

Abstract
The Sigirr gene (also known as Tir8) encodes for an orphan receptor of the Toll-like receptor (TLR)/interleukin 1 receptor family that inhibits TLR-mediated pathogen recognition in dendritic cells. Here, we show that Sigirr also inhibits the activation of dendritic cells and B cells upon exposure to RNA and DNA lupus autoantigens. To evaluate the functional role of Sigirr in the pathogenesis of systemic lupus erythematosus (SLE), we generated Sigirr-deficient C57BL/6-lpr/lpr mice. These mice developed a progressive lymphoproliferative syndrome followed by severe autoimmune lung disease and lupus nephritis within 6 mo of age as compared with the minor abnormalities observed in C57BL/6-lpr/lpr mice. Lack of Sigirr was associated with enhanced activation of dendritic cells and increased expression of multiple proinflammatory and antiapoptotic mediators. In the absence of Sigirr, CD4 T cell numbers were increased and CD4+CD25+ T cell numbers were reduced. Furthermore, lack of Sigirr enhanced the activation and proliferation of B cells, including the production of autoantibodies against multiple nuclear lupus autoantigens. These data identify Sigirr as a novel SLE susceptibility gene in mice.
Systemic autoimmunity means losing tolerance against ubiquitous autoantigens. Genetic factors are important in the pathogenesis of systemic autoimmunity (1), e.g., genetic variants in major tolerance-regulator genes like FOXP3 can cause fatal neonatal autoimmunity in non-autoimmune–prone mice and humans (2, 3). In contrast, the lupus erythematosus (LE) encompasses a variety of clinical manifestations, including serious autoimmune tissue injury that develops almost always after the neonatal phase (4). LE rather results from a combination of variants in genes that control lymphoproliferation and immune regulation at multiple levels (5). Recently, systematic genome-wide studies on multiple multiethnical cohorts of lupus patients have identified genetic variants in genes like IRF5, BANK1, ITGAM, TNFSF4, and STAT4 by mapping genomic regions associated with human systemic LE (SLE) (6–11). Combinations of genetic polymorphisms in either weak or potent susceptibility genes seem to account for the variability of time of disease onset and clinical manifestation patterns in human lupus (1, 4, 5). In mice, single loss-of-function mutations in potent susceptibility genes like Tgf-β1, DNase1, Lyn, Fas, or C1q are sufficient to cause late-onset lupus-like autoimmunity (12–18). Mutations in some susceptibility genes do not trigger autoimmunity in the absence of a second genetic factor, e.g., Sle1, Tlr7, or Tlr9 (19–21). Weaker disease modifier genes like IL-10 or IL-27R enhance LE only in the context of multiple susceptibility genes, e.g., being provided by the specific autoimmune genetic background of MRL mice (22, 23).
Single Ig IL-1–related receptor (SIGIRR), also known as Toll–IL-1 receptor 8 (TIR8), is a member of the Toll-like receptor (TLR)/IL-R family (24). Both the extracellular and intracellular domains of SIGIRR differ from the other members of the TLR/IL-1R superfamily (24). Its small single extracellular Ig domain does not support ligand binding. Furthermore, the intracellular domain of SIGIRR cannot activate NF-κB because it lacks two essential amino acids (Ser447 and Tyr536) in its highly conserved TIR domain (24). SIGIRR rather acts as an endogenous inhibitor of TLR and IL-1 signaling because overexpression of SIGIRR in Jurkat or HepG2 cells substantially reduced LPS or IL-1–induced activation of NF-κB (25–27). Pathogen challenge or damaging the intestinal epithelial barrier surfaces in mice with impaired SIGIRR function resulted in severe immunity-mediated tissue damage (25, 28–31). Lack of Sigirr enhanced LPS signaling in dendritic cells and intestinal epithelia. Hence, SIGIRR is one of several negative regulators that suppress TLR-mediated antimicrobial defense (32). The SIGIRR gene is localized at the p15 region of chromosome 11, a region to which linkage analyses have mapped yet unknown lupus susceptibility genes (33, 34). SIGIRR might contribute to the control of autoimmunity because SIGIRR suppresses TLR signaling in dendritic cells, a recently discovered pathomechanism of lupus (35). Immune complexes containing the lupus autoantigens U1snRNP or nucleosomes activate dendritic cells (and autoreactive B cells) in vitro via TLR7 and TLR9, respectively (36–41). In vivo studies with TLR7 antagonists (42), Tlr7-deficient mice (43), or TLR7 overexpression confirm this concept for TLR7 (20, 44). In contrast, data from studies using TLR9 antagonists (45, 46) and Tlr9-deficient autoimmune mice remain inconsistent (21, 43, 47).
We hypothesized a role for SIGIRR beyond the control of microbial defense, namely, suppressing inadequate activation of antigen-presenting cells in autoimmunity. We therefore characterized the phenotype of Sigirr-deficient C57BL/6lpr/lpr (B6lpr/lpr) mice in which the lpr mutation causes delayed autoimmunity and hardly detectable autoimmune tissue injury within 6 mo of age (12).
RESULTS
Lack of Sigirr induces severe lymphoproliferation in B6lpr/lpr mice
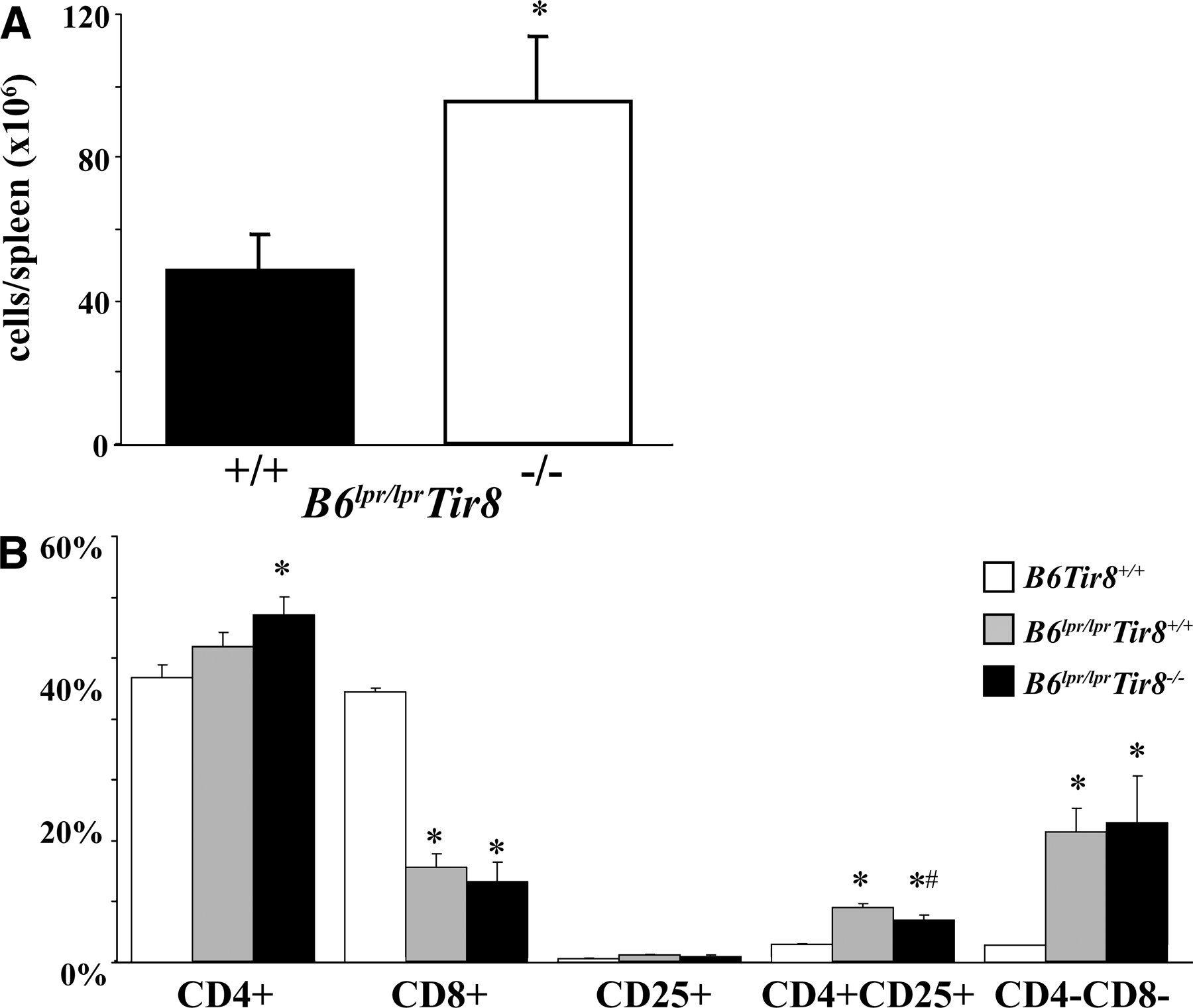
To evaluate the role of SIGIRR in autoimmunity we first carefully evaluated Sigirr-deficient B6 mice for signs of spontaneous autoimmunity, e.g., autoantibodies against double-stranded DNA (dsDNA) or rheumatoid factor. In Sigirr-deficient B6 mice up to 12 mo of age such antibodies could not be detected (Table S1, available at http://www.jem.org/cgi/content/full/jem.20072642/DC1). Furthermore, no antibodies binding to Critidae luciliae kinetoplast DNA could be detected in either of the two mouse strains (not depicted), indicating that lack of Sigirr alone does not induce autoimmunity against DNA in B6 mice. Next, we backcrossed Sigirr-deficient mice into autoimmune MRLlpr/lpr mice, but we were unable to continue backcrossing beyond the F4 generation because even the heterozygous female MRLlpr/lpr/Tir8−/+ died from accelerated SLE at an early age (not depicted). To avoid the impact of the multiple lupus susceptibility genes of the MRL genetic background, we generated Sigirr-deficient B6lpr/lpr mice. The autoimmune phenotype of B6lpr/lpr mice is introduced only by a single mutated LE susceptibility gene that impairs Fas-induced apoptosis of autoreactive B and T cells (12). B6lpr/lpr mice represent a rather mild model of lupus autoantibody production and hardly detectable autoimmune tissue injury late in life; therefore, B6lpr/lpr/Tir8−/− mice could be generated without the problems noted with MRLlpr/lpr/Tir8−/+ mice. For SLE phenotype analysis, we first evaluated the size of spleens and lymph nodes in 6-mo-old B6lpr/lpr and B6lpr/lpr/Tir8r−/− mice. Spleens and lymph nodes were massively enlarged in B6lpr/lpr/Tir8−/− mice as compared with B6lpr/lpr mice (Fig. 1 A). This was evident from spleen and bulk mesenteric lymph node weights (Fig. 1 B) and from total numbers of spleen cells quantified by flow cytometry (Fig. S1 A). Spleen histomorphology revealed lymph follicle hyperplasia in 6-mo-old Sigirr-deficient B6lpr/lpr mice (Fig. 1 C). Thus, lack of Sigirr causes excessive lymphoproliferation in mice when introduced into the context of a single additional lupus susceptibility gene (lpr).
Figure 1.

Lack of Sigirr is associated with massive lymphoproliferation in B6lpr/lpr mice. (A and B) At 6 mo of age, Sigirr-deficient B6lpr mice revealed massive hyperplasia of cervical, axillar (A), and mesenteric lymph nodes (B) as well as splenomegaly (B). Data are means ± SEM from 12 mice in each group. *, P < 0.05. (C) PAS staining of spleen sections indicates lymph follicle hyperplasia in Sigirr-deficient B6lpr/lpr mice. Images in A and C are representative of at least 12 mice in each group. Bars: (B) 1 cm; (C) 100 μm.
Sigirr suppresses dendritic cell activation upon exposure to lupus autoantigens
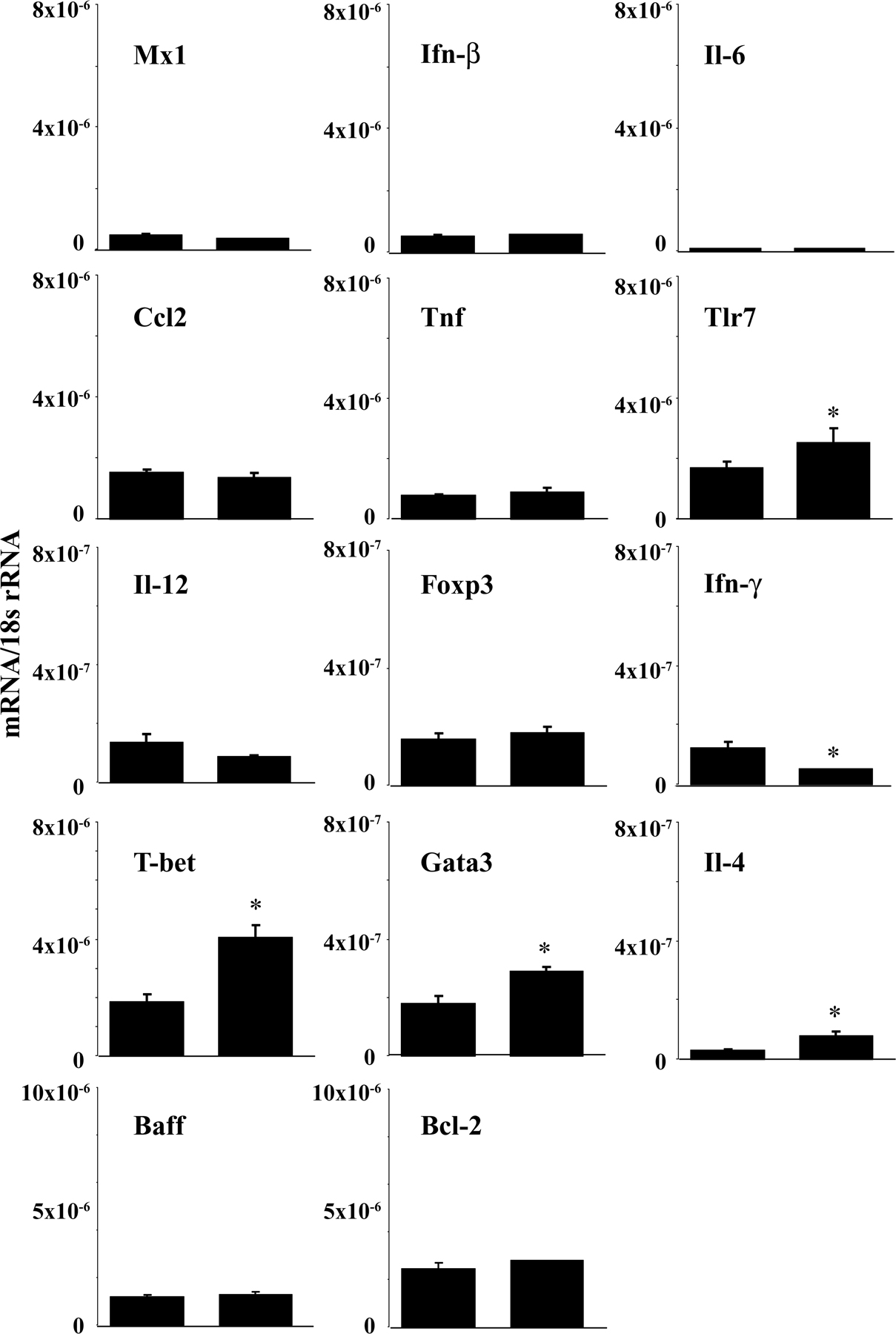
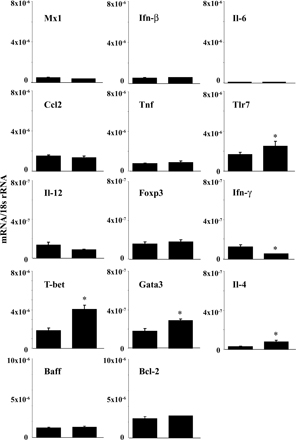
Sigirr modulates Tlr/Il-1 signaling in dendritic cells (25, 26), but does Sigirr also modulate the activation of dendritic cells upon exposure to lupus autoantigens? We prepared bone marrow dendritic cells from B6lpr/lpr and B6lpr/lpr/Tir8−/− mice and exposed them either to U1snRNP or nucleosome immune complexes. These classical lupus autoantigens are known to activate dendritic cells via Tlr7 and Tlr9 (37, 39). Homozygous deletion of the Tir8 gene was associated with a significant increase of Il-12p40 production by the dendritic cells upon exposure to such immune complexes as well as to LPS, imiquimod, and CpG-DNA (Fig. 2 A). In contrast, exposure to U1snRNP and nucleosomes (not depicted) or to anti-Smith (Sm) IgG (Y12) and nucleosome antibodies alone did not induce Il-12p40 production, respectively (Fig. 2 A). Similar results were obtained for mRNA expression levels of Mx1, an Ifn-α–responsive gene, also showing that Fc receptors facilitate the immunostimulatory effects of RNA and DNA immune complexes in Ftl3 dendritic cells (Fig. 2 B). Does Sigirr also modulate the activation of dendritic cells in B6lpr/lpr mice? We performed flow cytometry to quantify and characterize the activation state of CD11c+ dendritic cells without additional stimuli directly after the spleen harvest at 6 mo of age. The total number of spleen CD11c+ dendritic cells was significantly higher in B6lpr/lpr/Tir8−/− mice as compared with B6lpr/lpr mice (Fig. 2 C). 50% of CD11c+ cells were positive for the activation marker CD40 in B6lpr/lpr mice compared with 90% in Sigirr-deficient B6lpr/lpr mice (Fig. 2 C). Lack of Sigirr was also associated with increased mRNA levels of Mx1, Ifn-β, and Tnf (Fig. 2 D). Sigirr-deficient spleen dendritic cells also expressed higher levels of Baff and Bcl2 (Fig. 2 D), which support the survival of B and/or T cells (48). Consistent with increased dendritic cell activation B6lpr/lpr/Tir8−/− mice had higher serum levels of Il-12p40 as compared with B6lpr/lpr mice (Fig. 2 E). Thus, Sigirr suppresses dendritic cell activation upon exposure to complexed lupus autoantigens.
Figure 2.

Sigirr and dendritic cell activation. (A and B) Dendritic cells were prepared from bone marrow cells of B6lpr/lpr/Tir8−/− and B6lpr/lpr mice and stimulated in vitro with various TLR agonists, including immune complexes formed by U1snRNP and the anti-U1snRNP antibody Y12 (Y12) or a nucleosome-specific antibody and nucleosomes for 24 h. Supernatants were analyzed for IL-12p40 (A) by ELISA. MX1 mRNA levels, as a marker of type I IFN expression, were quantified by real-time PCR after 8 h of stimulation (B). Data are shown as mean ± SEM. *, P < 0.05 versus B6lpr/lpr/Tir8−/− mice. (C) The total number of CD11c+ dendritic cells in the spleens of B6lpr/lpr/Sigirr−/− and B6lpr/lpr wild-type mice was quantified by flow cytometry as described in Materials and methods. Data represent means ± SEM from five mice in each group. *, P < 0.05 versus B6lpr/lpr mice. (D) RNA was isolated from the spleens of CD11b+ cells from B6lpr/lpr/Tir8−/− and B6lpr/lpr mice for real-time PCR analysis. Data are expressed as means of the ratio of the specific mRNA versus that of 18S rRNA ± SEM. *, P < 0.05 versus B6lpr/lpr mice. (E) Serum samples from 6-mo-old B6lpr/lpr/Tir8−/− and B6lpr/lpr mice were analyzed for IL-12p40 by ELISA. Data are means ± SEM from at least 10 mice in each group. *, P < 0.05 versus B6lpr/lpr mice.
Sigirr suppresses CD4 T cells and maintains CD4+CD25+ T cells in B6lpr/lpr mice
How does lack of Sigirr affect T cell populations in B6lpr/lpr mice? The numbers of CD4+ but not of CD8+ T cells and CD4/CD8 double negative “autoreactive” T cells were increased in the spleens of Sigirr-deficient B6lpr/lpr mice (Fig. 3 A), consistent with the relative percentage of these populations among all spleen cells (Fig. S1 B). The total numbers and the percentage of splenic CD4+/CD25+ T cells were reduced as compared with B6lpr/lpr mice (Fig. 3 B and Fig. S1 B). The reduced numbers of CD4+CD25+ T cells were consistent with lower Foxp3 mRNA expression levels in these cells in B6lpr/lpr/Tir8−/− versus B6lpr/lpr mice (Fig. 3 C). In CD4+CD25− T cells, lack of Sigirr was associated with lower mRNA expression levels of the Th1 markers T-bet and Ifn-γ, and the Th2 markers Gata and Il-4 (Fig. 3 C). Collectively, Sigirr suppresses the expansion of CD4 T cells but maintains the CD4+CD25+ T cell population in B6lpr/lpr mice.
Figure 3.

Sigirr and T cell subsets in B6lpr/lpr mice. (A and B) Flow cytometry was used to determine the total number of distinct T cell subsets in the spleens of 6-mo-old B6lpr/lpr/Tir8−/− and B6lpr/lpr mice. The histogram presents means ± SEM of at least five mice in each group. *, P < 0.05 versus B6lpr/lpr mice. (C) Cellular mRNA was prepared from CD4+ and CD4+CD25+ spleen cell isolates from B6lpr/lpr/Tir8−/− and B6lpr/lpr mice and analyzed by real-time PCR. Data are expressed as means of the ratio of the specific mRNA versus that of 18S rRNA ± SEM. *, P < 0.05 versus B6lpr/lpr mice.
Sigirr suppresses the proliferation of B cells upon exposure to immune complexes containing RNA or DNA lupus autoantigens
Nothing is known about the role of Sigirr in B cells. In the spleens of 6-mo-old Sigirr-deficient B6lpr/lpr mice, B220 immunostaining revealed an expansion of spleen B cell areas and an increased number of B cells was found on quantitative spleen cell flow cytometry (Fig. 4, A and B). Immune complexes containing lupus autoantigens have been reported to stimulate B cell proliferation via Tlr7 and Tlr9 in vitro (35–37, 49). To test whether Sigirr modulates lupus autoantigen recognition in B cells, we prepared CD19+ B cells from B6lpr/lpr and B6lpr/lpr/Tir8−/− mice and exposed them to U1snRNP or nucleosome immune complexes. RNA and DNA immune complexes stimulated B cell proliferation just like CpG-DNA, LPS, or imiquimod, and lack of Sigirr significantly increased B cell proliferation upon exposure to all of these TLR agonists (Fig. 4 C). In contrast, anti-IgM, a Tlr-independent way of activating B cells, had a similar effect on B cells of either genotype. Anti-CD19 blocked the Sigirr-regulated effect of RNA and DNA immune complexes, indicating that lupus immune complexes activate B cells via a CD19-dependent mechanism at the cell surface. In contrast, the single components of RNA immune complexes, i.e., U1snRNP or nucleosomes (not depicted) and anti-Sm IgG (Y12) or anti-nucleosomes (13G10) did not enhance B cell proliferation (Fig. 4 C). Baff/BlyS and Bcl-2 B cell activation pathways may contribute to B cell proliferation in SLE (48). Baff/BLyS mRNA levels in CD11b+ cells and Bcl-2 mRNA levels were increased in Sigirr-deficient dendritic cells (Fig. 2 D) consistent with the increased numbers of B cells in the spleens of Sigirr-deficient B6lpr/lpr mice (Fig. 4 B). Thus, Sigirr specifically suppresses the lupus autoantigen complex–induced activation and proliferation of B cells in vitro and in vivo.
Figure 4.

Sigirr suppresses the proliferation of B cells from B6lpr/lprmice. (A) Immunostaining of spleen sections for B220 illustrates the expansion of B cell areas in Tir8-deficient B6lpr/lpr mice. Images are representative of at least 12 mice in each group. (B) The total number of spleen B220+ cells was quantified in B6lpr/lpr/Tir8−/− and B6lpr/lpr wild-type mice by flow cytometry. Data represent means ± SEM from five mice in each group. *, P < 0.05 versus B6lpr/lpr mice. (C) Spleen B cells from B6lpr/lpr/Tir8−/− and B6lpr/lpr mice were stimulated in vitro with anti-IgM antibodies, various TLR agonists, including immune complexes formed by U1snRNP, and the anti-U1snRNP antibody Y12 (Y12) or a nucleosome-specific antibody and nucleosomes for 72 h. B cell proliferation was assessed as described in Materials and methods. Data are shown as mean ± SEM of the OD at 490 nm. Data are shown as mean ± SEM. *, P < 0.05 versus B6lpr/lpr mice. Bar: (A) 100 μm.
Sigirr suppresses the production of autoantibodies in B6lpr/lpr mice
The role of Sigirr in modulating the activation of dendritic cells and B cells may affect Ig production, especially the evolution and production of antibodies against lupus autoantigens in B6lpr/lpr mice. Hence, serum samples were obtained at monthly intervals from all mice and antibody levels were determined by ELISA. Sigirr-deficient B6lpr/lpr mice developed more hypergammaglobulinemia as compared with B6lpr wild-type mice, which was evident from 4 mo of age and almost reached the level of hypergammaglobulinemia in 6-mo-old female MRLlpr/lpr mice (Fig. 5 A). Lack of Sigirr also increased the production of DNA autoantibodies from IgG isotypes, reaching the respective levels in 6-mo-old female MRLlpr/lpr mice, except that for IgG2c (Fig. 5 B). This effect antedated the development of hypergammaglobulinemia by 2–3 mo. The specificity of dsDNA autoantibodies was confirmed by using the C. luciliae assay. Diluted serum from B6lpr/lpr/Tir8−/− mice showed a much more intense binding to the dsDNA of the flagellate's kinetoplast as compared with serum of B6lpr/lpr mice (Fig. 5 C). In addition, lack of Sigirr was associated with increased production of anti-Sm IgG, anti-SnRNP IgG, anti-nucleosome IgG, and rheumatoid factor as compared with B6lpr/lpr mice, of which Sm antibodies and rheumatoid factor were already elevated from 2 mo of age (Fig. 5 D). Lack of Sigirr increased rheumatoid factor but not anti-Sm levels in B6lpr/lpr mice to the levels seen in age-matched MRLlpr/lpr mice. Thus, Sigirr suppresses the production of antibodies against numerous lupus autoantigens in B6lpr/lpr mice.
Figure 5.

Sigirr and the production of Igs and DNA autoantibodies in B6lpr/lprmice. Mice from both groups were bled at monthly intervals to determine serum levels of IgG (A) and dsDNA autoantibody isotypes (B) by ELISA. Serum from 6-mo-old female MRLlpr/lpr mice served as positive control. Data represent means ± SEM from at least 10 mice in each group. *, P < 0.05 versus B6lpr/lpr mice of the same time point; #, P < 0.05 versus month 1 of mice from the same strain. (C) C. luciliae slides were incubated with 1:50 diluted serum of 6-mo-old mice from both strains, and autoantibody binding to the flagellate's kinetoplast was detected using an FITC-labeled anti-mIgG. Signal intensity was scored applying a semiquantitative score from 0 to 3. Images on the left show anti-dsDNA IgG in green (top), and staining the kinetoplast DNA itself with DAPI is in blue (not depicted). The merged pictures are shown below demonstrating that FITC positivity matches with the kinetoplast at the flagella pole of C. luciliae. Data on the right show mean scores ± SEM from at least 6–10 mice in each group. *, P < 0.05 versus B6lpr/lpr mice. (D) Serum levels of autoantibodies against Sm antigen, U1snRNP, nucleosomes, or rheumatoid factor (RF) were determined at monthly intervals by ELISA. Serum from 6-mo-old female MRLlpr/lpr mice served as positive control. *, P < 0.05 versus B6lpr/lpr mice of the same time point; #, P < 0.05 versus month 1 of mice from the same strain; N.d., not done.
Sigirr protects B6lpr/lpr mice from autoimmune tissue injury
Systemic autoimmunity may or may not be associated with autoimmune tissue injury, which remains the ultimate definition of SLE. Autoimmune tissue injury in murine (or human) SLE commonly affects lungs and kidneys (4). B6lpr/lpr mice do not develop major autoimmune tissue lesions, although mild glomerulonephritis develops at an advanced age in B6lpr/lpr mice (12). Sigirr-deficient B6lpr/lpr mice revealed significant peribronchial inflammation characterized by mononuclear cell infiltrates and edema (Fig. 6; semiquantitative lung injury score for B6lpr mice, 0.5 ± 0.2 vs. B6lpr/Tir8−/− mice, 2.0 ± 0.5; P = 0.007). Furthermore, Sigirr deficiency was associated with diffuse mesangio-proliferative glomerulonephritis, as indicated by glomerular hypercellularity, periodic acid-Schiff (PAS)+ matrix expansion, and glomerular macrophage infiltrates (Fig. 6). The composite activity score for lupus nephritis was 6.8 ± 1.0 in B6lpr/lpr/Tir8−/− mice and 2.3 ± 0.2 in B6lpr/lpr mice (P = 0.003). The observed induction of hypergammaglobulinemia and lupus autoantibodies in B6lpr/lpr/Tir8−/− mice raised the question of whether these contribute to renal autoimmune tissue injury. Glomerular IgG deposits were increased in B6lpr/lpr/Tir8−/− mice (Fig. 6; B6lpr mice, 0.9 ± 0.1 vs. B6lpr/lpr/Tir8−/− mice, 1.8 ± 0.1; P = 0.006). Immune complex deposition mediates tissue injury via complement activation; hence, renal sections were also stained for complement factor C3c. In Sigirr-deficient B6lpr/lpr mice, increased glomerular IgG deposits were associated with increased glomerular positivity for C3c (Fig. 6; B6lpr/lpr mice, 1.2 ± 0.2 vs. B6lpr/lpr/Tir8−/− mice, 2.1 ± 0.2; P = 0.03). Does Sigirr also control intrarenal inflammation? Lack of Sigirr had no major impact on the mRNA expression levels of most of the aforementioned cytokines, chemokines, and transcription factors in the kidneys of B6lpr/lpr mice (Fig. S1). Collectively, Sigirr protects B6lpr/lpr mice not only from systemic autoimmunity, but also from autoimmune tissue injury, mainly by suppressing the activation of antigen-presenting cells.
Figure 6.

Lupus nephritis and lung injury in B6lpr/lprmice. Lung and renal sections were stained with PAS. On renal sections, immunostaining was also performed for IgG and complement factor C3c. Images are representative for 10 mice in each group. Bar, 200 μm.
DISCUSSION
Sigirr is known to suppress antimicrobial immunity (25–31). Our data identify autoimmunity control as a novel function of Sigirr, e.g., Sigirr inhibits the activation of dendritic cells upon exposure to lupus immune complexes. Such immune complexes contain, for example, U1snRNP or nucleosomes that can ligate Tlr7 and Tlr9 and activate the maturation of dendritic cells and B cells (36–41). Several avenues of experimental evidence support the concept that autoantigen recognition via Tlr7 is another important pathomechanism for murine SLE, i.e., use of Tlr7 antagonists (42), Tlr7-deficient mice (43), or Tlr7 overexpression (20, 43). In contrast, data from TLR9-deficient autoimmune mice revealed inconsistent outcomes as compared with late onset of TLR9 antagonism (43, 45–47), suggesting additional roles for TLR9 during the early phase of autoimmunity (35). B6lpr/lpr/Tir8−/− mice revealed enhanced activation of dendritic cells, type I Ifn signaling, and production of proinflammatory cytokines like Ccl2, Il-6, and Il-12p40, and the B cell survival factors Baff/BlyS and Bcl-2, which are associated with more severe murine and human SLE (50–54). However, type I Ifn induction was only detectable at the transcriptional level in the spleens of B6lpr/lpr mice, and we could not detect significant type I Ifn levels in the sera of either mouse strain. The pathogenic role of type I Ifn varies among different murine SLE models (20, 43, 47, 55–57) and has not yet been clearly defined in B6lpr/lpr mice. Sigirr also directly inhibits the proliferation of B cells upon exposure to RNA and DNA immune complexes, a process that also depends on the interaction with CD19. This finding supports the concept that a surface receptor–mediated uptake of lupus immune complexes into intracellular endosomes precedes the Sigirr-regulated Tlr signaling (35–37). Consistent with direct and indirect Sigirr-mediated effects on B cells, Sigirr-deficient B6lpr/lpr mice revealed increased total numbers and expansion of B cell areas in the spleen in association with enhanced production of lupus autoantibodies. The latter effect was not restricted to a specific class of autoantibodies because lack of Sigirr massively enhanced the production of autoantibodies against multiple nuclear autoantigens as well as hypergammaglobulinemia. Regulatory T cells control autoreactive B cell populations, and regulatory T cells are controlled by dendritic cells via secretion of Il-6 (58). In fact, the induction of Il-6 in B6lpr/lpr/Tir8−/− mice was associated with decreased Foxp3 expression and less CD4+CD25+ T cells in the spleens of B6lpr/lpr/Tir8−/− mice. The functional effect of this observation on autoreactive T cells remains uncertain because the CD4/CD8–double negative T cell population was not significantly affected by the Tir8 genotype. Collectively, we identified a novel function of the TLR/IL-1R family member Sigirr, i.e., the control of autoimmunity, lymphoproliferation, and autoimmune tissue injury mainly by inhibiting the endogenous activation of dendritic cells and B cells.
SLE results from a combination of genetic abnormalities that affects the handling of nuclear autoantigens, persistence of autoreactive lymphocytes, and immunoregulatory factors (1, 5–11, 59). The first two mechanisms affect the loss of tolerance, which becomes clinically detectable by the presence of serum antinuclear antibodies. However, the presence of (low titers of) antinuclear antibodies in humans or in lpr-deficient B6 mice is not generally associated with autoimmune tissue injury (12). Autoimmune tissue injury does not develop unless additional genetic abnormalities support the expansion of autoreactive lymphocytes, immune complex disease, and tissue pathology (1, 5). Many genes have been identified that promote autoimmune tissue injury in complex autoimmune genetic backgrounds in mice (22, 23, 59). However, mutant Sigirr is sufficient to cause a severe SLE-like lymphoproliferative syndrome and autoimmune tissue injury in B6lpr/lpr mice because Sigirr is required to, for example, suppress the activation of antigen-presenting cells that handle autoantigens (35). We therefore conclude that Sigirr is a novel susceptibility gene for murine SLE. Interestingly, the Sigirr gene is localized at the p15.5 region of human chromosome 11, a region to which linkage analyses have mapped a yet unknown lupus susceptibility gene in African-Americans with a LOD score of 3.3 (34). Our studies propose to elucidate the role of SIGIRR in human SLE.
In summary, lack of functional Sigirr is associated with severe autoimmune tissue injury in B6lpr/lpr mice. This represents a previously unknown function of Sigirr in autoimmunity control. The involvement of this as well as of other negative regulators in human lupus and autoimmunity in general deserves careful scrutiny.
MATERIALS AND METHODS
Animal studies.
Sigirr-deficient mice were generated as described previously (28) and backcrossed to the C57BL/6 strain (B6; Charles River Laboratories) to the F6 generation. B6lpr/lpr/Tir8−/− and B6lpr/lpr (Charles River Laboratories) were mated to generate B6lpr/lpr/Sigirr−/+ mice, which were then mated among each other to generate B6lpr/lpr/Tir8−/−, B6lpr/lpr/Tir8−/+, and B6lpr/lpr/Tir8+/+ mice. Littermates were used for all in vivo and in vitro experimental procedures. In each individual mouse the genotype was assured by PCR. Mice were housed in groups of five mice in sterile filter top cages with a 12-h dark/light cycle and unlimited access to autoclaved food and water. All experimental procedures were performed according to the German animal care and ethics legislation and had been approved by the local government authorities (government of Upper Bavaria). All mice were killed by cervical dislocation at 24 wk of age.
In vitro experiments.
U1snRNP was purified from HeLa cell nuclear extracts (60). The anti-Sm (B/D) antibody clone Y12, mouse IgG3 isotype, was purified from Y12 hybridoma supernatant (MWG Biotech). Bone marrow cells from wild-type and knockout mice were cultured with 20 ng/ml human recombinant Flt3L (R&D Systems) in complete medium for 7 d to generate >90% CD11c+ dendritic cells with 40–50% CD11blow/CD86low/B220high plasmacytoid dendritic cells and 40–50% CD11bhigh/B220low dendritic cells. On day 7, cells were harvested, resuspended in fresh medium, and seeded at 4 × 105 cells/well (100 μl/well in 96-well plates). RNAs and the isolated U1snRNP were preincubated with 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) cationic lipid (Carl Roth) for 30 min at room temperature. Y12 antibody was incubated with U1snRNP in PBS for 15 min on ice plus 5 min at 37°C. Stimuli were added in 100-μl volume per well (20 μg/ml final concentration) of 0.5 μg/ml of ultrapure LPS (InvivoGen), 0.5 μM CpG-DNA 1668 (TibMolbiol), and 0.5 μg/ml imiquimod (Sequoia Research Products Ltd) for 24 h. 13G10 anti-nucleosome antibodies (BD Biosciences) were incubated with dsDNA–histone complexes for 30 min at 37°C and added in 100-μl volume per well (1 μg/ml final concentration). B cells were isolated from the spleens of female C57BL/6 lpr/lpr mice using the B Cell Isolation kit (Miltenyi Biotec) according to the manufacturer's instructions. Purity as determined by FACS analysis using CD45/B220-PE or rat IgG2a as an isotype (BD Biosciences) revealed 97% B cells after each isolation. Proliferation of B cells was assessed using CellTiter 96 Proliferation Assay (Promega). In brief, 105 B cells were incubated in 96-well plates in 100 μl RPMI medium that contained 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Biochrom KG), with U1snRNP (20 μg/ml final concentration) for 72 h. Goat anti-IgM (Jackson ImmunoResearch Laboratories) was used for measuring the capacity of B cells to undergo proliferation. To each well, 20 μl of CellTiter96 Aqueous One Solution (Promega) was added and incubated at 37°C for 4 h. The OD was measured at 492 nm. Monoclonal anti-CD19 antibodies (clone 1D3; BD Biosciences) and anti-CD16/CD32 antibodies (clone 2.4G2; BD Biosciences) were used 60 min before stimulation (10 μg/ml final concentration).
Flow cytometry.
Anti–mouse CD3, CD4, CD8, and CD25 (BD Biosciences) antibodies were used to detect CD3+CD4−CD8− double negative T cell and CD4+CD25+ regulatory T cell populations in the spleens. CD11c has been stained to identify plasmacytoid and myeloid dendritic cells, and their activation was assed by costaining for CD40 (BD Biosciences). Respective isotype antibodies were used to demonstrate specific staining of cell subpopulations.
Evaluation of autoimmune tissue injury.
Spleens, lymph nodes, lungs, and kidneys from all mice were fixed in 10% buffered formalin, processed, and embedded in paraffin. 2-μm sections for PAS stains were prepared after routine protocols. The severity of the renal lesions was graded using the indices for activity and chronicity as described for human lupus nephritis (61). Immunostaining was either performed on paraffin-embedded or frozen sections as described previously (46) using the following primary antibodies: anti–mouse IgG (1:100; Caltag Laboratories), anti–mouse C3c (1:200; complement; GAM/C3c/FITC; Nordic Immunological Laboratories), or anti–mouse B220 (BD Biosciences). Negative controls included incubation with a respective isotype antibody. For quantitative analysis, glomerular cells were counted in 10 cortical glomeruli per section. Semiquantitative scoring of glomerular IgG and C3c deposits from 0 to 3 plus was performed on 15 cortical glomerular sections as described previously (42).
Autoantibody analysis.
Serum antibody levels were determined by ELISA as follows. Anti-dsDNA antibodies: NUNC maxisorp ELISA plates were coated with poly-l-lysine (Trevigen) and mouse embryonic stem cell dsDNA. After incubation with mouse serum, dsDNA-specific IgG, IgG1, IgG2a/c, IgG2b, IgG3, and serum IgG levels were detected by ELISA (Bethyl Laboratories). C. luciliae assay: 1:50 diluted serum was applied to fixed C. luciliae slides (Bio-Rad Laboratories). Binding to C. luciliae kinetoplast was detected with FITC-conjugated goat anti-mIgG (1:1,000; Invitrogen). DAPI staining (Vector Laboratories) allowed colocalization with kinetoplast dsDNA. For quantitation of kinetoplast staining intensity, a semiquantitative score from 0 to 3 was used. Anti-Sm: NUNC maxisorp ELISA plates were coated with Sm antigen (Immunovision). The Sm IgG (Y12) antibody (GeneTex) was used for standard. A horseradish peroxidase–conjugated goat anti–mouse IgG (Rockland) was used for detection. The same procedure was followed for anti-Sm RNP and anti-nucleosome antibodies as for anti-Sm, except the ELISA plates were captured with Sm–RNP complex (Immunovision) or dsDNA together with histones (USB Corporation), respectively, instead of Sm antigen. Rheumatoid factor: ELISA plates were coated with 10 μg/ml rabbit IgG (Jackson ImmunoResearch Laboratories) overnight at 4°C. Serum samples were diluted at 1:100, and C57BL/6 10-wk mouse serum was used as negative control. Horseradish peroxidase–conjugated anti–mouse IgG was used as secondary antibody. Serum cytokine levels and cell culture supernatants were determined using commercial ELISA kits for Il-6 and Il-12p40 (OptEiA; BD Biosciences) according to the manufacturer's instructions.
Real-time quantitative (TaqMan) RT-PCR.
Real-time RT-PCR was performed on total spleen mRNA as described previously (42). Controls consisting of ddH2O were negative for target and housekeeper genes. 300 nM of oligonucleotide primer and 100 nM of probes were from PE Biosystems and used as follows: 18S rRNA was used as a housekeeper. Controls consisting of ddH2O were negative for target and housekeeper genes. 300 nM of oligonucleotide primer and 100 nM of probes were from Applied Biosystems and used as follows: IL-4: ID Mm00445259_m1 FAM 5′-ACGAAGAACACCACAGAGAGTGAGC-3′; IL-6: ID Mm00446190_m1 FAM 5′-AAATGAGAAAAGAGTTGTGCAATGG-3′; IL-12: ID Mm00434165_m1 FAM 5′-TGACATGGTGAAGACGGCCAGAGAA-3′; Mx1: ID Mm00487796_m1 FAM 5′-TGTACTGCTAAGTCCAAAATTAAAG-3′; IFN-β: ID Mm00439546_s1 FAM: 5′-TCCACGCTGCGTTCCTGCTGTGCTT-3′; IFN-γ: ID Mm00801778_m1 FAM 5′-CTATTTTAACTCAAGTGGCATAGAT-3′; Tnf: ID Mm00443258_m1 FAM 5′-GTCCCCAAAGGGATGAGAAGTTCCC-3′; TLR7: ID AY035889 FAM: 5′-CCAAGAAAATGATTTTAATAAC-3′; Gata3 ID Mm00484683_m1 FAM: 5′-CCCACCACGGGAGCCAGGTATGCCG-3′; Tbx21: ID Mm00450960_m1 FAM: 5′-GCAAGGACGGCGAATGTTCCCATTC-3′; Ccl2: ID Mm00441242_m1 FAM 5′-GCTCAGCCAGATGCAGTTAACGCCC-3′; Foxp3: ID Mm00475156_m1 FAM 5′-ACCCAGCCACTCCAGCTCCCGGCAA-3′; IL-23: ID Mm00518984_m1 FAM 5′-CAAGGACAACAGCCAGTTCTGCTTG-3′; Baff/BLyS: ID Mm00446347_m1 FAM 5′-ACTCGGCTGGCATCGCGAGGCTGGA-3′; Bcl-2: ID Mm00477631_m1 FAM 5′-GATAACGGAGGCTGGGATGCCTTTG-3′.
Statistical analysis.
One-way ANOVA followed by post-hoc Bonferroni's test was used for multiple comparisons using GraphPad Prism software (version 4.03). Single groups were compared by unpaired two-tailed Student's t test. Data were expressed as mean ± SEM. Statistical significance was assumed at a p-value of <0.05.
Online supplemental material.
Table S1 shows production of Igs and dsDNA autoantibodies in B6Tir8−/− and B6Tir8+/+ mice. Fig. S1 shows spleen cell subsets and Fig. S2 shows the mRNA expression of various genes in the kidneys of B6lpr/lpr/Tir8−/− and B6lpr/lpr/Tir8+/+, respectively. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20072646/DC1.
Supplementary Material
Acknowledgments
The expert technical assistance of Ewa Radomska, Dan Draganovic, and Jana Mandelbaum is gratefully acknowledged.
This work was supported by a grant from the Fritz Thyssen Foundation (no. 10.04.2.140), the Deutsche Forschungsgemeinschaft (AN372/8-1 and GRK 1202 to H.-J. Anders), the EU Integrated Project “INNOCHEM” (FP6-518167 to H.-J. Anders and A. Mantovani), and “MUGEN” (LSHG-CT-2005-005203 to C. Garlanda and A. Mantovani). A. Mantovani also received support from the Italian Ministero Università e Ricerca (MIUR) and Ministero della Salute.
The authors have no conflicting financial interests.
Abbreviations used: dsDNA, double-stranded DNA; LE, lupus erythematosus; PAS, periodic acid-Schiff; SIGIRR, single IG IL-1–related receptor; SLE, systemic LE; Sm, Smith; TIR8, Toll–IL-1 receptor 8; TLR, Toll-like receptor.
References
- 1.Goodnow, C.C. 2007. Multistep pathogenesis of autoimmune disease. Cell. 130:25–35. [DOI] [PubMed] [Google Scholar]
- 2.Bennett, C.L., J. Christie, F. Ramsdell, M.E. Brunkow, P.J. Ferguson, L. Whitesell, T.E. Kelly, F.T. Saulsbury, P.F. Chance, and H.D. Ochs. 2001. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27:20–21. [DOI] [PubMed] [Google Scholar]
- 3.Brunkow, M.E., E.W. Jeffery, K.A. Hjerrild, B. Paeper, L.B. Clark, S.A. Yasayko, J.E. Wilkinson, D. Galas, S.F. Ziegler, and F. Ramsdell. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27:68–73. [DOI] [PubMed] [Google Scholar]
- 4.D'Cruz, D.P., M.A. Khamashta, and G.R. Hughes. 2007. Systemic lupus erythematosus. Lancet. 369:587–596. [DOI] [PubMed] [Google Scholar]
- 5.Rahman, A., and D.A. Isenberg. 2008. Systemic lupus erythematosus. N. Engl. J. Med. 358:929–939. [DOI] [PubMed] [Google Scholar]
- 6.Graham, R.R., S.V. Kozyrev, E.C. Baechler, M.V. Reddy, R.M. Plenge, J.W. Bauer, W.A. Ortmann, T. Koeuth, M.F. González Escribano, Argentine and Spanish Collaborative Groups, et al. 2006. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 38:550–555. [DOI] [PubMed] [Google Scholar]
- 7.Hom, G., R.R. Graham, B. Modrek, K.E. Taylor, W. Ortmann, S. Garnier, A.T. Lee, S.A. Chung, R.C. Ferreira, P.V. Pant, et al. 2008. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 358:900–909. [DOI] [PubMed] [Google Scholar]
- 8.Graham, D.S., R.R. Graham, H. Manku, A.K. Wong, J.C. Whittaker, P.M. Gaffney, K.L. Moser, J.D. Rioux, D. Altshuler, T.W. Behrens, and T.J. Vyse. 2008. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 40:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozyrev, S.V., A.K. Abelson, J. Wojcik, A. Zaghlool, M.V. Linga Reddy, E. Sanchez, I. Gunnarsson, E. Svenungsson, G. Sturfelt, A. Jönsen, et al. 2008. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 40:211–216. [DOI] [PubMed] [Google Scholar]
- 10.International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), J.B. Harley, M.E. Alarcón-Riquelme, L.A. Criswell, C.O. Jacob, R.P. Kimberly, K.L. Moser, B.P. Tsao, T.J. Vyse, and C.D. Langefeld. 2008. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 40:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remmers, E.F., R.M. Plenge, A.T. Lee, R.R. Graham, G. Hom, T.W. Behrens, P.I. de Bakker, J.M. Le, H.S. Lee, F. Batliwalla, et al. 2007. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 357:977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 13.Botto, M., C. Dell'Agnola, A.E. Bygrave, E.M. Thompson, and H.T. Cook. 1998. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19:56–59. [DOI] [PubMed] [Google Scholar]
- 14.Manderson, A.P., M. Botto, and M.J. Walport. 2004. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22:431–456. [DOI] [PubMed] [Google Scholar]
- 15.Napirei, M., H. Karsunky, B. Zevnik, H. Stephan, H.G. Mannherz, and T. Moroy. 2000. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 25:177–181. [DOI] [PubMed] [Google Scholar]
- 16.Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, and D. Calvin. 1992. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasutomo, K., T. Horiuchi, S. Kagami, H. Tsukamoto, C. Hashimura, M. Urushihara, and Y. Kuroda. 2001. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28:313–314. [DOI] [PubMed] [Google Scholar]
- 18.Hibbs, M.L., D.M. Tarlinton, J. Armes, D. Grail, G. Hodgson, R. Maglitto, S.A. Stacker, and A.R. Dunn. 1995. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 83:301–311. [DOI] [PubMed] [Google Scholar]
- 19.Mohan, C., E. Alas, L. Morel, P. Yang, and E.K. Wakeland. 1998. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J. Clin. Invest. 101:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian, S., K. Tus, Q.Z. Li, A. Wang, X.H. Tian, J. Zhou, C. Liang, G. Bartov, L.D. McDaniel, X.J. Zhou, et al. 2006. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA. 103:9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lartigue, A., P. Courville, I. Auquit, A. Francois, C. Arnoult, F. Tron, D. Gilbert, and P. Musette. 2006. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J. Immunol. 177:1349–1354. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu, S., N. Sugiyama, K. Masutani, A. Sadanaga, Y. Miyazaki, Y. Inoue, M. Akahoshi, R. Katafuchi, H. Hirakata, M. Harada, et al. 2005. Membranous glomerulonephritis development with Th2-type immune deviations in MRL/lpr mice deficient for IL-27 receptor (WSX-1). J. Immunol. 175:7185–7192. [DOI] [PubMed] [Google Scholar]
- 23.Yin, Z., G. Bahtiyar, N. Zhang, L. Liu, P. Zhu, M.E. Robert, J. McNiff, M.P. Madaio, and J. Craft. 2002. IL-10 regulates murine lupus. J. Immunol. 169:2148–2155. [DOI] [PubMed] [Google Scholar]
- 24.Thomassen, E., B.R. Renshaw, and J.E. Sims. 1999. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine. 11:389–399. [DOI] [PubMed] [Google Scholar]
- 25.Wald, D., J. Qin, Z. Zhao, Y. Qian, M. Naramura, L. Tian, J. Towne, J.E. Sims, G.R. Stark, and X. Li. 2003. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4:920–927. [DOI] [PubMed] [Google Scholar]
- 26.Polentarutti, N., G.P. Rol, M. Muzio, D. Bosisio, M. Camnasio, F. Riva, C. Zoja, A. Benigni, S. Tomasoni, A. Vecchi, et al. 2003. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur. Cytokine Netw. 14:211–218. [PubMed] [Google Scholar]
- 27.Qin, J., Y. Qian, J. Yao, C. Grace, and X. Li. 2005. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem. 280:25233–25241. [DOI] [PubMed] [Google Scholar]
- 28.Garlanda, C., F. Riva, N. Polentarutti, C. Buracchi, M. Sironi, M. De Bortoli, M. Muzio, R. Bergottini, E. Scanziani, A. Vecchi, et al. 2004. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA. 101:3522–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, X., L.D. Hazlett, W. Du, and R.P. Barrett. 2006. SIGIRR promotes resistance against Pseudomonas aeruginosa keratitis by down-regulating type-1 immunity and IL-1R1 and TLR4 signaling. J. Immunol. 177:548–556. [DOI] [PubMed] [Google Scholar]
- 30.Xiao, H., M.F. Gulen, J. Qin, J. Yao, K. Bulek, D. Kish, C.Z. Altuntas, D. Wald, C. Ma, H. Zhou, et al. 2007. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 26:461–475. [DOI] [PubMed] [Google Scholar]
- 31.Garlanda, C., D. Di Liberto, A. Vecchi, M.P. La Manna, C. Buracchi, N. Caccamo, A. Salerno, F. Dieli, and A. Mantovani. 2007. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J. Immunol. 179:3119–3125. [DOI] [PubMed] [Google Scholar]
- 32.Liew, F.Y., D. Xu, E.K. Brint, and L.A. O'Neill. 2005. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5:446–458. [DOI] [PubMed] [Google Scholar]
- 33.Gaffney, P.M., G.M. Kearns, K.B. Shark, W.A. Ortmann, S.A. Selby, M.L. Malmgren, K.E. Rohlf, T.C. Ockenden, R.P. Messner, R.A. King, et al. 1998. A genome-wide search for susceptibility genes in human systemic lupus erythematosus sib-pair families. Proc. Natl. Acad. Sci. USA. 95:14875–14879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintero-Del-Rio, A.I., J.A. Kelly, J. Kilpatrick, J.A. James, and J.B. Harley. 2002. The genetics of systemic lupus erythematosus stratified by renal disease: linkage at 10q22.3 (SLEN1), 2q34-35 (SLEN2), and 11p15.6 (SLEN3). Genes Immun. 3:S57–S62. [DOI] [PubMed] [Google Scholar]
- 35.Marshak-Rothstein, A., and I.R. Rifkin. 2007. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 25:419–441. [DOI] [PubMed] [Google Scholar]
- 36.Lau, C.M., C. Broughton, A.S. Tabor, S. Akira, R.A. Flavell, M.J. Mamula, S.R. Christensen, M.J. Shlomchik, G.A. Viglianti, I.R. Rifkin, and A. Marshak-Rothstein. 2005. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 202:1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leadbetter, E.A., I.R. Rifkin, A.M. Hohlbaum, B.C. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 38.Boule, M.W., C. Broughton, F. Mackay, S. Akira, A. Marshak-Rothstein, and I.R. Rifkin. 2004. Toll-like receptor 9–dependent and –independent dendritic cell activation by chromatin-immunoglobulin G complexes. J. Exp. Med. 199:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savarese, E., O.W. Chae, S. Trowitzsch, G. Weber, B. Kastner, S. Akira, H. Wagner, R.M. Schmid, S. Bauer, and A. Krug. 2006. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 107:3229–3234. [DOI] [PubMed] [Google Scholar]
- 40.Means, T.K., E. Latz, F. Hayashi, M.R. Murali, D.T. Golenbock, and A.D. Luster. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vollmer, J. 2005. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J. Exp. Med. 202:1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pawar, R.D., A. Ramanjaneyulu, O.P. Kulkarni, M. Lech, S. Segerer, and H.J. Anders. 2007. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J. Am. Soc. Nephrol. 18:1721–1731. [DOI] [PubMed] [Google Scholar]
- 43.Christensen, S.R., J. Shupe, K. Nickerson, M. Kashgarian, R.A. Flavell, and M.J. Shlomchik. 2006. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 25:417–428. [DOI] [PubMed] [Google Scholar]
- 44.Pisitkun, P., J.A. Deane, M.J. Difilippantonio, T. Tarasenko, A.B. Satterthwaite, and S. Bolland. 2006. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 312:1669–1672. [DOI] [PubMed] [Google Scholar]
- 45.Dong, L., S. Ito, K.J. Ishii, and D.M. Klinman. 2005. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB x NZW mice. Arthritis Rheum. 52:651–658. [DOI] [PubMed] [Google Scholar]
- 46.Patole, P.S., D. Zecher, R.D. Pawar, H.J. Grone, D. Schlondorff, and H.J. Anders. 2005. G-rich DNA suppresses systemic lupus. J. Am. Soc. Nephrol. 16:3273–3280. [DOI] [PubMed] [Google Scholar]
- 47.Wu, X., and S.L. Peng. 2006. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 54:336–342. [DOI] [PubMed] [Google Scholar]
- 48.Mackay, F., P.A. Silveira, and R. Brink. 2007. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr. Opin. Immunol. 19:327–336. [DOI] [PubMed] [Google Scholar]
- 49.Barrat, F.J., T. Meeker, J. Gregorio, J.H. Chan, S. Uematsu, S. Akira, B. Chang, O. Duramad, and R.L. Coffman. 2005. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baccala, R., K. Hoebe, D.H. Kono, B. Beutler, and A.N. Theofilopoulos. 2007. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 13:543–551. [DOI] [PubMed] [Google Scholar]
- 51.Lipsky, P.E. 2006. Interleukin-6 and rheumatic diseases. Arthritis Res. Ther. 8:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kyogoku, C., and N. Tsuchiya. 2007. A compass that points to lupus: genetic studies on type I interferon pathway. Genes Immun. 8:445–455. [DOI] [PubMed] [Google Scholar]
- 53.Theofilopoulos, A.N., R. Baccala, B. Beutler, and D.H. Kono. 2005. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23:307–336. [DOI] [PubMed] [Google Scholar]
- 54.Vielhauer, V., H.J. Anders, and D. Schlondorff. 2007. Chemokines and chemokine receptors as therapeutic targets in lupus nephritis. Semin. Nephrol. 27:81–97. [DOI] [PubMed] [Google Scholar]
- 55.Nacionales, D.C., K.M. Kelly-Scumpia, P.Y. Lee, J.S. Weinstein, R. Lyons, E. Sobel, M. Satoh, and W.H. Reeves. 2007. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 56:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jørgensen, T.N., E. Roper, J.M. Thurman, P. Marrack, and B.L. Kotzin. 2007. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F(1) mice. Genes Immun. 8:653–662. [DOI] [PubMed] [Google Scholar]
- 57.Schwarting, A., K. Paul, S. Tschirner, J. Menke, T. Hansen, W. Brenner, V.R. Kelley, M. Relle, and P.R. Galle. 2005. Interferon-beta: a therapeutic for autoimmune lupus in MRL-Faslpr mice. J. Am. Soc. Nephrol. 16:3264–3272. [DOI] [PubMed] [Google Scholar]
- 58.Pasare, C., and R. Medzhitov. 2003. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 59.Zhu, J., and C. Mohan. 2007. SLE 1, 2, 3…genetic dissection of lupus. Adv. Exp. Med. Biol. 601:85–95. [DOI] [PubMed] [Google Scholar]
- 60.Bochnig, P., R. Reuter, P. Bringmann, and R. Lührmann. 1987. A monoclonal antibody against 2,2,7-trimethylguanosine that reacts with intact, class U, small nuclear ribonucleoproteins as well as with 7-methylguanosine-capped RNAs. Eur. J. Biochem. 168:461–467. [DOI] [PubMed] [Google Scholar]
- 61.Austin, H.A. 3rd., L.R. Muenz, K.M. Joyce, T.T. Antonovych, and J.E. Balow. 1984. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney Int. 25:689–695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}