

Abstract
In contrast to most amyloidogenic proteins or peptides that do not contain any significant post-translational modifications, the prion protein (PrP) is modified with either one or two polysaccharides and a GPI anchor which attaches PrP to the plasma membrane. Like other amyloidogenic proteins, however, PrP adopts a fibrillar shape when converted to a disease-specific conformation. Therefore, PrP polymerization offers a unique opportunity to examine the effects of biologically relevant non-peptidic modifications on conversion to the amyloid conformation. To test the extent to which a long hydrophobic chain at the C-terminus affects the intrinsic amyloidogenic propensity of PrP, we modified recombinant PrP with a N-myristoylamido-maleimidyl group, which can serve as a membrane anchor. We show that while this modification increases the affinity of PrP for the cell membrane, it does not alter the structure of the protein. Myristoylation of PrP affected amyloid formation in two ways: (i) it substantially decreased the extent of fibrillation, presumably due to off-pathway aggregation, and (ii) it prohibited assembly of filaments into higher-order fibrils by preventing their lateral association. The negative effect on lateral association was abolished if the myristoylated moiety at the C-terminus was replaced by a polar group of similar size or by a hydrophobic group of smaller size. When preformed PrP fibrils were provided as seeds, myristoylated PrP supported fibril elongation and formation of higher-order fibrils composed of several filaments. Our studies illustrate that, despite a bulky hydrophobic moiety at C-terminus, myristoylated PrP can still incorporate into fibrillar structure, and that the C-terminal hydrophobic substitution does not affect the size of the proteinase K resistant core, but controls the mode of lateral assembly of filaments into higher-order fibrils.
Conversion of proteins and peptides into amyloid fibrils is linked to several neurodegenerative or conformational maladies including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and prion diseases [1;2]. Most amyloidogenic proteins involved in these diseases are cytosolic and do not contain any significant post-translational modifications. Prion protein is unique in this respect. PrP is modified with either one or two polysaccharides [3] and a GPI anchor, which attaches PrP to the plasma membrane [4]. However, like other amyloidogenic proteins, PrP adopts a fibrillar shape, referred to as scrapie associated fibrils or prion rods, when converted into the disease-associated form [5-7]. Therefore, PrP fibril formation offers a unique opportunity to evaluate the effect of biologically relevant natural modifications consisting of polysaccharides and/or hydrophobic anchor, both of non-peptide nature, on acquiring the amyloid conformation.
GPI anchor can impact prion polymerization in several ways. First, GPI anchor attaches PrPC to the plasma membrane; this results in specific orientation of PrPC molecules on the two-dimensional surface and significantly increases its effective concentration. Second, the GPI anchor might impose steric constrains on packing of the polypeptide chain into the fibrillar structure. Specifically, the GPI anchor would preclude formation of structures in which C-termini are buried within the fibrillar interior (Fig. 1A). Other arrangements may leave the C-terminus more exposed to the solvent and would thus be less affected by modifications at that position (Fig. 1B). It is even possible that presence of the GPI anchor can redirect the preferred packing of PrP molecules into an alternative fibrillar conformation where PrP molecules are arranged in a manner different from that found for GPI-deficient PrP fibrils.
Figure 1.
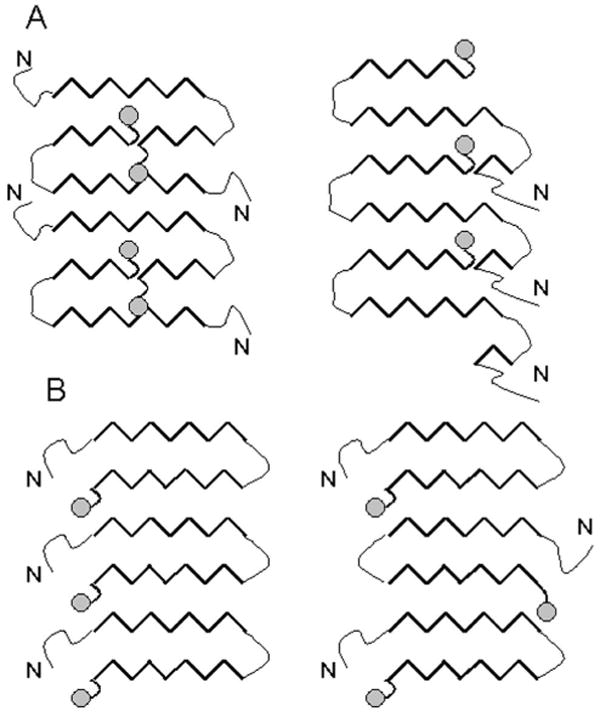
Schematic representation of possible packing of PrP molecules within amyloid fibrils. Hypothetical arrangements of PrP molecules within fibrillar form, where GPI anchor (gray circle) is buried inside the fibrils and interferes with the structure of fibrillar core (A), and where GPI anchor does not interfere with the structure of the fibrils (B).
While removal of the GPI anchor from mature PrPSc does not reduce prion infectivity [8], it is unclear whether the lack of GPI anchor in PrPC results in formation of fibrils that may be biologically irrelevant. The limited infectivity of fibrils produced in vitro from GPI-deficient recombinant PrP could be attributed to the lack of GPI-imposed steric constrains essential to infectious fibril formation [9;10].
In the current study, we examined whether a hydrophobic moiety, which is attached to the C-terminal amino acid residue and mimics a GPI anchor, interferes with or alters the amyloid structure adopted by PrP in solution. Previously we developed an experimental procedure for in vitro conversion of recombinant full-length PrP (rPrP) into amyloid fibrils [11]. Being a bacterially expressed recombinant protein, the rPrP used in our studies lacked a GPI anchor. Absence of the anchor removes steric constrains that might have been imposed on the packing of the polypeptide chain in the cross β-sheet fibrillar core. Therefore, in the fibrillar state, GPI-deficient PrP polypeptides may adopt a conformation different from the disease-specific conformations formed by PrPC in the cell. To mimic a GPI anchor, we mutated the C-terminal residue of rPrP into cysteine whose thiol side chain was chemically modified with a hydrophobic myristoyl-conjugated maleimide 1 (Fig. 2). Fibril formation from this modified protein (myr-PrP(S230C)) was partially disrupted at the early stages, possibly because the hydrophobic modification enhanced off-pathway aggregation. But myr-PrP(S230C) supported fibril elongation when preformed rPrP fibrils were used as seeds. These results illustrate that despite the bulky hydrophobic moiety at its C-terminus, myr-PrP(S230C) is able to incorporate into the fibrillar structure formed by GPI-deficient rPrP.
Figure 2.
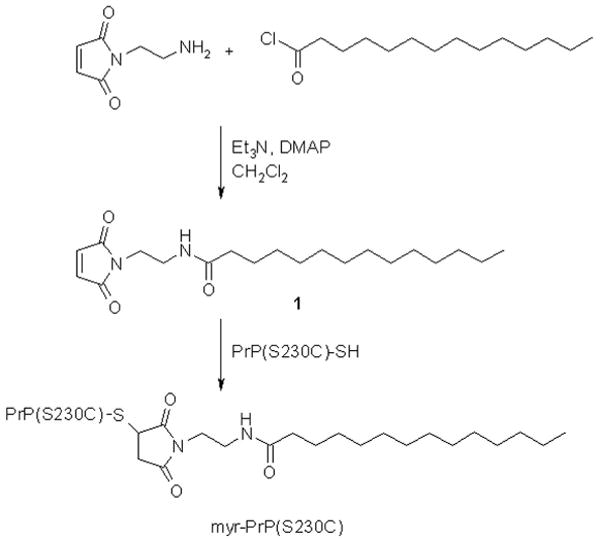
Synthesis of myr-PrP(S230C). Maleimide derivative 1 was prepared by acylation of N-(2-aminoethyl)maleimide with myristoyl chloride in the presence of triethylamine and 4-dimethylaminopyridine (DMAP). The resulting maleimide was attached to PrP(S230C) to yield myr-PrP(S230C).
Materials and Methods
Reagents and supplies were purchased from the following companies and were of the highest quality available: N-ethylmaleimide, USB (Cleveland, OH); oxidized glutathione, Calbiochem (San Diego, CA); HPLC grade acetonitrile, J. T. Baker (Phillipsburg, NJ); other chemicals and buffer salts, Sigma-Aldrich (St. Louis, MO); Nupage gels, secondary antibodies, Dulbecco's modified Eagle medium, Glutamax, fetal bovine serum, Invitrogen (Carlsbad, CA); Permanox 8-well Lab-Tek chamber slides, Nalge Nunc (Rochester, NY).
Expression and purification of wild type rPrP and PrP(S230C)
Wild-type mouse rPrP23-230 (Mo rPrP) was expressed and purified as described earlier [11;12]. The purified rPrP was confirmed by SDS-PAGE and electrospray mass spectrometry to be a single species with an intact disulfide bond and correct molecular weight. Refolding of rPrP to amyloid fibrils and electron microscopy were performed as previously described [12]. Fibril formation was conducted at 1 μM (automatic conversion format) [13] or 10 μM (manual conversion format) rPrP in the presence of 2 M GdnHCl, 50 mM MES and 10 mM thiourea (used as antioxidant) at pH 6, 37 °C with shaking at 600 RPM. For seeded fibril formation in the manual format the solution of rPrP was rotated in a Nutator (VWR) in the presence of 5% of preformed fibrils. Seeds were sonicated for 10 sec in a bath-type sonicator prior to reaction.
S230C point mutation was introduced into the mouse PrP23-230 gene using the QuickChange procedure, and the results were verified by sequencing. The mutant protein was expressed and purified as previously described [11;12] with the following modifications. For solubilization of inclusion bodies and purification on an IMAC column, β-mercaptoethanol was replaced with 10 mM reduced glutathione. After gel filtration, the PrP(S230C) solution (0.3 mg/mL in 6 M urea, 100 mM Tris-HCl, pH 7.5) was supplemented with 5 mM EGTA and 0.2 mM L-cystine, oxidized at 23 °C for 1 hour, and dialyzed against 3 M urea, 100 mM Tris-HCl, 5 mM EGTA, 0.2 mM L-cystine, pH 7.5 overnight for further oxidation. All subsequent steps were as previously described [11;12].
Synthesis of N-(2-myristoylamidoethyl)maleimide 1
N-(2-aminoethyl)maleimide trifluoroacetate salt (100 mg, 0.393 mmol), 4-dimethylaminopyridine (< 1 mg), and triethylamine (170 μL) were dissolved in 5 mL dichloromethane under dry argon atmosphere. Myristoyl chloride (110 μL, 0.405 mmol) was then added in a single portion. The reaction mixture was stirred under argon at 23 °C for 2.5 hr. The mixture was extracted with 0.5 M sodium citrate buffer (pH 4.5; 4×2mL). The organic phase was extracted once with 2 mL saturated NaHCO3, then washed with 2 mL of the citrate buffer, and finally washed with 2 mL saturated NaCl. The organic phase was dried over anhydrous MgSO4 containing a pinch of activated carbon, and then filtered. Solvent removal under reduced pressure on a rotary evaporator yielded 0.101 g of 1 as a white foam (73% yield). HRMS (ESI): m/z calculated for C20H35N2O3 (M+H+) 351.2648, found 351.2639.
Protein modification with maleimide 1
In a typical reaction, to a solution of the S230C mutant of rPrP (400 μM, 50 μL) in 6 M GdnHCl were added Hepes buffer (1 M, pH 7.3, 50 μL), 6 M GdnHCl (50 μL), DMSO (20 μL), TCEP (10 mM in water, 5 μL) and water (255 μL). The reaction mixture was shaken at 23 °C for 10 min and a solution of the maleimide 1 (2.5 mM in DMSO, 70 μL) was added. The reaction was shaken at 23 °C for 5 h. The reaction mixture was purified by HPLC (Symmetry 300 C4, 4.6×250 mm; Waters Corp. Milford MA). The eluant gradient consisted of 1→20% acetonitrile in water for 10 min, followed by 20→60% acetonitrile in water over 55 min; flow rate was 0.8 mL/min, and eluant contained 0.1% TFA. The product eluted at 37% acetonitrile and was lyophilized. The purity of modified rPrP was confirmed by SDS-PAGE followed by silver staining and electrospray mass spectrometry. The masses of WT and myristoylated rPrP were 23062 and 23422 Da, respectively. This procedure yielded 135 μg of modified protein (29% yield).
Protein modification with N-ethylmaleimide (NEM)
In a typical reaction, to a solution of the S230C mutant of rPrP (1 mg/mL, 42 μM, 300 μL) in water were added ammonium acetate (1 M, pH 7.0, 6 μL), TCEP (5 mM in water, 8 μL), and a solution of NEM (25 mM in DMSO, 10 μL, 25 eq.). The reaction mixture was shaken at 23 °C for 18 h and purified by HPLC as described above. The product eluted in 37% acetonitrile and was lyophilized. The yield of the product was about 180 μg (60%). The identity and purity of the modified protein was verified by ESI-MS and SDS-PAGE.
Estimation of hydrodynamic dimensions
Dynamic light scattering was used to determine the hydrodynamic radii of particles in solution with a DynaPro Molecular Sizing Instrument (Protein Solutions, Lakewood, NJ) and Dynamics software using a 12-μl quartz cuvette with 1.5-mm path length.
FTIR spectroscopy
FTIR spectra were measured with a Bruker Tensor 27 FTIR instrument (Bruker Optics, Bullerica, MA) equipped with a multiple internal reflection cell BioATR II (effective pathlength is ∼1 μm) and a MCT detector cooled with liquid nitrogen. Fibrils were dialyzed against 10 mM sodium acetate buffer (pH 5.0), and 10 μL of each sample (0.25 mg/mL) were loaded into a BioATR II cell. A total of 1024 scans at 2 cm-1 resolution were collected for each sample under constant purging with nitrogen. Spectra were corrected for water vapor, and background spectra of the same buffer were subtracted. The bands were resolved by Fourier self-deconvolution in the Opus 4.2 software package using a Lorentzian line shape and parameters equivalent to 20 cm-1 bandwidth at half height and a noise suppression factor of 0.3.
Maturation and proteinase K digestion of amyloid fibrils
For detergent-mediated maturation, preformed rPrP fibrils were dialyzed from denaturants and heated in a water bath at 80 °C in 100 mM Tris-HCl buffer (pH 7.5) and 0.05% Triton X-100 for 20 min as described earlier [14]. The solution was then spun down in a tabletop centrifuge for 2 s to restore the original volume. The amyloid fibrils of rPrP (0.1 mg/mL, 10 μL) were then treated with PK at 37 °C for 1 h in 100mM Tris-HCl buffer (pH 7.5, 1:100 PK:rPrP ratio). Digestion was stopped by quenching with PMSF (0.5 μL, 50 mM in acetonitrile), followed by addition of 4× sample buffer and 9 M urea to the final urea concentration of 2.25 M. Samples were heated at 95 °C for 10 min and analyzed by SDS-PAGE on precast 12% NuPage gels (Invitrogen) followed by silver staining.
Atomic force microscopy
The samples were imaged with a PicoSPM LE AFM (Molecular Imaging, Phoenix, AZ), operating in the AAC (acoustic alternative current) AFM mode and using a silicon cantilever PPP-NCH (Nanoscience, Phoenix, AZ) with a tip radius < 7 nm and a spring constant of 42 N/m. α-rPrP, α-myr-PrP(S230C) or myr-PrP(S230C) fibrils (10 μL) were deposited onto glass cover slips (25 mm × 25 mm, Fisher) at 25 μg/mL (for rPrP monomers) or 5 μg/mL (for fibrils). The coverslips were cleaned with 30% HNO3 before use. Samples were washed with distilled H2O 30 min after deposition and then dried with nitrogen. The images (512×512 pixel scans) were collected at a scan rate of 1-2 lines/s and a drive frequency of 285 kHz.
Immunoconformational assay
Dual-color staining was performed as described earlier [15]. Briefly, amyloid fibrils were incubated for 1 h in Permanox 8-well Lab-Teks in 10 mM sodium acetate buffer (pH 5.0), then fixed with 4% formaldehyde for 15 min, washed with Tris-buffered saline (TBS), incubated with 3 M or 6 M of GdnHCl in 50 mM TrisHCl (pH 7.5) for 1 h, and washed with TBS prior to assaying. Samples were incubated for 1 h at 23 °C with humanized anti-PrP Fab R1 (2 mg/mL, 1:400), followed by incubation with a secondary goat anti-human IgG (1:700) labeled with Alexa-488. Then, the samples were incubated for 1 h with a reference Ab: mouse IgG AG4 (2 mg/mL, 1:1000) that recognizes epitope 37-50 in PrP, followed by incubation with secondary Ab: goat anti-mouse IgG (1:700) labeled with Alexa-546. Fluorescence microscopy was carried out on an inverted microscope (Nikon Eclipse TE2000-U; Plan Fluor 100×, N.A. 1.3 objective) with an illumination system X-Cite 120 (EXFO Photonics Solutions Inc.) connected through fiber-optics.
Cell culture and immunostaining
Human SH-SY5Y neuroblastoma cells (ATCC) were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM Glutamax, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cultured cells were plated at a density ∼8,000 cells/cm2 in 8-well Permanox Lab-Tek chamber slides and incubated at 37 °C in humidified 5% CO2/95% air. Wild type α-rPrP or myr-α-PrP(S230C) were administered to the culture medium at 10 μg/mL and incubated for 1 h. Cells were fixed with 4% formaldehyde in PBS and incubated for 1 h at 23 °C with anti-PrP AG4 Ab (1:1000) followed by incubation with secondary Ab: goat anti-mouse IgG (1:700) labeled with Alexa-546.
Results
C-terminal epitope 224-230 of rPrP is buried within fibrils
We have observed earlier that the C-terminus of rPrP encompassing residues 152-230 is a part of the protease-resistant, β-sheet-rich core in amyloid fibrils [16]. Using a recently developed immunoconformational assay we confirmed that the epitopes located within the most stable PK-resistant core 152-230, including the extreme C-terminal region (residues 224-230), are buried in the fibril interior (Fig 1) [15]. The accessibility of the 224-230 epitope was probed using dual staining with a pair of antibodies, AG4 and R1, where antibody AG4, which recognizes the unstructured N-terminal region of PrP, was used as an internal control. We found that R1 antibody (epitope 224-230) does not bind to WT rPrP fibrils under native conditions. Incubation of fibrils in 6 M GdnHCl, however, resulted in denaturation and partial exposure of the R1 epitope (Fig. 3). Global denaturation of fibrils at 6 M GdnHCl seems to be required for this epitope to become immunoreactive, as incubation in 3 M GdnHCl had only minor effect. These data indicate that the C-terminus of rPrP is buried in the fibrillar core. In light of this, it seems likely that attaching a bulky hydrophobic moiety at position 230 could interfere with amyloid formation or have a significant effect on amyloid structure. To test this hypothesis, we have introduced a point mutation at position 230 followed by chemical modification of this residue and investigated the consequent effect on the amyloid structure.
Figure 3.

Immunoconformational assay on WT rPrP fibrils. Fluorescence microscopy images of fibrils stained with a pair of anti-PrP antibodies: AG4 (recognizing residues 37-50) and R1 (rresidues 224-230). The fibrils were kept under native conditions (top panel) or exposed to 3 M GdnHCl (central panel) or to 6 M GdnHCl (bottom panel) prior to staining. The secondary Ab to AG4 was labeled with Alexa-546 (red), and secondary Ab to R1 was labeled with Alexa-488 (green). Unstained fibrils have no detectable fluorescence.
Design and synthesis of myr-PrP(S230C)
In order to introduce modifications at the C-terminus of rPrP23-230, we mutated the C-terminal serine residue (S230) to cysteine. The mutant was expressed and purified using the same procedure previously described for WT rPrP with minor modifications. We took special precautions regarding formation of the proper disulfide bond in this mutant by performing oxidative refolding of the protein in partially denaturing conditions (3 M urea; see Materials and Methods). ESI-MS, SDS-PAGE, CD, and size-exclusion chromatography confirmed that the final product of purification is the correctly folded monomeric α-helical rPrP(S230C) (data not shown), which is present as a mixture of cysteine and glutathione adducts at cysteine 230 (Fig. 4A).
Figure 4.
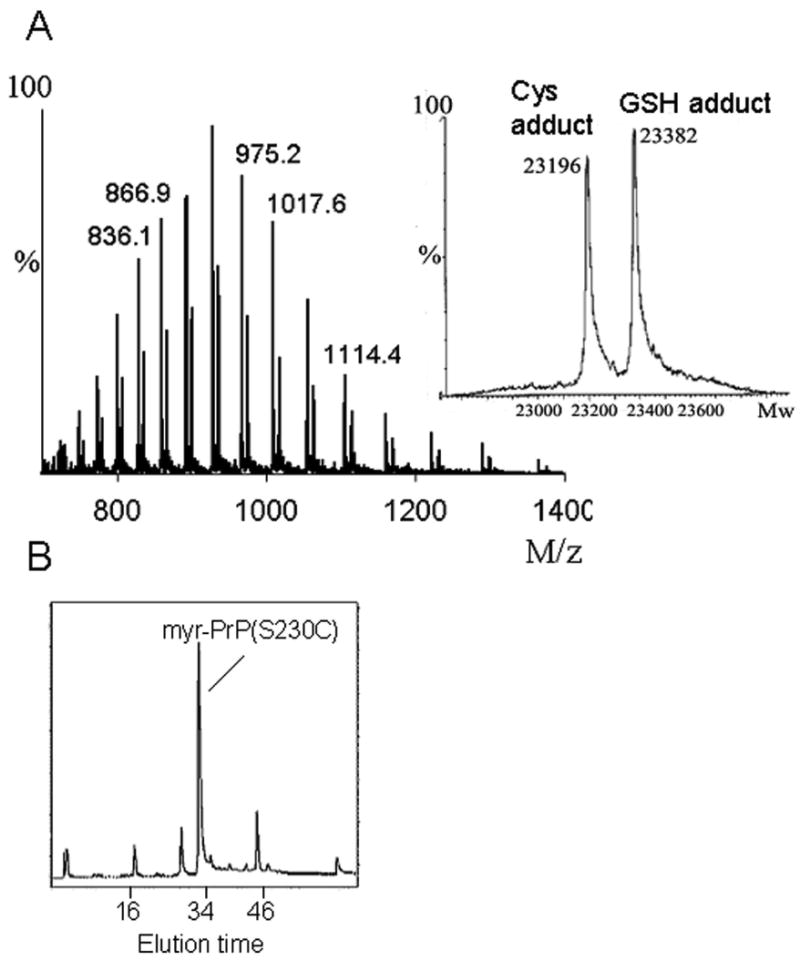
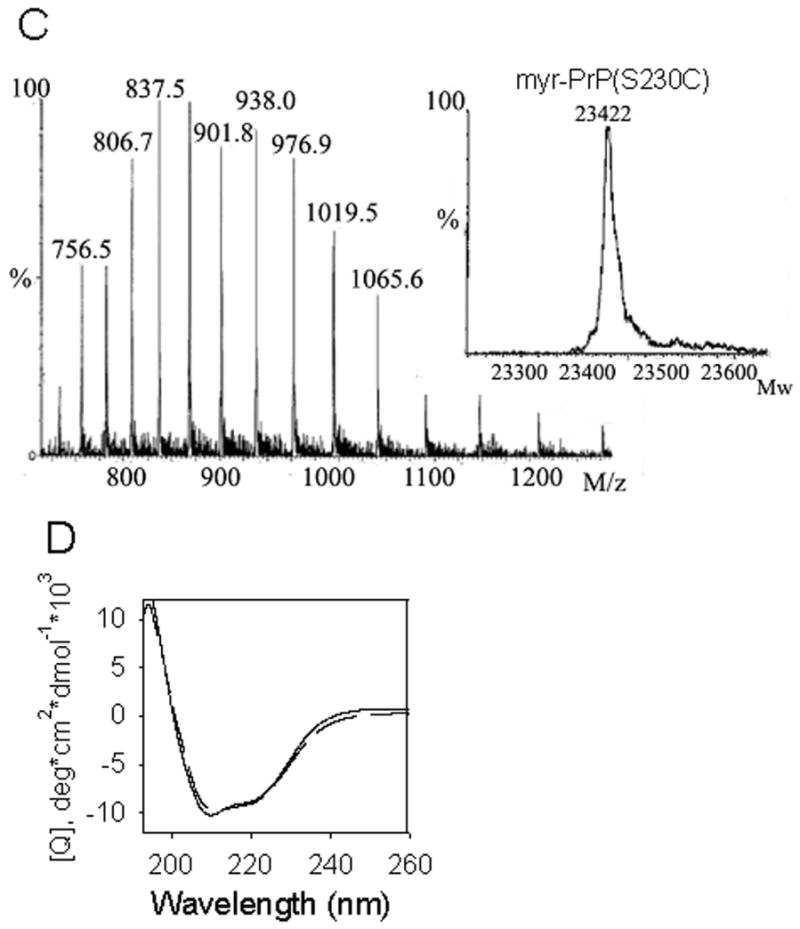
Preparation of myr-PrP(S230C). (A) ESI-MS of PrP(S230C) displays a mixture of two adducts. In the inset, deconvoluted mass spectrum shows two peaks at 23196 Da and 23382 Da that correspond to cysteine and glutathione adducts of PrP(S230C), respectively. (B) PrP(S230C) was modified with myristoylated maleimide 1 in ∼80% yield and purified by HPLC. (C) ESI-MS of myr-PrP(S230C) displays single component with a correct molecular weight of 23422 Da as calculated from the deconvoluted mass spectrum (inset). (D) CD spectra of myr-PrP(S230C) (dashed line) and WT rPrP (solid line) are similar and both show predominantly α-helical structure.
The high reactivity of free cysteine with electrophiles allows selective modification of this amino acid [17;18]. We prepared a maleimide derivative 1 with a myristoyl (C14) acyl chain by coupling of N-(2-aminoethyl)maleimide with myristoyl chloride (Fig. 2 and Materials and Methods). The resulting substituted maleimide was covalently attached to cysteine 230 of rPrP(S230C). Two natural cysteines in rPrP(S230C) are involved in the disulfide bond buried in the interior of the protein, and the newly introduced cysteine could be reduced and modified with good selectivity. In the optimized conditions (40 μM protein, 100 μM TCEP, 350 μM 1, 1.2 M GdnHCl, pH 7.3, 23 °C, 5 h) cysteine 230 in rPrP(S230C) was modified with myristoylated maleimide 1 in ∼80% yield. The product (myr-PrP(S230C)) was purified by reverse-phase HPLC (Fig. 4B). Only 30% yield of the modified protein was obtained after purification, possibly due to aggregation of the product during purification. Purity and identity of the modified protein was verified by electrospray mass spectrometry (MW 23422 Da) (Fig. 4C). Using CD spectroscopy we confirmed that myr-PrP(S230C) maintains an α-helix-rich conformation similar to that of the WT α-rPrP (Fig. 4D).
Myr-PrP(S230C) binds to the cell membrane
While myristoylation had no visible effect on the secondary structure of rPrP, we suspected that hydrophobic substitution on the C-terminus of rPrP might change other biophysical properties, such as the ability of rPrP to interact with the cell membrane. Because the GPI anchor is normally responsible for attachment of PrPC to the cellular membranes [19-21], we were interested to know whether myr-PrP(S230C) could mimic the localization of PrPC to lipid interfaces. Indeed, we observed that myristoylation of rPrP significantly increased its binding to the cell surface of SH-SY5Y neuroblastoma cells (Fig. 5A). This is not surprising since protein myristoylation is known to promote protein-membrane interactions [22]. WT rPrP did not bind efficiently to the cell surface under the same experimental conditions (Fig. 5B). Our data are consistent with previous observations that rPrP with a synthetic hydrophobic modification at the C-terminus binds to phospholipid membranes [23;24].
Figure 5.

Myr-PrP(S230C) (A) but not WT rPrP (B) binds to cell membranes. Human SH-SY5Y neuroblastoma cells were incubated with rPrPs (10 μg/mL) for 1 hour and stained with the anti-PrP antibody. Top panels represent immunofluorescence images using anti-PrP AG4 antibody (green). Bottom panels represent different fields of view, where phase-contrast images were superimposed with the immunofluorescence images (antibody staining is shown in red). Cells treated with WT rPrP (B, left top panel) and control cells that were not treated with rPrPs (B, right top panel) display only dim fluorescence, which was due to endogenously expressed PrPC.
Myristoylation of rPrP interferes with spontaneous fibril formation
To test whether myristoylation at the C-terminus interferes with conversion into fibrillar form, we subjected myr-PrP(S230C) to conditions that readily convert WT protein to amyloid fibrils (2 M GdnHCl, 50 mM MES, pH 6.0, 10 mM thiourea, 37 °C, shaking) [12]. Under these conditions, myristoylated protein did form fibril-like elongated aggregates but the reaction proceeded with 3-5 fold lower yield as determined by ThT binding assay (Fig. 8A, B). As judged from atomic force and electron microscopy, the end products of the conversion reaction resembled fibrils (Fig. 6 A,B). However, the majority of aggregates were short (100-500 nm in length, less than 8 nm in height) and bent or kinked (Fig. 6A, panels 1, 4, 6-8) or composed of single filaments (Fig. 6 A, panels 2,5 and B, panels 2-8). These filaments were strikingly different from the highly ordered fibrils formed from WT rPrP, which were found to be composed of several laterally assembled filaments [11;25]. In addition to fibril-like structures, a large number of non-fibrillar amorphous aggregates (2 nm height, 200-500 nm radius) were observed by atomic force microscopy at the end-point of conversion of myr-PrP(S230C) (Fig. 6A, panels 1,3); such aggregates were not observed in WT rPrP samples (data not shown, [25]). For the samples taken at the end-point of the conversion reaction, second derivatives of FTIR spectra showed significantly smaller peaks corresponding to β-sheets (1616-1626 cm-1) and β-turns and loops (1662 cm-1) and a bigger peak corresponding to α-helices (1651 cm-1) for myr-PrP(S230C) compared to the WT rPrP (Fig. 6C). These data illustrate a decrease in the yield of fibril formation. PK digestion revealed a smaller amount of protease-resistant fragments compared to that produced from WT fibrils (Fig. 6D). The PK-resistant fragments, however, had the same size for fibrils formed from WT rPrP and myr-PrP(S230C). A new PK-resistant band of 16kDa was observed upon brief incubation of WT rPrP and myr-PrP(S230C) fibrils at high temperature (a procedure referred to as maturation) (Fig. 6D). Temperature-induced maturation consists of brief heating at 80 °C and results in elongation of the proteinase K-resistant core [14]. Maturation was shown to be specific for amyloid rPrP structures, but not for β-sheet-rich non-amyloid oligomers [14]. Taken together, these results strongly suggest that myr-PrP(S230C) is capable of forming amyloid structures with amyloid cores that are similar in size to the cores of wild type fibrils. However, myristoylation at the C-terminus of rPrP23-230 reduces the efficiency of spontaneous conversion into fibrils and seems to interfere with the complex process of lateral assembly of filaments into higher-order fibrils [25].
Figure 8.
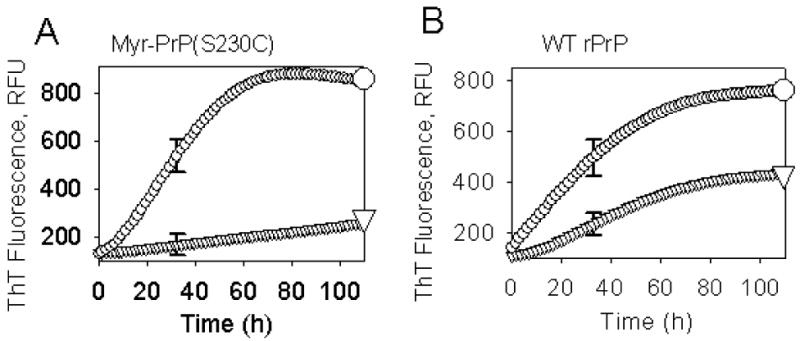
Kinetics of fibril formation from myr-PrP(S230C) (A) or WT rPrP (B) in the absence (▽) or presence (∓) of 1% WT rPrP fibrils used as seeds. The conversion reactions were carried out in automated format at 37 °C at rPrP concentration of 1 μM in 50 mM MES (pH 6.0) in the presence of 2 M GdnHCl and 10 mM thiourea and monitored by ThT fluorescence in a Fluoroscan Ascent CF microplate reader using 444 nm excitation filter with a half-bandwidth of 12 nm and 485 nm emission filter with a half-bandwidth of 14 nm (RFU corresponds to relative fluorescence units). The measurements were conducted in triplicate, and the results averaged; error bars represent standard deviation for triplicates. Data were acquired at 10-min intervals.
Figure 6.

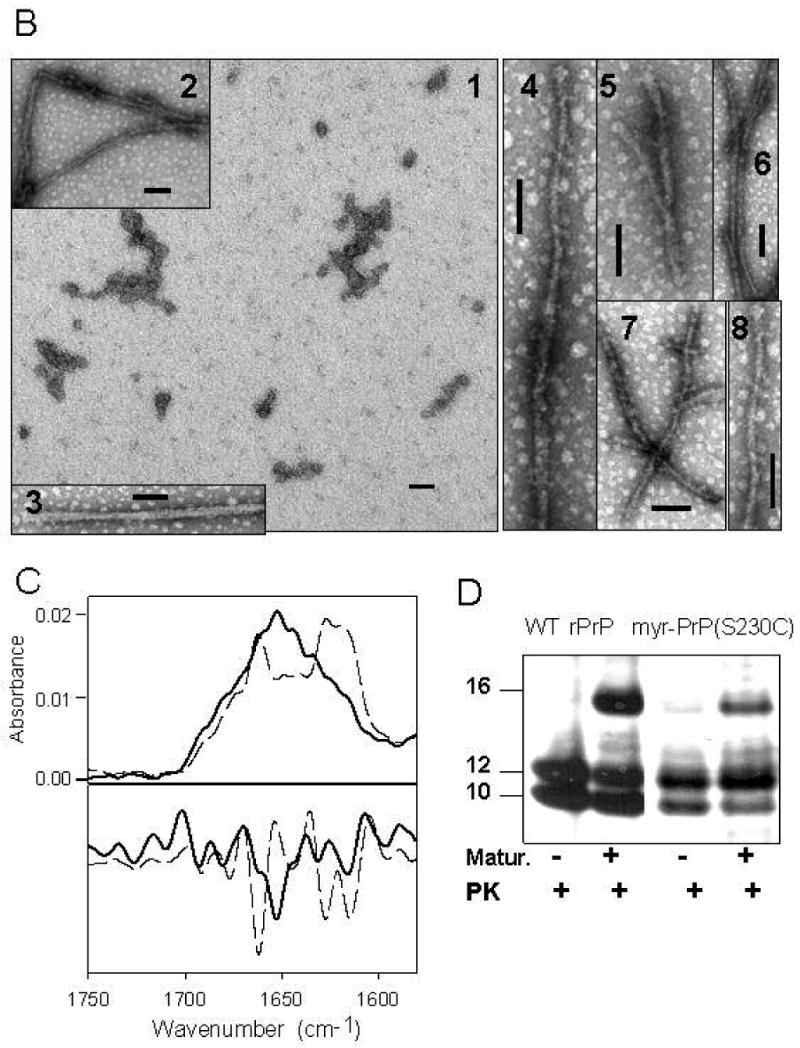
Myristoylation of rPrP impairs fibril formation. (A) AFM images of myr-PrP(S230C) fibrils taken at the end-point of growth: low-magnification (panels 1,3), high-magnification (panels 4-8) and 3D image (panel 2). Green arrows show non-fibrillar aggregates, black arrows show fibril-like structures. Scale bars = 0.1 μm. (B) Electron micrographs of myr-PrP(S230C) fibrils taken at the end-point of growth. Scale bars = 0.1 μm. (C) Deconvoluted FTIR spectra (top panel) and their second derivatives (bottom panel) of myr-PrP(S230C) (solid) and WT rPrP (dashed) fibrils show that conversion of myr-PrP(S230C) into fibrillar isoform is incomplete. (D) Proteinase K digestion assay of WT rPrP and myr-PrP(S230C) fibrils performed before and after maturation of fibrils.
To understand why myristoylation interferes with spontaneous fibrillation, we suggest that this substitution either changes physical properties of the conversion substrate, i.e. monomeric rPrP, or prohibits certain stages of polymerization.
Myr-PrP(S230C) forms multimers in solution
Formation of off-pathway aggregates is one of the possible explanations for the low yield of fibrils from myr-PrP(S230C). We addressed this issue directly by examining the oligomeric state of the modified protein. Dynamic light scattering of myr-PrP(S230C) (0.3 mg/ml, 0.5 M GdnHCl, 50 mM MES, pH 6) showed a significant population of large (Stokes radius > 20 nm) aggregates that were not observed for WT rPrP under the same conditions (Fig. 7A). Atomic force microscopy (10 mM acetate, pH 5) showed that while WT rPrP formed a few aggregates (Fig. 7B, panel 1), myr-PrP(S230C) formed a large number of spherical aggregates of variable sizes (Fig. 7B, panel 2). These results indicate the tendency of myr-PrP(S230C) to form multimeric aggregates under solvent conditions similar to that used for the conversion reaction. Addition of large hydrophobic substituents such as myristoyl is known to facilitate formation of protein-protein complexes [26;27]. Lack of secondary structural changes upon myristoylation of rPrP indicates that binding between monomers in myr-PrP(S230C) multimers is unlikely to be tight, and the multimers may be considered protein micelles. Off-pathway aggregation of myr-PrP(S230C) is consistent with the low yield observed in fibril formation. However, the aggregation does not explain the deficiency in assembling of filaments into high-order fibrils.
Figure 7.
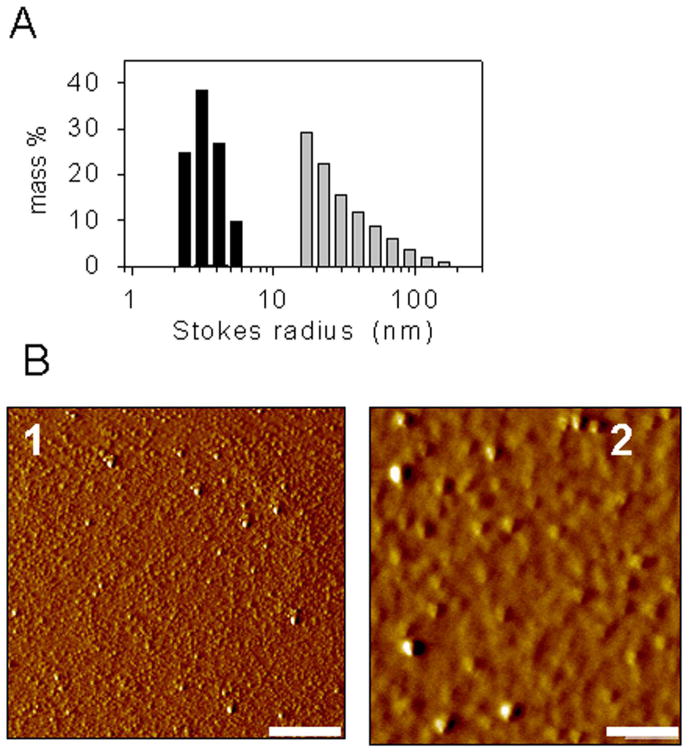
Myristoylated rPrP forms multimers in solution. Size of the particles formed by WT rPrP (black bars) and myr-PrP(S230C) (gray bars) in solution was examined by dynamic light scattering (A) and atomic force microscopy (B): WT rPrP - panel 1, myr-PrP(S230C) - panel 2. Scale bar = 1 μm.
Mature fibrils can be formed from myristoylated rPrP upon seeding
In order to evaluate whether myr-PrP(S230C) is competent to form amyloid structure, we initiated fibril formation from this protein by addition of preformed WT rPrP fibrils as seeds. Unlike spontaneous fibril formation from myr-PrP(S230C), which proceeded with low yield (see above), in fibril formation initiated by addition of 1% WT rPrP fibrils, the final ThT fluorescence increased to the level observed when WT rRrP was used as a substrate (Fig. 8). The lag phase, however, did not disappear completely even in the presence of the seeds (compare curves marked by ∓ in Fig. 8A and 8B). This is possible indication that the physical properties of myr-PrP(S230C) in solution differ from those of WT rPrP. Specifically, slightly longer lag-phase can be attributed to the high oligomerization state of myr-PrP(S230C) in solution.
To test whether fibrils formed from myr-PrP(S230C) are structurally different from the fibrils produced from WT rPrP, we prepared seeded fibrils of myr-PrP(S230C) on a larger scale. FTIR spectra of seeded fibrils were very similar to those formed from WT rPrP (Fig. 9A). Fibrils produced from both proteins displayed double bands at 1618 and 1628 cm-1 that correspond to intramolecular and intermolecular β-sheet structures and a band at 1662 cm-1 that accounts for β-turns and loops. Proteinase K digestion pattern was identical for seeded fibrils of myr-PrP(S230C) and for WT fibrils (data not shown). As judged from electron microscopy, the seeded fibrils of myr-PrP(S230C) were composed of several laterally assembled filaments that displayed relatively regular twisting patterns (Fig. 9B). These data indicate that myr-PrP(S230C) was successfully recruited by preformed fibrils to support elongation of higher-order fibrils.
Figure 9.
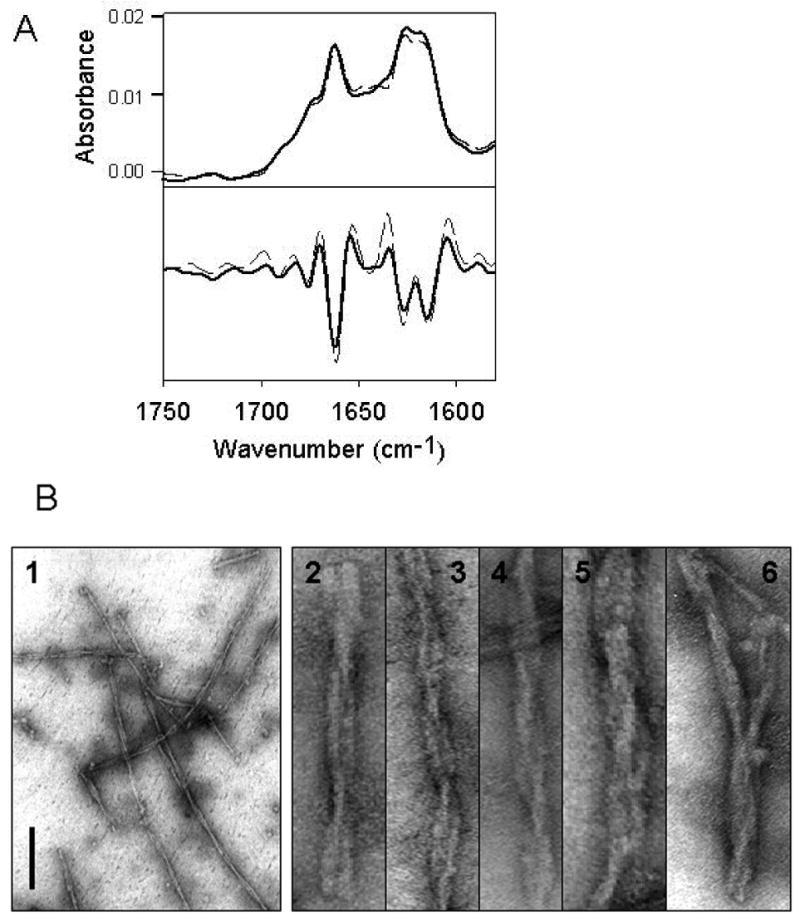
Fibril formation from myr-PrP(S230C) in the presence of 5% WT PrP seeds. (A) Deconvoluted FTIR spectra (top panel) and their second derivatives (bottom panel) of WT PrP fibrils (dashed), and fibrils formed from myr-PrP(S230C) seeded with WT rPrP fibrils (solid). (B) Electron microscopy images of seeded myr-PrP(S230C) fibrils. Scale bar = 0.2 μm. The images in panels 2-6 were expanded perpendicular to the fibril axis to visualize the details in the intertwining pattern.
Smaller hydrophobic NEM adduct and a hydrophilic glutathione adduct interfere less with PrP fibril formation
To test whether the size or polarity of the modification at the C-terminus influences spontaneous fibril formation from rPrP, we prepared adducts of S230C rPrP mutant with the small and hydrophobic N-ethylmaleimide (NEM-PrP(S230C)) and the larger but hydrophilic glutathione (GS-PrP(S230C)). NEM-PrP(S230C) was prepared by reacting rPrP(S230C) with NEM in the presence of TCEP at neutral pH. GS-PrP(S230C) was produced by modification of purification procedure for rPrP(S230C) where oxidized glutathione was used instead of cystine for oxidative refolding of the protein.
Both NEM-PrP(S230C) and GS-PrP(S230C) were used in our standard non-seeded conversion conditions (2 M GdnHCl, pH 6, 37 °C, shaking). In comparison to the fibrils produced from WT rPrP, fibrilar samples obtained from NEM-PrP(S230C) showed lower ThT fluorescence (data not shown) and somewhat larger proportion of α-helical conformation (shoulder at 1651 cm-1) as judged from FTIR spectra (Fig. 10A). A prominent peak at 1626 cm-1 with a shoulder at 1616 cm-1 indicated presence of intermolecular β-sheets (Fig. 10A). Electron microscopy showed high-order twisted fibrils composed of several filaments (Fig. 10C). These fibrils exhibited complex ultrastructures suggesting that NEM did not interfere with lateral association.
Figure 10.
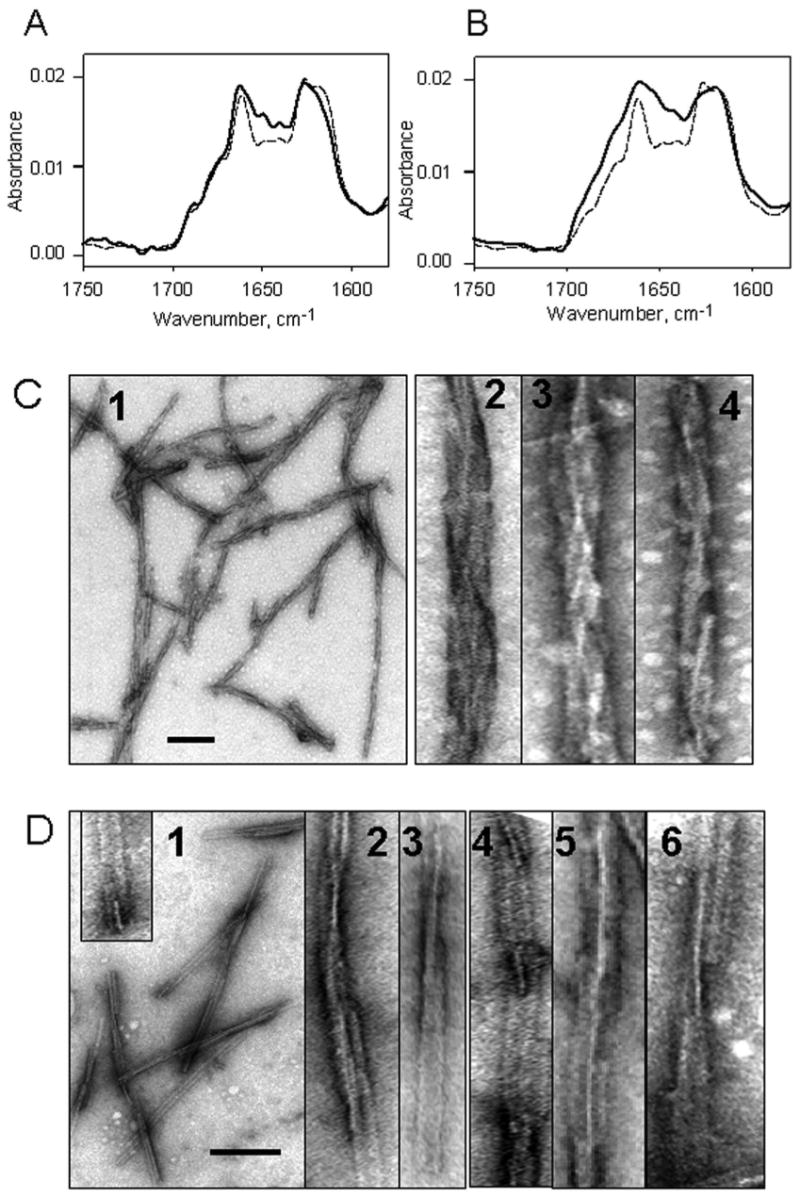
Fibril formation from NEM-PrP(S230C) and GS-PrP(S230C). (A) Deconvoluted FTIR spectra of NEM-PrP(S230C) (solid) and WT rPrP (dashed) fibrils. (B) Deconvoluted FTIR spectra of GS-PrP(S230C) (solid) and WT rPrP (dashed) fibrils. Electron micrographs of NEM-PrP(S230C) (C) and GS-PrP(S230C) (D) fibrils. Scale bars = 0.2 μm. The images of individual fibrils were expanded perpendicular to the fibril axis to visualize the details in fibrillar ultrastructures.
Kinetics of fibril formation from GS-PrP(S230C) and final ThT fluorescence were similar to WT rPrP (data not shown). FTIR spectra of GS-PrP(S230C) fibrils resembled the spectra of WT rPrP fibrils with the exception of an extra shoulder at 1651 cm-1 (α-helices) and a slightly decreased intensity at 1626 cm-1 (β-sheets) (Fig. 10B). When examined by electron microscopy, the resulting fibrils were composed of several filaments and displayed straight and flat ribbon-like morphologies (Fig. 10D). Both substituents at the C-terminus of rPrP, small hydrophobic NEM and larger hydrophilic glutathione, appeared to impact the capacity of the filaments for lateral association much less than the myristoyl group. NEM and glutathione, however, seem to favor different modes of lateral association to produce morphologically different populations of high-order fibrils. NEM-PrP(S230C) formed twisted fibrils that displayed relatively regular twisting patterns, whereas GS-PrP(S230C) produced predominantly flat ribbon-like fibrils, which occasionally showed some twisting.
Discussion
The GPI anchor is an integral part of the prion protein expressed in vivo. It has a significant effect on the properties of the protein by enabling attachment to the cell membrane. There is strong evidence that conversion of PrPC to PrPSc occurs on the cell membrane, possibly in lipid rafts [28-32].
In an attempt to mimic membrane-bound PrPC, several laboratories have labeled recombinant or synthetic PrP with synthetic surrogates of the GPI anchor. Ball and coworkers chemically synthesized a ‘mini-prion’ (lacking resides 23-88 and 141-176 of WT PrP) and attached a lipophilic group to the C-terminal lysine residue [33]. The protein bound to the cell surface in an orientation resembling that of PrPC. Glockshuber and coworkers added a short cysteine-containing peptide to the C-terminus of full-length rPrP [23]. The cysteine was the site of covalent attachment of the protein to liposomes containing the thiol-reactive lipid PDP-DHPE. The structure of rPrP remained unchanged upon its modification and coupling to the liposomes as judged by CD and PK digestion [23]. Pinheiro and coworkers mutated the C-terminal residue of rPrP to cysteine and attached a synthetic GPI mimic [24]. This modification resulted in reversible increase of rPrP β-sheet content as judged by FTIR and CD. When modified rPrP was incorporated into POPC or raft liposomes, its secondary structure reverted back to that of unmodified PrP as judged by FTIR [24]. These results show that a variety of nonpolar substituents at the C-terminus of rPrP can mimic a GPI anchor and mediate protein attachment to phospholipid membranes without causing significant structural changes. However, none of the existing studies attempted to examine the effect of GPI surrogates on conversion of rPrP into disease-related isoforms. In the current study, we introduced a nonpolar myristoylated group at the C-terminus of rPrP as a modification designed to mimic a GPI anchor. We did not attempt to directly emulate prion polymerization on a surface as it occurs in vivo, where the membrane-attached PrPC is recruited by membrane-bound PrPSc. Rather, we explored the effects of C-terminal hydrophobic substitution on the intrinsic ability of rPrP to form amyloid fibrils. Previous studies using other cell-free conversion protocols demonstrated that lipid-raft-bound PrPC resisted conversion into PK-resistant conformation (referred to as PrP-res) until PrPC was released from rafts by phospholipase digestion or PrPSc seeds were fused with the membranes [31;34]. It seems that both PrPC and PrPSc need to be aligned in a similar orientation for the conversion reaction to occur on a surface.
To test the extent to which the intrinsic ability of rPrP to form fibrils is affected, we introduced a hydrophobic modification into the C-terminus of rPrP by covalent coupling to the maleimide 1 prepared by acylation of N-(2-aminoethyl)maleimide with myristoyl chloride. This substituent is somewhat shorter than the natural GPI anchor or its previously used synthetic analogs [23;24]. Nonetheless, it was sufficient to significantly increase the binding of myr-PrP(S230C) to cell membranes compared to WT rPrP (Fig. 5). Moreover, the secondary structure of rPrP was unchanged by the modification (Fig. 4). These results suggest that our modification is a satisfactory functional analog of the GPI anchor.
Our experiments show a significant decrease in the yield of fibrils from myr-PrP(S230C) (Fig. 6). It appears that the rest of the protein is present in the form of non-fibrillar aggregates, which account for a substantial drop in the yield of fibrillation (Fig. 6, 7). The low yield of spontaneous fibrillation observed for myr-PrP(S230C) is consistent with the results of Riesner and coworkers on conversion of PrPC into fibrillar form in vitro [35]. Using different solvent conditions for the in vitro conversion reaction (0.01% SDS, pH 7.2), they observed that PrPC formed a mixture of fibrils and amorphous aggregates. Aggregates accounted for the majority of the protein (over 85%). Thus, formation of amorphous aggregates may be a general property of PrP with a hydrophobic modification at the C-terminus in the absence of any cellular components or cellular membrane.
As judged from PK-digestion pattern, the length of the cross-β amyloid core was not affected by the myristoylated modification at the C-terminus. However, the ability of filaments to assemble into higher-order fibrils was significantly impaired for myr-PrP(S230C). The negative effect of the large hydrophobic modification on fibrillation was abolished when WT rPrP fibrils were provided as seeds. These data suggest that (i) the hydrophobic moiety at the C-terminus prevents filament interaction and their lateral association, (ii) fibrils formed from unmodified rPrP served as efficient seeds for polymerization of myr-PrP(S230C). The differences in the mechanism of spontaneous versus seeded conversion could account for dramatic differences in the impact of myristoylation observed in these two experiments. Spontaneous polymerization is a highly hierarchical process that consists of consecutive steps of filament formation followed by their lateral assembly into higher-order fibrils [25;36]. Seeded conversion, on the other hand, seems to proceed via binding of single PrP molecules to fibrillar edges. Therefore, the negative effect of myristoylation on seeded conversion is minimized as this effect appears to be counteracted by the energy of binding of myr-PrP(S230C) polypeptide to the fibrillar cross β-structure. These results are consistent with a report by Chesebro and coworkers that PrPSc fibrils formed in mice that express GPI-deficient PrPC were capable of initiating prion replication and induced prion disease in WT mice by recruiting normal GPI-anchored PrPC [37].
In our study, smaller (NEM) or less hydrophobic (glutathione) substitutions at the C-terminus affected spontaneous fibrillation to a lesser extent than myristoylated modification. High-order fibrils composed of several filaments were observed for both NEM- and glutathione-modified rPrPs (Fig. 10). The mode of lateral assembly, however, was significantly affected by the size and the chemical nature of the group attached to the C-terminus. Together with our former studies on mapping of PK-resistant regions and epitope presentation, these data suggest that the C-terminus is not only involved in formation of cross-β-structure but might also contributes to formation of the interface between filaments [15;16].
Chesebro and coworkers recently showed that infection of mice expressing GPI-deficient PrPC with PrPSc led to accumulation of protease-resistant PrPSc deposits [37]. These deposits (containing anchorless PrPSc) were infectious towards mice expressing wild-type PrPC, which suggest that GPI-anchor is not required for generating infectivity in the PrPSc-dependent conversion reaction in vivo [37]. Also, Jackson and coworkers have shown that enzymatic removal of the GPI anchor from PrPSc did not lead to any decrease in infectivity as tested in cultured cells and in mice expressing wild-type PrPC [8]. These experiments clearly indicate that conversion of PrPC to PrPScin vivo produces infectious material independently of the presence of GPI anchor on either of the participants. It is not clear, however, whether the GPI anchor is important for generating infectivity in the absence of PrPSc seeds, the way it occurs in sporadic CJD. Our conversion reaction is designed to mimic spontaneous formation of prions, when PrPSc seeds are not provided. Future investigation of fibril formation from myr-PrP(S230C) bound to the cellular or synthetic membrane will provide further insight into the mechanism of PrP polymerization.
Acknowledgments
This work was supported by National Institute of Health grant NS045585 to I.V.B. and GM56481 to J.P.Y.K
Abbreviations
- PrP
prion protein
- rPrP
recombinant full-length prion protein
- PrPC
cellular isoform of the prion protein
- PrPSc
disease associated isoform of the prion protein
- GPI
glycosylphosphatidylinisotol
- PrP-res
proteinase K resistant form of the prion protein
- WT
wild type
- ThT
Thioflavin T
- PK
proteinase K
- GS
glutathione
- GdnHCl
guanidine hydrochloride
- NEM
N-ethylmaleimide
- AFM
atomic force microscopy
References
- 1.Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350:134–138. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Shattuck Lecture -- Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 3.Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry. 1989;28:8380–8388. doi: 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- 4.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 6.Somerville RA, Ritchie LA, Gibson PH. Structural and biochemical evidence that scrapie-associated fibrils assemble in vivo. J Gen Virol. 1989;70:25–35. doi: 10.1099/0022-1317-70-1-25. [DOI] [PubMed] [Google Scholar]
- 7.Liberski PP, Brown P, Xiao SY, Gajdusek DC. The ultrastructural diversity of scrapie-associated fibrils isolated from experimental scrapie and Creutzfeldt-Jakob disease. J Comp Pathol. 1991;105:377–386. doi: 10.1016/s0021-9975(08)80107-7. [DOI] [PubMed] [Google Scholar]
- 8.Lewis PA, Properzi F, Prodromidou K, Clarke AR, Collinge J, Jackson GS. Removal of the clycosylphosphotidyl inositol anchor from PrPSc by cathepsin-D does not reduce prion infectivity. Biochem J. 2006;395:443–448. doi: 10.1042/BJ20051677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legname G, Baskakov IV, Nguyen HOB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 10.Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway Complexity of Prion Protein Assembly into Amyloid. J Biol Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 11.Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. J Mol Biol. 2005;346:645–659. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 12.Breydo L, Bocharova OV, Makarava N, Salnikov VV, Anderson M, Baskakov IV. Methionine Oxidation Interferes with Conversion of the Prion Protein into the Fibrillar Proteinase K-Resistant Conformation. Biochemistry. 2005;44:15534–15543. doi: 10.1021/bi051369+. [DOI] [PubMed] [Google Scholar]
- 13.Breydo L, Bocharova OV, Baskakov IV. Semiautomated cell-free conversion of prion protein: Applications for high-throughput screening of potential antiprion drugs. Anal Biochem. 2005;339:165–173. doi: 10.1016/j.ab.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Bocharova OV, Makarava N, Breydo L, Anderson M, Salnikov VV, Baskakov IV. Annealing PrP amyloid firbils at high temperature results in extension of a proteinase K resistant core. J Biol Chem. 2006;281:2373–2379. doi: 10.1074/jbc.M510840200. [DOI] [PubMed] [Google Scholar]
- 15.Novitskaya V, Makarava N, Bellon A, Bocharova OV, Bronstein IB, Williamson RA, Baskakov IV. Probing the conformation of the prion protein within a single amyloid fibril using a novel immunoconformational assay. J Biol Chem. 2006;281:15536–15545. doi: 10.1074/jbc.M601349200. [DOI] [PubMed] [Google Scholar]
- 16.Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease PrPSc. Prot Science. 2005;14:1222–1232. doi: 10.1110/ps.041186605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scozzafava A, Casini A, Supuran CT. Targeting cysteine residues of biomolecules: new approaches for the design of antiviral and anticancer drugs. Curr Med Chem. 2002;9:1167–1185. doi: 10.2174/0929867023370077. [DOI] [PubMed] [Google Scholar]
- 18.Carne AF. Chemical modifications of proteins as an aid to sequence analysis. Methods Mol Biol. 2003;211:333–354. doi: 10.1385/1-59259-342-9:333. [DOI] [PubMed] [Google Scholar]
- 19.Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3:319–329. doi: 10.1093/glycob/3.4.319. [DOI] [PubMed] [Google Scholar]
- 20.Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry. 1990;29:5405–5412. doi: 10.1021/bi00474a028. [DOI] [PubMed] [Google Scholar]
- 21.Caughey B, Neary K, Butler R, Ernst D, Perry L, Chesebro B, Race RE. Normal and scrapie-associated forms of prion protein differ in their sensitivities to phospholipase and proteases in intact neuroblastoma cells. J Virol. 1990;64:1093–1101. doi: 10.1128/jvi.64.3.1093-1101.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peitzsch RM, McLaughlin S. Binding of acylated peptides and fatty acids to phospholipid vesicles pertinence to myristoylated proteins. Biochemistry. 1993;32:10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- 23.Eberl H, Tittman P, Glockshuber R. Characterization of recombinant, membrane-attached full-length prion protein. J Biol Chem. 2004;279:25058–25065. doi: 10.1074/jbc.M400952200. [DOI] [PubMed] [Google Scholar]
- 24.Hicks MR, Gill AC, Bath IK, Rullay AK, Sylvester ID, Crout DH, Pinheiro TJ. Synthesis and structural characterization of a mimetic membrane-anchored prion protein. FEBS J. 2006;273:1285–1299. doi: 10.1111/j.1742-4658.2006.05152.x. [DOI] [PubMed] [Google Scholar]
- 25.Anderson M, Bocharova OV, Makarava N, Breydo L, Salnikov VV, Baskakov IV. Polymorphysm and Ultrastructural Organization of Prion Protein Amyloid Fibrils: An Insight from High Resolution Atomic Force Microscopy. J Mol Biol. 2006;358:580–596. doi: 10.1016/j.jmb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi N, Nakagawa C, Ito Y, Takasaki A, Jinbo Y, Yamakava Y, Titani K, Hashimoto K, Izumi Y, Matsushima N. Myristoylation-regulated direct interaction between calcium-bound calmodulin and N-terminal region of pp60v-src. J Mol Biol. 2004;338:169–180. doi: 10.1016/j.jmb.2004.02.041. [DOI] [PubMed] [Google Scholar]
- 27.Chow M, Newman JFE, Filman D, Hogle JM, Rowlands DJ, Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 1987;327:482–486. doi: 10.1038/327482a0. [DOI] [PubMed] [Google Scholar]
- 28.Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA. 1996;93:14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibits formation of the scrapie isoform. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 31.Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raf associated prion protein to the protease-resistant state requires insertion of PrP-res(PrP(Sc)) into contiguous membrane. EMBO J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris DA. Cellular Biology of Prion Diseases. Clin Microbiol Rev. 1999;12:429–444. doi: 10.1128/cmr.12.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ball HL, King DS, Cohen FE, Prusiner SB, Baldwin MA. Engineering the prion protein using chemical synthesis. J Pept Res. 2001;58:357–374. doi: 10.1034/j.1399-3011.2001.00943.x. [DOI] [PubMed] [Google Scholar]
- 34.Baron GS, Caughey B. Effect of Glycosylphosphatidylinositol Anchor-dependent and -independent Prion Protein Associated with Model Raft Membranes on Conversion to the Protease-resistant Isoform. J Biol Chem. 2003;278:14883–14892. doi: 10.1074/jbc.M210840200. [DOI] [PubMed] [Google Scholar]
- 35.Leffer KW, Wille H, Stohr J, Junger E, Prusiner SB, Riesner D. Assembly of natural and recombinant prion protein into fibrils. Biol Chem. 2005;386:569–580. doi: 10.1515/BC.2005.067. [DOI] [PubMed] [Google Scholar]
- 36.Makarava N, Bocharova OV, Salnikov VV, Breydo L, Anderson M, Baskakov IV. Dichotomous versus palm-type mechanisms of lateral assembly of amyloid fibrils. Prot Science. 2006;15:1334–1341. doi: 10.1110/ps.052013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chesebro B, Trifilo M, Race M, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein result in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]