (Chart 1). At this stage we did not separate the homodimers from the active heterodimers in the mixtures. For the oxime dimers derived from 5-formyluracil (2) and 2, 4-dihydroxybenzaldehyde (13), a broad dependence on linker length was observed with length n = 2 being most favorable for inhibitory activity (i.e. mixed oxime 2-(2)-13, Chart 1). In contrast, a very stringent linker length of n = 3 was required for maximal inhibitory activity with the oxime mixture derived from 6-formyluracil (3) and 2, 4-dihydroxybenzaldehyde (13) to form mixed oxime 3-(3)-13 (Chart 1). To confirm these results, 2-(2)-13 and 3-(3)-13 were separated from their respective homodimers using reversed phase HPLC (see Methods), and the concentration dependence of inhibition was determined. The measured IC50 values for 2-(2)-13 and 3-(3)-13 were 5.8 and 1.1 μM, respectively (Fig. 5).
Chart 1.
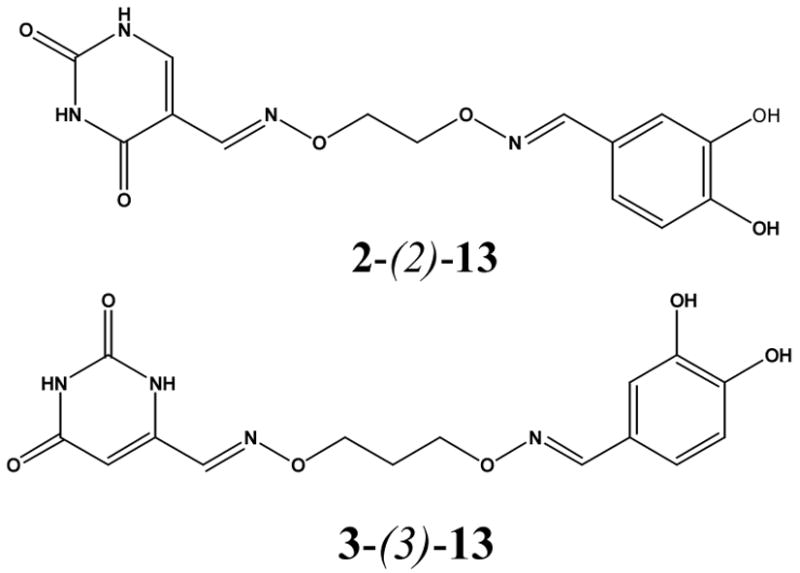
Heterodimer Oximes Identified from Deconvolution of Active Mixtures
Figure 5.
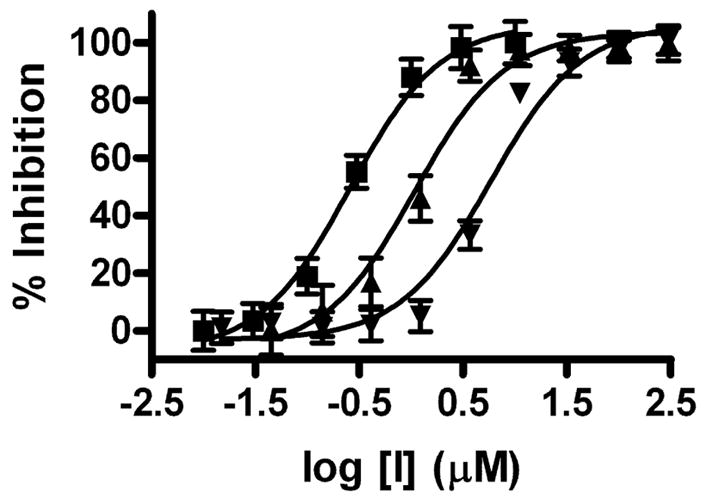
IC50 analysis of for 2-(2)-13 (▼), 3-(3)-13 (▲) and 3-(3)-27 (■).
Structure Activity Relationships
In an effort to find more potent inhibitors based on the 3-(3)-13 scaffold, twenty-five commercially available benzaldehyde precursors were purchased (18–42, cf. Supporting Information Table S2). The HTP screen was then performed on this set of oxime mixtures (3-(3)-R) in an identical fashion as described above. This structure-activity study established that the 3- and 4-hydroxyl groups of 3-(3)-13 were essential for activity because alkylation or halogen substitution at these positions had a substantial deleterious effect on inhibitory activity (see Supporting Information Table S2). Thus, hydrogen bond donating groups at the 3- and 4-positions of the benzyl ring appear to be essential.
One compound in this series with an additional hydroxyl group at the 2-position of the benzyl ring (3-(3)-27), showed a 3-fold greater potency than 3-(3)-13 (Figure 5, ■)(IC50 = 0.3 μM). To further investigate SARs based around the 3-(3)-27 scaffold, we synthesized four more 3, 4-dihydroxybenzaldehyde analogues (43–46, Table 2), where the substituent at the 2-position was varied (R = F, Cl, Br or NO2). Within this series there was a strong trend correlating with atomic size for the halogens, with the smaller fluorine substituent binding 16-fold more tightly than bromine. However, no substituent in this series was more effective than the 2-hydroxyl group. In conclusion, the binding pocket for the 2-substituent favors a hydrogen bond donating group with a van der Waals radius smaller than chlorine.
Table 2.
Inhibitory activity for structural variants of 3-(3)-27a
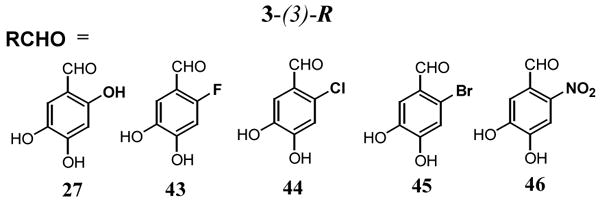 | ||
|---|---|---|
| IC50 (μM) | 2-R | |
| 3-(3)-27 | 0.26 | OH |
| 3-(3)-43 | 2.7 | F |
| 3-(3)-44 | 16 | Cl |
| 3-(3)-45 | 40 | Br |
| 3-(3)-46 | 40 | NO2 |
The concentration dependence of inhibition was determined using 50 nM substrate.
Inhibition Mechanisms of 3-(3)-27, 2-(2)-13 and Uracil
Although the uracil-directed ligand tethering strategy is expected to produce competitive inhibitors of UNG, we thoroughly investigated whether this assumption was true. The detailed mode of inhibition by 3-(3)-27 and 2-(2)-13 was evaluated by varying both substrate and inhibitor concentrations (Fig. 6A and 6B). Standard double reciprocal plots of 1/kobsd against 1/[DNA] at increasing concentrations of 3-(3)-27 showed no significant intercept effects establishing a competitive aspect to the inhibition (Figure 6A). However, a secondary plot of the Lineweaver-Burk slopes against [3-(3)-27] showed a parabolic response consistent with the presence of at least two inhibitor binding sites (Figure 6A, inset)36. Global discrimination fitting of the inhibition data by computer simulation with the program Dynafit using competitive, noncompetitive, uncompetitive, mixed-type, two-site competitive-noncompetitive, and two-site competitive-uncompetitive inhibition mechanisms unambiguously confirmed the presence of two inhibitory binding sites for 3-(3)-27 (see Supplemental Information)37. Simulations clearly indictated that the first tight site is competitive with respect to substrate. Although the simulations indicated a slight statistical advantage for a partial mixed-type inhibition mode for the second weaker site, it was difficult to eliminate an uncompetitive mode for this site. Using the criterion of Occam’s razor, the inhibition parameters for 3-(3)-27 are reported in Table 3 using the simulation results for the competitive-partial uncompetitive mechanism (Scheme 1).
Figure 6.
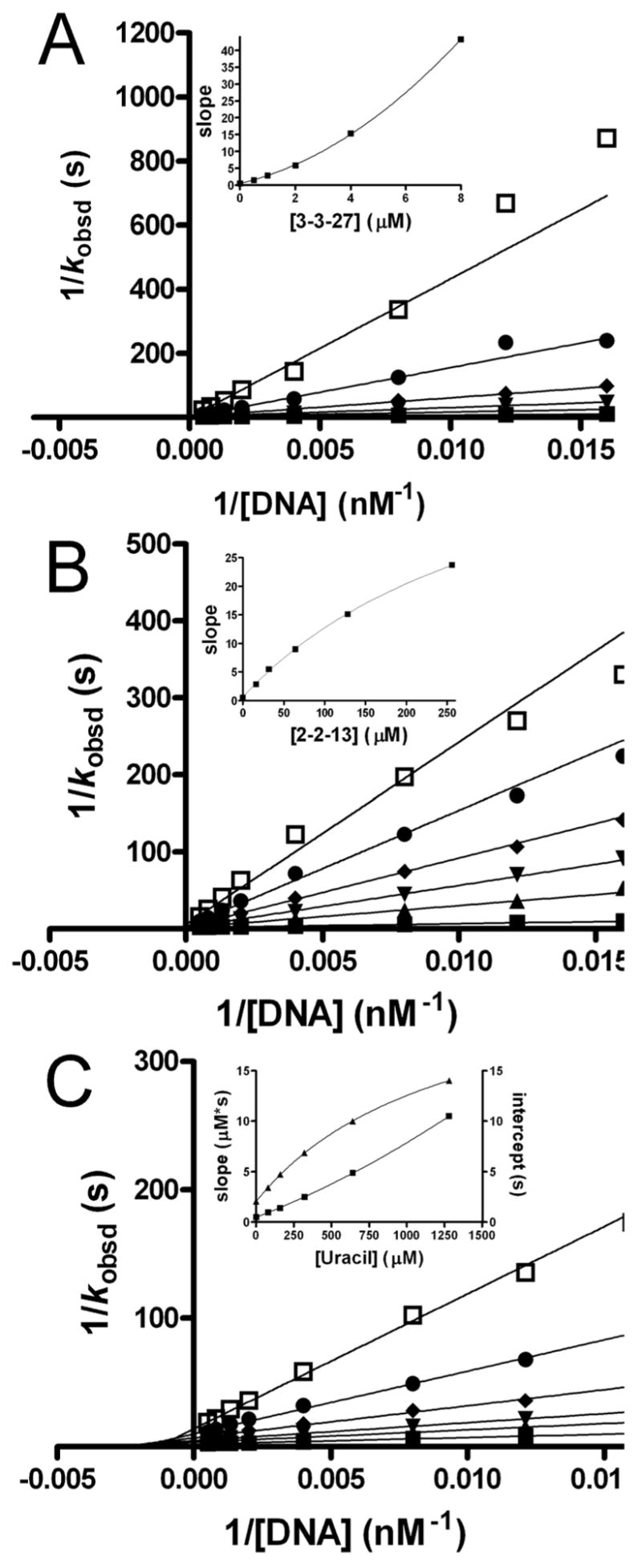
Mode of inhibition analysis. Double reciprocal plots and secondary slope and intercept replots for inhibition by increasing concentrations of (A) 3-(3)-27, (B) 2-(2)-13, and (C) uracil. Slope and intercept effects in the inset to (C) are shown as squares and triangles, respectively.
Table 3.
Inhibition Constants for Uracil and its Derivativesa
| Parameter | 3-(3)-27 | 2-(2)-13 | Uracil | 51 |
|---|---|---|---|---|
| Ks (μM) | 0.19 ± 0.02 | 0.23 ± 0.03 | 0.23 ± 0.02 | 0.16 ± 0.01 |
| kcat (s−1) | 0.41 ± 0.01 | 0.50 ± 0.02 | 0.47 ± 0.01 | 0.33 ± 0.01 |
| kcat′(s−1) | 0.16 ± 0.04 | 0.012 ± 0.02 | 0.06 ± 0.01 | - |
| Kc (μM) | 0.32 ± 0.02 | - | 80 ± 7 | 45 ± 2 |
| Kn (μM) | - | 2.8 ± 0.1 | - | - |
| Knc (μM) | 1.2 ± 0.2 | - | 300 ± 55 | - |
| Kns (μM) | 1 ± 0.3 | 125 ± 46 | 104 ± 7 | - |
|
| ||||
| Mode of Inhibition | Two sites, competitive, partial uncompetitive | One site, partial mixed-type | Two sites, competitive, partial uncompetitive | One site, competitive |
Parameters correspond to the mechanisms shown in Scheme 1. Kc and Kn represent dissociation constants for inhibitor binding sites that are competitive and noncompetitive with substrate, respectively. Knc and Kns represent the dissociation constants for inhibitor binding to the noncompetitive site when the active site is occupied by the competitively bound inhibitor or substrate, respectively. In these simulations the Michaelis-Menten parameters for the substrate were fixed using values from nonlinear regression fits (Fig. 6). Other parameters were obtained from simulations to the data using the program Dynafit (cf. Supplemental Information)
Scheme 1.

Inhibition Mechanisms for 3-(3)-27 and 2-(2)-13 and Uracila
aOnly 3-(3)-27, 2-(2)-13 and uracil have mechanisms that include the kcat′ step. The mechanism for 3-(3)-and uracil do not include the equilibrium constant Kn, and the mechanism for 2-(2)-13 does not include the equilibria Kc or Knc.
Like its 6-substituted analog, initial inspection of the Lineweaver-Burk analysis of 2-(2)-13 indicates mixed-type inhibition with a strong preference for binding to the free enzyme (i.e. slope effects, Figure 6B). However, in contrast to 3-(3)-27, the secondary plot of the Lineweaver-Burk slopes versus 2-(2)-13 concentration is hyperbolic, indicating that binding of 2-(2)-13 results in partial inhibition (Figure 6B, inset)36. Because binding to the active site would result in complete inhibition, 2-(2)-13 most likely binds to the noncompetitive site observed for 3-(3)-27. Global discrimination fitting of the inhibition data by computer simulation confirmed this inhibition mechanism (Scheme 1), and provided the inhibition constants reported in Table 3. These observations strongly indicate that 2-(2)-13 binds to a site distinct from the active site, although DNA binding is strongly antagonistic to inhibitor binding (Table 3). In summary, the inhibition mechanisms of 3-(3)-27 and 2-(2)-13 indicate that two inhibition modes exist for these uracil derivatives: one mode competitively targets the active site, the second weaker mode is noncompetitive or uncompetitive with respect to substrate binding. These data, quite surprisingly, suggested the presence of two uracil binding sites on human UNG.
To further investigate the interesting possibility of two uracil binding sites on UNG we performed a mode of inhibition analysis for uracil itself (Fig. 6C). Confirming this initial expectation, inhibition by uracil involves two sites. The first site is competitive and the second is partially uncompetitive. Accordingly, the Lineweaver-Burk slope replot was slightly parabolic indicating that inhibition involved binding of more than one molecule of uracil, and the intercept replot was hyperbolic indicating a partial uncompetitive mode. These characteristics of the inhibition by uracil combine the features observed for 3-(3)-27 and 2-(2)-13, and establish that the two site binding of 3-(3)-27 is not attributable to the trihydroxybenzaldoxime moiety, but instead, arises from the uracil functionality itself.
Implications for Two Uracil Binding Sites
Why would UNG have a second uracil binding site? Although the answer to this question cannot be firmly established by inhibition data alone, an intriguing role for this site during the mechanism of uracil base flipping is supported by several different experimental findings. First, kinetic experiments following the pathway of uracil flipping from duplex DNA have detected a weakly bound intermediate state of uracil that precedes its attainment of the final extrahelical state seen in the crystal structure (Figure 1A)15, 38–40. Solution and solid state NMR studies of uracil flipping support the existence of a weak uracil binding site because UNG is found to transiently stabilize thymine and other uracil congeners in an extrahelical conformation, without these bases gaining full access to the uracil active site pocket41, 42. Relevant to these observations, the crystal structure of herpesvirus UDG bound to pTTTp shows that the 5′ T is bound in the mouth of the active site pocket in a manner that is consistent with a transient state on the pathway for base flipping of uracil 43. Finally, the crystal structure of another base flipping DNA repair enzyme, human 8-oxoguanine DNA glycosylase, suggests that this related enzyme can flip the normal base guanine into a discrimination pocket that was distinct from the active site pocket that only accommodates 8-oxoguanine44. These combined data provide a compelling case for a generalized pathway for base flipping involving transient enzyme stabilization of at least one extrahelical intermediate state before the base is docked into the active site. Based on the observation that 2-(2)-13 is excluded from the active site but that 3-(3)-27 and uracil can occupy both sites, we surmise that the relative binding affinities for each site might depend on the bulkiness of the substituent at the 5-position of uracil. In other words, uracil congeners with small substituents at the five position (such as hydrogen in the case of 3-(3)-27) would favor binding to the active site, and uracil derivatives with bulkier substituents (such as the dihydroxybenzaldoxime of 2-(2)-13) would be sterically excluded from the active site, but could gain access to the weaker less selective site. Indeed, it is well known that the active site of UNG uses the bulky side chain of a tyrosine to exclude thymidine (5-methyluracil) 14, 45–47, yet 6-substituted uracil derivatives such as 3-(3)-27 have been generally observed to bind to the active site 14. Thus, the uracil-based inhibitors found here have revealed a possible pyrimidine discrimination site that may be employed during the multistep extrahelical uracil recognition mechanism. It should be noted that the noncompetitive inhibition mode for 2-(2)-13 requires that the final extrahelical state can be attained, albeit inefficiently, even when the transient uracil binding site is occupied by the inhibitor. In contrast, the partial uncompetitive mechanism for binding of 3-(3)-27 to its second site does not present the same apparent discrepancy, because for uncompetitive inhibition, the compound binds after the substrate is fully inside the active site pocket (see above).
Inhibition by the Untethered Parts
It is of interest to ask how well uracil-directed ligand tethering has performed. To dissect the energetic contributions of the formyluracil and hydroxybenzaldoxime binding elements of 3-(3)-27 and 2-(2)-13, we synthesized the methyl oxime derivatives of aldehydes 2, 3, 27 and 13 as shown in Scheme 2. These methyl oxime derivatives are reasonable mimics of the two individual binding elements and in principle could provide an energetic analysis of the binding affinities of the two separate elements. If the sum of the binding energies of each element equals the entire binding free energy of the whole tethered molecule, then it may be concluded that (i) the tether is energetically inert with respect to binding, and (ii) the binding of one element does not affect the other by induced strain or forcing a tighter fit. If the whole tethered molecule binds much more weakly or tightly than expected from the summation of the binding free energies of the two individual binding elements, then nonadditive energetic effects are present. Such effects would indicate either an energetic penalty for tethering (antagonistic binding of the parts), or alternatively, a nonadditive energetic benefit (synergistic binding of the parts)48, 49.
Scheme 2.
O-methyl Oxime Derivatives of the Aldehyde Binding Elements of 2-(2)-13 and 3-(3)-27
Comparison of the binding affinity of 3-(3)-27 to its competitive site (Kc 3-(3)-27 = 0.32 μM) with that of the 6-formyluracil O-methyl oxime binding element alone (51) allows estimation of the free energy benefit of tethering the trihydroxybenzaldoxime binding element to the 6-formyluracil oxime part. Conversely, comparison of the binding affinity of 3-(3)-27 with that of the trihydroxybenzaldoxime O-methyl ether (48) allows estimation of the free energy benefit of tethering the 6-formyluracil oxime binding element to the trihydroxybenzaldoxime part. The 6-formyluracil O-methyl oxime 51 shows a cleanly competitive mode of inhibition with Kc51 = 45 ± 2 μM (Table 4, data not shown). Thus, the enhancement in the free energy of binding upon addition of the trihydroxybenzaldoxime (THB) part to the 6-formyluracil oxime element is ΔΔGTHB = −RT ln (Ki 3-(3)-27/Ki51) = −3 kcal/mol. We were unable to perform a similar energetic analysis with the trihydroxybenzaldoxime O-methyl ether (48) due to its extremely weak binding (9 % inhibition at 1 mM concentration, data not shown). Similarly, an energetic analysis of the binding elements comprising 2-(2)-13 was not possible because of the extremely weak inhibition by the 5-formyluracil O-methyl oxime (50) and the dihydroxybenzaldoxime O-methyl ether (47). Nevertheless, the 140-fold greater binding affinity of 3-(3)-27 as compared to the 6-formyluracil O-methyl oxime binding element (51) alone indicates that a large benefit can be derived from tethering 50.
Experimental Section
Reagents and General Methods
All chemicals were purchased from commercial sources without further purification unless otherwise stated. The 1H, 13C and 19F NMR spectra were recorded on a 400 MHz Varian Innova instrument. The spectra were recorded in deuteriochloroform (CDCl3) or in hexadeuteriodimethylsulfoxide (DMSO-d6). The chemical shifts of protons are given in ppm with TMS as internal standard. The chemical shifts of carbons are obtained in ppm with solvents as internal standards. That of fluorine is given in ppm with 1% trifluoroacetic acid in DMSO-d6 as an external standard. Most of oximes were purified by HPLC using aqueous triethylammonium acetate (TEAA) as a running buffer. Therefore, TEAA was not completely removed and it appeared in the NMR spectra. Accordingly, proton and carbon chemical shifts of TEAA were not listed during the characterizations of the oximes. During the purification of the oxime 3-(3)-27, 2-mercaptoethanol was used as an anti-oxidant. Therefore, small amounts of this compound and its oxidation product are also present in the oxime 3-(3)-27. Flash chromatographies were performed with silica (70–230 mesh from Sorbent Technologies) and monitored by thin layer chromatography (TLC) with silica plates (Merck, Kieselgel 60 F254).
Synthesis of Alkyl Hydroxyamines
O, O′-Diaminoalkanediol linkers of variable length (ethyl, propyl, butyl, pentyl, hexyl) were prepared from the corresponding dibromoalkanes in two steps according to literature procedures (Figure 2A) 33, 51, 52.
General Synthesis of Tethered Oxime Dimers
A set of 14 aryl aldehydes (4–17, cf. Supplementary Information, Table S1) was selected for library synthesis for coupling to the three uracil containing aldehydes (1, 2, 3, Figure 2) using the O, O′-diaminoalkanediol linkers as follows. To each 0.5-ml well of a Matrix microtiter plate was added a DMSO stock solution of AcOH (20 μl, 150 mM, 3 μmol), uracil aldehyde 1, 2 or 3 (20 μl, 150 mM, 3 μmol) and a single aryl aldehyde (20 μl, 150 mM, 3 μmol). The plate was carefully agitated to make the solutions homogenous. To each of the uracil-aryl aldehyde mixture was added a DMSO solution of theO, O′-diaminoalkanediol linkers containing each of the five linker lengths in equal proportion (22 μl, 150 mM, 3.3 μmol total amine equivalents). The plate was sealed, further agitated and incubated in an oven for 12 h at 37°C.
The most potent inhibitors from this first screen 2-(2)-13 and 3-(3)-13) were synthesized in larger scale and thoroughly characterized after HPLC purification of the heterodimers:
2-(2)-13: 1H NMR (400 MHz, DMSO-d6): δ 8.05 (s, 1 H), 7.91 (s, 1 H), 7.78 (s, 1 H), 7.04 (s, J = 2.4 Hz, 1 H), 6.86 (m, 1 H), 6.74 (d, J = 8.0 Hz, 1 H), 4.26 (s, 1 H); 13C NMR (100 MHz, DMSO-d6): δ 162.40, 151.04, 149.25, 147.92, 145.75, 142.74, 140.66, 123.05, 119.88, 115.74, 113.10, 104.31, 71.82, 71.54. UV/Vis: λmax 275 nm; HRMS (m/z): [M+Na]+ calcd for C14H14N4O6Na, 357.08; found, 357.08.
3-(3)-13: 1H NMR (400 MHz, DMSO-d6): δ 9.10 (bs, H), 8.01 (s, 1 H), 7.94 (s, 1 H), 7.04 (d, J = 1.6 Hz, 1 H), 6.82 (d, J = 7.6 Hz, 1 H), 6.74 (d, J = 7.6 Hz, 1 H), 5.78 (s, 1 H), 4.26 (t, J = 6.8 Hz, 2 H), 4.12 (t, J = 6.0 Hz, 2 H), 2.06 (t, J = 6.8 Hz, 2 H); 13C NMR (125 MHz, DMSO-d6): δ 163.95, 151.15, 148.96, 148.04, 145.89, 144.73, 142.23, 123.12, 119.83, 115.81, 113.15, 101.60, 71.94, 69.76, 28.46; UV/Vis: λmax 273 nm; HRMS (m/z): [M+H]+ calcd for C15H17N4O6, 349.11; found, 349.11.
The second set of oxime dimers based on the 3-(3)-13 hit discovered in the first screening round were synthesized in an identical fashion as described above using uracil aldehyde 3 and hydroxybenzaldehydes 18 through 42, and the O, O′-diaminopropanediol linker (cf. Supplementary Information, Table S2). The most potent inhibitor identified from this second round of screening (3-(3)-27) was synthesized in larger scale and thoroughly characterized. 3-(3)-27: 1H NMR (400 MHz, DMSO-d6): δ 9.10 (bs, H), 8.21 (s, 1 H), 7.94 (s, 1 H), 6.88 (s, 1 H), 6.31 (s, 1 H), 5.78 (s, 1 H), 4.28 (t, J = 6.0 Hz, 2 H), 4.10 (t, J = 6.0 Hz, 2 H), 2.06 (m, 2 H); 13C NMR (125 MHz, DMSO-d6): δ 163.91, 151.04, 150.25, 149.17, 146.90, 144.63, 142.20, 138.73, 112.76, 107.78, 103.56, 101.69, 71.90, 69.77, 28.38; UV/Vis: λmax 286 nm; ESI (m/z): [M+H]+ calcd for C15H18N4O7, 366; found, 366. [M+Na]+ calcd for C15H17N4O7Na, 388; found, 388. [M−H]− calcd for C15H16N4O7, 364; found, 364.
Isolation and Purification of Oxime Dimers using HPLC
All of the most active oxime heterodimers were purified by HPLC using a Phenomenex Aqua reversed phase C-18 HPLC column (250 mm, 10 mm, 5 μm). Most of the oximes were purified using gradient elution from 0 to 30% CH3CN in 0.1 M aqueous TEAA over the course of 2 h using UV detection at 254 nm. An exception was oxime 3-(3)-27, which is prone to air oxidation. In this case, 25 mM 2-mercaptoethanol was added to both of the running buffers. The oximes all eluted with baseline resolution in the order: U-U homodimer, U-R heterodimer, followed by the R-R homodimer. This HPLC method was also used to confirm the expected 1:2:1 stoichiometries of homodimer and heterodimer oxime formation, using ten representative uracil and aryl aldehydes from the library (see Supplementary Information, Figure S1). Additional NMR evidence supporting the expected stoichiometries is detailed in the Supplementary Information, Figures S2 and S3.
Synthesis of 2-R Substituted 3, 4-Dihydroxybenzaldehydes and the Corresponding Mixed Oximes with 3
Aldehyde 43 was synthesized by removing the methyl groups of the commercially available 3, 4-dimethoxy-6-fluorobenzaldehyde using BBr3 in CH2Cl2 53. The aldehydes 44 and 45 were synthesized by removing the methylene group of the corresponding 2-halogenated piperonal using AlCl3 and 6N HCl 54. Aldehyde 46 was commercially available. These four aldehydes (43 through 46) were reacted with 6-formyluracil 3 and the O, O′-diaminopropanediol linker using the procedure described above, and the mixed oxime dimer was obtained after HPLC purification.
3-(3)-43: 1H NMR (400 MHz, DMSO-d6): δ 8.11 (s, 1 H), 7.94 (s, 1 H), 7.05 (d, J = 7.2 Hz, 1 H), 6.58 (d, J = 6.8 Hz, 1 H), 5.77 (s, 1 H), 5.10 (bs, H), 4.28 (t, J = 6.8 Hz, 2 H), 4.16 (t, J = 6.0 Hz, 2 H), 2.07 (m, 2 H); 13C NMR (125 MHz, DMSO-d6): δ 163.91, 155.50, 153.09, 151.08, 149.87, 149.76, 144.65, 142.72, 142.30, 142.21, 110.91, 110.87, 108.62, 108.49, 103.21, 102.96, 101.67, 71.85, 70.06, 28.38; 19F NMR (DMSO-d6): δ −54.33, −54.35, −54.36, −54.38; UV/Vis: λmax 268 nm; HRMS (m/z): [M+Na]+ calcd for C15H15FN4O6Na, 389.09; found, 389.09.
3-(3)-44: 1H NMR (400 MHz, DMSO-d6): δ 8.23 (s, 1 H), 7.94 (s, 1 H), 7.18 (s, 1 H), 6.77 (s, 1 H), 5.78 (s, 1 H), 4.27 (t, J = 5.6 Hz, 2 H), 4.16 (t, J = 6.0 Hz, 2 H), 2.08 (m, 2 H); 13C NMR (125 MHz, DMSO-d6): δ163.90, 151.08, 149.71, 145.48, 145.16, 144.64, 142.21, 123.00, 119.09, 116.08, 112.33, 101.67, 71.85, 70.22, 28.37; UV/Vis: λmax 275 nm; HRMS (m/z): [M+Na]+ calcd for C15H15ClN4O6Na, 405.06; found, 405.06.
3-(3)-45: 1H NMR (400 MHz, DMSO-d6): δ 8.18 (s, 1 H), 7.94 (s, 1 H), 7.19 (s, 1 H), 6.93 (s, 1 H), 5.78 (s, 1 H), 4.27 (t, J = 6.4 Hz, 2 H), 4.16 (t, J = 6.4 Hz, 2 H), 2.08 (m, 2 H); 13C NMR (125 MHz, DMSO-d6): δ 163.91, 151.10, 150.07, 147.41, 146.02, 144.66, 142.22, 120.58, 119.15, 112.89, 112.29, 101.69, 71.86, 70.24, 28.38; UV/Vis: λmax 278 nm; HRMS (m/z): [M+Na]+ calcd for C15H15BrN4O6Na, 449.01; found, 449.01.
3-(3)-46: 1H NMR (400 MHz, DMSO-d6): δ 8.56 (d, J = 1.2 Hz, 1 H), 7.95 (s, 1 H), 7.37 (s, 1 H), 6.74 (s, 1 H), 6.26 (bs, H), 5.78 (d, J = 1.2 Hz, 1 H), 4.28 (t, J = 6.0 Hz, 2 H), 4.16 (t, J = 6.0 Hz, 2 H), 2.09 (m, 2 H); 13C NMR (125 MHz, DMSO-d6): δ 163.88, 160.27, 151.05, 148.62, 148.00, 144.62, 142.22, 133.36, 122.38, 113.80, 109.23, 101.73, 71.85, 70.07, 28.39; UV/Vis: λmax 269 nm; HRMS (m/z): [M+H]+ calcd for C15H16N5O8, 394.10; found, 394.10.
Synthesis of Methyl Oxime Derivatives of 1–3, 13 and 27
The O-methyl oxime of 3, 4-dihydroxybenzaldehyde (47) is known, and was synthesized using 13 and O-methylhydroxylamine hydrochloride 55. O-methyloximes 48–51 were made using a similar method.
48: To a solution of 27 (308 mg, 2.0 mmol) in 4.0 ml of EtOH-H2O-THF (0.45/0.3/0.25) were added sodium acetate (264 mg) and O-methylhydroxylamine hydrochloride (183 mg), and the solution was stirred at room temperature for overnight. The solvents were removed in vacuo and the residue was extracted with chloroform three times. The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/hexanes) to give product 347 mg in 95% yield. 1H NMR (400 MHz, DMSO-d6): δ 9.38 (s, 1 H), 9.21 (s, 1 H), 8.52 (s, 1 H), 8.18 (s, 1 H), 6.88 (s, 1 H), 6.30 (s, 1 H), 3.80 (s, 3 H); 13C NMR (125 MHz, DMSO-d6): δ 150.17, 148.88, 146.52, 138.61, 112.51, 107.77, 103.46, 61.28; UV/Vis: λmax 239, 274 nm; HRMS (m/z): [M+H]+ calcd for C8H10NO4, 184.06; found, 184.06.
49: To a solution of 1 (10.8 mg, 0.063 mmol) in hot DMF (0.5 ml), were added sodium acetate (5.2 mg, 0.063 mmol) solution in water (0.1 ml) and O-methylhydroxylamine hydrochloride (5.3 mg, 0.063 mmol), and the solution was stirred at room temperature for overnight. The solvents were removed in vacuo and the residue was purified by column chromatography using 10–15% (v/v) methanol in CH2Cl2, resulting in 90% yield (10.3 mg 50/50 mixture of trans and cis geometric isomers). 1H NMR (400 MHz, chloroform-d): δ 9.56 (s, 1 H), 7.43 (t, J = 5.2 Hz,0.5 H), 7.20 (m, 1 H), 6.80 (t, J = 4.4 Hz,0.5 H), 5.78 (m, 1 H), 4.55 (d, J = 4.4 Hz,1 H), 4.48 (d, J = 5.6 Hz,1 H), 3.94 (s, 1.5 H), 3.87 (s, 1.5 H); 13C NMR (100 MHz, DMSO-d6): δ 163.92, 163.87, 151.06, 150.99, 144.72, 144.54, 143.93, 143.53, 103.14, 102.97, 62.68, 62.37, 46.52, 43.76. UV/Vis: λmax 263 nm; HRMS (m/z): [M+H]+ calcd for C7H10N3O3, 184.07; found, 184.07.
50: To a solution of 2 (70 mg, 0.5 mmol) in hot DMF (1 ml), were added sodium acetate (41 mg, 0.5 mmol) solution in water (0.5 ml) and O-methylhydroxylamine hydrochloride (42 mg, 0.5 mmol), and the solution was stirred at room temperature for 4 hrs. The solvents were removed in vacuo and the residue was collected by filtration and washed with cold water 2 × 1 ml, resulting in 76% yield (70 mg 87/13 mixture of trans and cis geometric isomers). 1H NMR (400 MHz, DMSO-d6): δ 11.40 (bs, 2 H), 8.52 (s, 0.13 H), 7.87 (s, 0.87 H), 7.74 (s, 0.87 H), 7.29 (s, 0.13 H), 3.89 (s, 0.39 H), 3.80 (s, 2.61 H); 13C NMR (100 MHz, DMSO-d6): δ 162.98, 162.36, 150.80, 150.27, 146.08, 142.26, 140.09, 137.31, 104.41, 103.43, 62.32, 61.44; UV/Vis: λmax 288 nm; HRMS (m/z): [M+H]+ calcd for C6H8N3O3, 170.06; found, 170.06.
51: To a solution of 3 (79 mg, 0.5 mmol) in hot DMF (2.0 ml), were added sodium acetate (46 mg, 0.5 mmol) solution in water (0.5 ml) and O-methylhydroxylamine hydrochloride (46 mg, 0.5 mmol), and the solution was stirred at 50°C for 4 h. The solvents were removed in vacuo and the residue was washed by cold water. After the filtration, product was obtained in 62% yield (53 mg). 1H NMR (400 MHz, DMSO-d6): δ 11.18 (s, 1 H), 10.77 (s, 1 H), 7.91 (s, 1 H), 5.77 (s, 1 H), 3.96 (s, 3 H); 13C NMR (125 MHz, DMSO-d6): δ 163.87, 151.02, 144.51, 142.17, 101.41, 62.84; UV/Vis: λmax 292 nm; HRMS (m/z): [M+Na]+ calcd for C6H7N3O3Na, 192.04; found, 192.04.
High-Throughput Inhibitor Screening
The substrate in this HTS assay was synthesized using standard phosphoramidite DNA solid phase chemistry using reagents purchased from Glen Research. The DNA was purified using anion exchange chromatography followed by desalting using reversed phase methods. The sequence and size was confirmed using analytical denaturing polyacrylamide gel electrophoresis and MALDI-MS. The substrate is a double-stranded 14-mer DNA containing nine U·A base pairs (5′-FAM-GCA CUU AAG AAU UG:3′-DABSYL-CA AUU CUU AAG UGC). The UNG HTS assay is performed as follows. To a 96-well microtiter plate was added 5 μL 2 mM total compound in DMSO, followed by 75 μL 33.3 pM human UNG in reaction buffer (10 mM Tris·HCl, pH 8.0, 20 mM NaCl, 7.5 mM MgCl2, 0.002% brij-35). The reactions were initiated by the addition of 20 μL 250 nM molecular beacon substrate in reaction buffer. The plates are incubated at ambient temperature in a fluorescence plate reader for 30 minutes, and the progress of the reaction was monitored every five minutes (Ex. 485 nm/Em. 520 nm). The final concentrations of the reagents in the assay are 10 mM Tris·HCl, pH 8.0, 20 mM NaCl, 7.5 mM MgCl2, 0.002% Brij-35, 25 pM human UNG, 50 nM molecular beacon substrate, 100 μM total compound, 5% DMSO. The MgCl2 is essential to increase the stability of the double-stranded DNA substrate, and thus decrease the initial fluorescence of the molecular beacon and increase the maximum signal of the assay. Addition of Brij-35, a non-ionic detergent, is essential to stabilize human UNG at the low concentration used in this assay. A similar assay has been described by Maksimenko et al. that utilizes a 39-mer hairpin DNA56. However, the synthesis and purification of this more complex substrate proceeds with low efficiency, and requires higher temperature to induce strand separation (Krosky and Stivers, unpublished). In contrast, the 14 mer double-stranded molecular beacon is routine, and allows screening to be performed conveniently at room temperature.
Mechanism of Inhibition
The substrate used in mechanism of inhibition studies was a modified DNA hairpin where the two strands described above are connected by a hexa-polyethylene glycol linker (PEG-U9). This substrate was easier to synthesize and purify than an all DNA hairpin, and unlike the double stranded DNA substrate, does not require MgCl2 to achieve minimum fluorescence. To a 96-well plate was added 5 μL compound in DMSO, followed by 75 μL PEG-U9 hairpin in reaction buffer (20 mM Tris HCl, pH 8.0, 50 mM KCl, 0.2 mM MgCl2, 0.002% Brij-35, 1 mM DTT). Eight different DNA concentrations were used in the range of 62.5 to 2000 nM. Reactions were initiated by the addition of 20 μL 0.5 nM human UNG in reaction buffer. The final concentrations of reagents in the assay are 20 mM Tris HCl, pH 8.0, 50 mM KCl, 0.2 mM MgCl2, 0.002% Brij-35, 1 mM DTT, 5% DMSO, 0.1 nM human UNG, 62.5–2000 nM PEG-U9 hairpin DNA, and variable amounts of inhibitor. The plates were incubated at ambient temperature in a fluorescence plate reader for 60 minutes, and the progress of each reaction was monitored every five minutes (Ex. 485 nm/Em. 520 nm). Afterwards,· E. coli UNG was added to each well to drive the reactions to completion, and the overall change in fluorescence values were measured. These values were used to convert initial velocities from units of fluorescence units/s to [product]/s. Mechanisms of inhibition and their corresponding inhibitor dissociation constants were determined by Lineweaver-Burk slope and intercept replot analysis and by computational simulations of the initial velocity against inhibitor concentration data using Dynafit v.3.28 (see Supplemental Information)
Conclusions
We have developed an efficient strategy to develop small molecule inhibitors of UNG that have the potential for activity in cell culture or in vivo. The method is quite general and could be adapted to target other enzymes that bind extrahelical bases or free nucleosides. Two future targets of the current uracil mixed oxime library would be the essential bacterial enzyme deoxyuridine nucleotidylhydrolase which converts dUTP to dUMP23, 57–60, and human thymidine phosphorylase, an enzyme implicated in vascularization of tumors61. Such inhibitors could serve as useful tools to study the life cycle of pathogenic human viruses, the biology of uracil base excision repair in normal cell lines and tissues, and mechanisms of tumor vascularization.
Supplementary Material
Tables of aryl aldehydes and hydroxybenzaldehydes used to construct oxime libraries, HPLC traces and NMR spectra to ascertain stoichiometries of oxime mixtures, 1H and 13C NMR spectra of 3-(3)-13 and 3-(3)-27, computer simulations of inhibition data, and analytical equations for each inhibition mechanism.
Acknowledgments
This work was supported by NIH grant GM56834-10 to J.T.S. D.J.K was supported by the DOD Breast Cancer Research Program (DAMD17-03-1-1251).
References
- 1.Lindahl T, Wood RD. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 2.Di Noia J, Neuberger MS. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 3.Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A. Nature Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 4.Storb U, Stavnezer J. Curr Biol. 2002;12:R725–727. doi: 10.1016/s0960-9822(02)01247-2. [DOI] [PubMed] [Google Scholar]
- 5.Chen R, Wang H, Mansky LM. J Gen Virol. 2002;83:2339–2345. doi: 10.1099/0022-1317-83-10-2339. [DOI] [PubMed] [Google Scholar]
- 6.Prichard MN, Duke GM, Mocarski ES. J Virol. 1996;70:3018–3025. doi: 10.1128/jvi.70.5.3018-3025.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Silva FS, Moss B. J Virol. 2003;77:159–166. doi: 10.1128/JVI.77.1.159-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart DT, Upton C, Higman MA, Niles EG, McFadden G. J Virol. 1993;67:2503–2512. doi: 10.1128/jvi.67.5.2503-2512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priet S, Gros N, Navarro JM, Boretto J, Canard B, Querat G, Sire J. Mol Cell. 2005;17:479–490. doi: 10.1016/j.molcel.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Ladner RD. Curr Protein Pept Sci. 2001;2:361–370. doi: 10.2174/1389203013380991. [DOI] [PubMed] [Google Scholar]
- 11.Tinkelenberg BA, Hansbury MJ, Ladner RD. Cancer Res. 2002;62:4909–4915. [PubMed] [Google Scholar]
- 12.Stivers JT, Drohat AC. Arch Biochem Biophys. 2001;396:1–9. doi: 10.1006/abbi.2001.2605. [DOI] [PubMed] [Google Scholar]
- 13.Bianchet MA, Seiple LA, Jiang YL, Ichikawa Y, Amzel LM, Stivers JT. Biochemistry. 2003;42:12455–12460. doi: 10.1021/bi035372+. [DOI] [PubMed] [Google Scholar]
- 14.Jiang YL, Cao C, Stivers JT, Song F, Ichikawa Y. Bioorg Chem. 2004;32:244–262. doi: 10.1016/j.bioorg.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Krosky DJ, Song F, Stivers JT. Biochemistry. 2005;44:5949–5959. doi: 10.1021/bi050084u. [DOI] [PubMed] [Google Scholar]
- 16.Sekino Y, Bruner SD, Verdine GL. J Biol Chem. 2000;275:36506–36508. doi: 10.1074/jbc.C000585200. [DOI] [PubMed] [Google Scholar]
- 17.Putnam CD, Shroyer MJN, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- 18.Bouhamdan M, Benichou S, Rey F, Navarro JM, Agostini I, Spire B, Camonis J, Slupphaug G, Vigne R, Benarous R, Sire J. J Virol. 1996;70:697–704. doi: 10.1128/jvi.70.2.697-704.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.BouHamdan M, Xue Y, Baudat Y, Hu B, Sire J, Pomerantz RJ, Duan LX. J Biol Chem. 1998;273:8009–8016. doi: 10.1074/jbc.273.14.8009. [DOI] [PubMed] [Google Scholar]
- 20.Chen R, Le Rouzic E, Kearney JA, Mansky LM, Benichou S. J Biol Chem. 2004;279:28419–28425. doi: 10.1074/jbc.M403875200. [DOI] [PubMed] [Google Scholar]
- 21.Klarmann GJ, Chen X, North TW, Preston BD. J Biol Chem. 2003;278:7902–7909. doi: 10.1074/jbc.M207223200. [DOI] [PubMed] [Google Scholar]
- 22.Mansky LM, Preveral S, Selig L, Benarous R, Benichou S. J Virol. 2000;74:7039–7047. doi: 10.1128/jvi.74.15.7039-7047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payne SL, Elder JH. Curr Protein Pept Sci. 2001;2:381–388. doi: 10.2174/1389203013381008. [DOI] [PubMed] [Google Scholar]
- 24.Selig L, Benichou S, Rogel ME, Wu LI, Vodicka MA, Sire J, Benarous R, Emerman M. J Virol. 1997;71:4842–4846. doi: 10.1128/jvi.71.6.4842-4846.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willetts KE, Rey F, Agostini I, Navarro JM, Baudat Y, Vigne R, Sire J. J Virol. 1999;73:1682–1688. doi: 10.1128/jvi.73.2.1682-1688.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansky LM, Le Rouzic E, Benichou S, Gajary LC. J Virol. 2003;77:2071–2080. doi: 10.1128/JVI.77.3.2071-2080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller RJ, Cairns JS, Bridges S, Sarver N. J Virol. 2000;74:7187–7195. doi: 10.1128/jvi.74.16.7187-7195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elder RT, Zhu X, Priet S, Chen M, Yu M, Navarro JM, Sire J, Zhao Y. Biochem Biophys Res Commun. 2003;306:693–700. doi: 10.1016/s0006-291x(03)01036-2. [DOI] [PubMed] [Google Scholar]
- 29.Lau A, Swinbank KM, Ahmed PS, Taylor DL, Jackson SP, Smith GC, O’Connor MJ. Nature Cell Biol. 2005;7:493–500. doi: 10.1038/ncb1250. [DOI] [PubMed] [Google Scholar]
- 30.Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Mol Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 31.Drohat AC, Stivers JT. Biochemistry. 2000;39:11865–11875. doi: 10.1021/bi000922e. [DOI] [PubMed] [Google Scholar]
- 32.Drohat AC, Stivers JT. J Am Chem Soc. 2000:1840–1841. [Google Scholar]
- 33.Maly DJ, Choong IC, Ellman JA. Proc Natl Acad Sci U S A. 2000;97:2419–2424. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winans KA, Bertozzi CR. Chem Biol. 2002;9:113–129. doi: 10.1016/s1074-5521(02)00093-5. [DOI] [PubMed] [Google Scholar]
- 35.Kwon K, Nagarajan R, Stivers JT. Biochemistry. 2004;43:14994–15004. doi: 10.1021/bi048801s. [DOI] [PubMed] [Google Scholar]
- 36.Segel IH. Enzyme Kinetics. John Wiley & Sons, Inc.; New York: 1993. [Google Scholar]
- 37.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 38.Jiang YL, Kwon K, Stivers JT. J Biol Chem. 2001;276:42347–42354. doi: 10.1074/jbc.M106594200. [DOI] [PubMed] [Google Scholar]
- 39.Jiang YL, Song F, Stivers JT. Biochemistry. 2002;41:11248–11254. doi: 10.1021/bi026227j. [DOI] [PubMed] [Google Scholar]
- 40.Jiang YL, Stivers JT. Biochemistry. 2002;41:11236–11247. doi: 10.1021/bi026226r. [DOI] [PubMed] [Google Scholar]
- 41.Cao C, Jiang YL, Stivers JT, Song F. Nat Struct Mol Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 42.Jiang YL, McDowell L, Poliks B, Studelska D, Cao C, Potter GS, Schaefer J, Song F, Stivers JT. Biochemistry. 2004;43:15429–15438. doi: 10.1021/bi0483864. [DOI] [PubMed] [Google Scholar]
- 43.Savva R, McAuley-Hecht K, Brown T, Pearl L. Nature. 1995;373:487–493. doi: 10.1038/373487a0. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee A, Yang W, Karplus M, Verdine GL. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 45.Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA, Krokan HE. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 46.Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 47.Kwon K, Jiang Y, Stivers J. Chem Biol. 2003;10:1–20. doi: 10.1016/s1074-5521(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 48.Page MI, Jencks WP. Proc Natl Acad Sci U S A. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jencks WP. Catalysis in Chemistry and Enzmology. Dover Publications, Inc.; New York: 1987. Binding Energy, Specificity, and Enzymic Catalysis: The Circe Effect; pp. 615–807. [Google Scholar]
- 50.The uracil N1-acetaldehyde O-methyl oxime (49) showed undetectable inhibition (Kc > 10 mM.
- 51.Kung PP, Bharadwaj R, Fraser AS, Cook DR, Kawasaki AM, Cook PD. J Org Chem. 1998;63:1846–1852. [Google Scholar]
- 52.Weiss RH, Furfine E, Hausleden E, Dixon DW. J Org Chem. 1984;49:4969–4972. [Google Scholar]
- 53.Kirk KL, Cantacuzene D, Nimitkitpaisan Y, Mcculloh D, Padgett WL, Daly JW, Creveling CR. J Med Chem. 1979;22:1493–1497. doi: 10.1021/jm00198a012. [DOI] [PubMed] [Google Scholar]
- 54.Reitz A, Avery MA, Verlander MS, Goodman M. J Org Chem. 1981;46:4859–4863. [Google Scholar]
- 55.Watanabe T, Suzuki T, Umezawa Y, Takeuchi T, Otsuka M, Umezawa K. Tetrahedron. 2000;56:741–752. [Google Scholar]
- 56.Maksimenko A, Ishchenko AA, Sanz G, Laval J, Elder RH, Saparbaev MK. Biochem Biophys Res Commun. 2004;319:240–246. doi: 10.1016/j.bbrc.2004.04.179. [DOI] [PubMed] [Google Scholar]
- 57.Grasser FA, Romeike BF, Niedobitek G, Nicholls J, Kremmer E. Curr Protein Pept Sci. 2001;2:349–360. doi: 10.2174/1389203013381053. [DOI] [PubMed] [Google Scholar]
- 58.Hidalgo-Zarco F, Gonzalez-Pazanowska D. Curr Protein Pept Sci. 2001;2:389–397. doi: 10.2174/1389203013381026. [DOI] [PubMed] [Google Scholar]
- 59.Studebaker AW, Balendiran GK, Williams MV. Curr Protein Pept Sci. 2001;2:371–379. doi: 10.2174/1389203013380946. [DOI] [PubMed] [Google Scholar]
- 60.Williams MV. Virology. 1988;166:262–264. doi: 10.1016/0042-6822(88)90171-7. [DOI] [PubMed] [Google Scholar]
- 61.Toi M, Atiqur Rahman M, Bando H, Chow LW. Lancet Oncol. 2005;6:158–166. doi: 10.1016/S1470-2045(05)01766-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables of aryl aldehydes and hydroxybenzaldehydes used to construct oxime libraries, HPLC traces and NMR spectra to ascertain stoichiometries of oxime mixtures, 1H and 13C NMR spectra of 3-(3)-13 and 3-(3)-27, computer simulations of inhibition data, and analytical equations for each inhibition mechanism.