

Abstract
How dihydropyridines modulate L-type voltage-gated Ca2+ channels is not known. Dihydropyridines bind cooperatively with Ca2+ binding to the selectivity filter, suggesting that they alter channel activity by promoting structural rearrangements in the pore. We used radioligand binding and patch-clamp electrophysiology to demonstrate that calcicludine, a toxin from the venom of the green mamba snake, binds in the outer vestibule of the pore and, like Ca2+, is a positive modulator of dihydropyridine binding. Data were fit using an allosteric scheme where dissociation constants for dihydropyridine and calcicludine binding, KDHP and KCaC, are linked via the coupling factor, α. Nine acidic amino acids located within the S5-Pore-helix segment of repeat III were sequentially changed to alanine in groups of three resulting in the mutant channels, Mut-A, Mut-B and Mut-C. Mut-A, whose substitutions are proximal to IIIS5, exhibits a 4.5-fold reduction in dihydropyridine binding and is insensitive to calcicludine binding. Block of Mut-A currents by calcicludine is indistinguishable from wild-type, indicating that KCaC is unchanged and that the coupling between dihydropyridine and calcicludine binding (i.e., α) is disrupted. Mut-B and Mut-C possess KDHP values that resemble wild-type. Mut-C, the most C-terminal of the mutant channels, is insensitive to calcicludine binding and block. KCaC values for the Mut-C single mutants, E1122A, D1127A and D1129A, increase from 0.3 (Wild-type) to 1.14, 2.00 and 20.5 μM, respectively. Together, these findings suggest that dihydropyridine antagonist and calcicludine binding to L-type Ca2+ channels promote similar structural changes in the pore that stabilize the channel in a nonconducting, blocked state.
The flow of Ca2+ ions through voltage-gated Ca2+ channels drives a variety of cellular processes including excitation-contraction coupling, neurotransmitter release and gene expression. Voltage activated Ca2+ channels are heteromultimeric complexes consisting of α1, β, α2/δ and sometimes γ subunits. The pore-forming α1 subunit contains all of the structural determinants required for voltage-dependent gating, drug binding and ion permeation. The membrane topology of the α1 subunit consists of four homologous repeats (I, II, III, IV), each consisting of six transmembrane segments (S1–S6). Each of the four S5/S6 connecting segments contains a highly conserved negatively charged glutamate residue that together form a binding site for Ca2+ ions called the selectivity filter. The selectivity filter is the narrowest region in the pore and is the site that enables the channel to conduct Ca2+ ions under physiological conditions where Na+ is in excess (1).
L-type CaV1.2 Ca2+ channels are targeted by numerous small organic molecules including the dihydropyridines (DHPs). DHPs are allosteric modulators of Ca2+ channels and can behave either as agonists or antagonists. Despite much research spanning over two decades, the molecular details that underlie DHP action are still a mystery. Localizing the DHP receptor site was an important step toward developing an understanding of how DHPs interact with and modulate L-type Ca2+ channels. DHPs are small lipophilic compounds that have a strong tendency to associate with lipid bilayers (2). Kass and colleagues found that the DHP receptor site lies within the lipid bilayer approximately 11–14 Å from the extracellular surface of the membrane (3). These conclusions are consistent with mutagenic studies where individual amino acid residues located on transmembrane segments IIIS5, IIIS6 and IVS6 were found to be critical for DHP binding and activity (4–8).
Important clues regarding the molecular basis for DHP action came from the finding that DHP binding to a site that lies outside the permeation pathway is cooperative with Ca2+ binding to the selectivity filter, suggesting that the binding of DHPs and Ca2+ promotes reciprocal structural rearrangements in the outer pore that alter the functional behavior of the channel (9, 10). Two recent studies using independent approaches, radioligand binding and whole-cell patch-clamp electrophysiology, suggest that DHPs block monovalent and divalent currents by stabilizing a nonconducting blocked state that is structurally and functionally analogous to a channel with a single Ca2+ ion in its selectivity filter (11, 12). It is expected that the elusive molecular mechanisms that underlie DHP activity can be revealed by gaining a deeper understanding of the structural features of the conducting and nonconducting blocked states of the Ca2+ channel.
Toxins from a wide range of organisms bind to voltage-gated Ca2+ channels (1, 13). Calcicludine, a toxin from the venom of the green mamba snake (Dendroaspis angusticeps), is a 60 amino acid peptide that consists of three disulfide bonds, twelve positively charged arginine or lysine residues and only two negatively charged glutamate residues (Fig. 1A). Because of its folding pattern, calcicludine is classified as a bovine pancreatic trypsin inhibitor (BPTI)/Kunitz-like toxin (14–17) and is structurally homologous to the K+ channel selective dendrotoxins (18).
FIGURE 1. Calcicludine is a positive allosteric modulator of DHP binding. (A) Primary sequence of calcicludine.
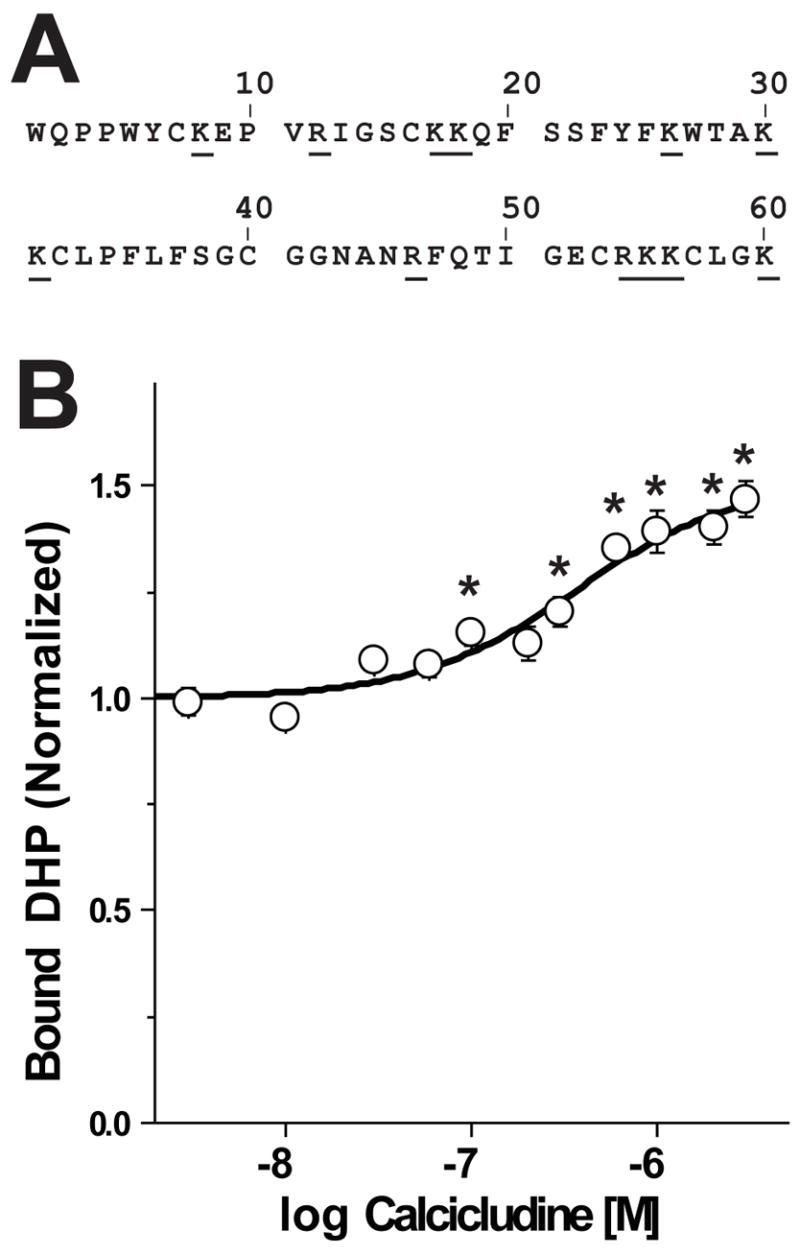
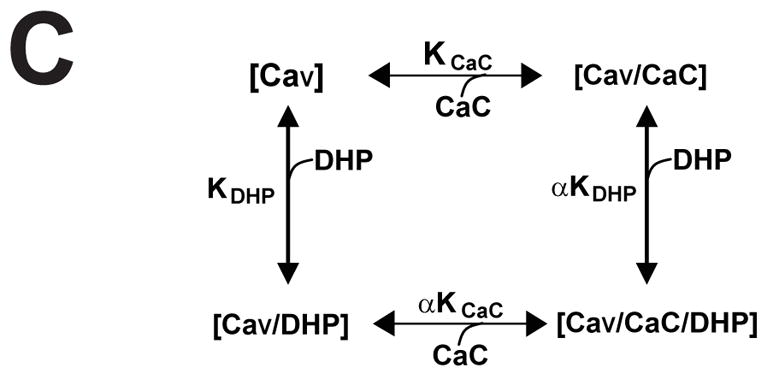
Calcicludine is a 60 amino acid peptide containing three disulfide bonds, 12 positively charged lysine or arginine residues (underlined) and two negatively charged glutamate residues. (B) Membranes from cells expressing wild-type CaV1.2 Ca2+ channels were incubated with ~350 pM [3H]PN200-110 and increasing concentrations of calcicludine. Each experiment was fit using Scheme 1 (see Equation 4, Materials and Methods) and normalized such that the occupancy in zero calcicludine is equal to 1.0. Note that [3H]PN200-110 binding increases as calcicludine is raised from 3 nM to 3 μM. Binding data are means ± SEM, and calcicludine concentrations where DHP binding differs significantly from that with no toxin (determined by ANOVA) are indicated with asterisks (*; P<0.05; n=6). Error bars smaller than symbols do not appear in figures. (C) Allosteric binding model for DHP and calcicludine binding. See Materials and Methods and text for details.
Schweitz et al. reported that N- and P/Q-type currents are effectively blocked by calcicludine (16). In contrast, Stotz et al. found that currents through CaV2.1, CaV2.2 and CaV2.3 Ca2+ channels are only modestly inhibited (<10%) by 100 nM calcicludine (19). Chimeric Ca2+ channels were constructed to identify which domain(s) of CaV1.2 contain calcicludine binding determinants (19). The level of block was reduced through chimeric channels where domains I, II and III of CaV1.2 were individually replaced with the corresponding regions of CaV2.3. These findings led the authors to conclude that calcicludine interacts with all three domains of CaV1.2. The greatest effects were observed using the domain III chimera (19).
We hypothesized that if calcicludine does indeed interact with the outer pore of CaV1.2, it may act as modulator of DHP binding, as was observed for the binding of a single Ca2+ ion to the selectivity filter (9, 10, 12). Thus, calcicludine could be used as a molecular probe to study the structural rearrangements associated with DHP binding and aid in the development of a deeper understanding of the molecular events that couple DHP binding to changes in channel block. Here, we 1) demonstrate that calcicludine and DHP antagonists bind cooperatively to CaV1.2 channels; 2) identify three acidic amino acid residues located in the outer vestibule of domain III that are critical calcicludine binding determinants; and 3) identify a cluster of acidic residues positioned at the carboxyl terminus of IIIS5 that are important for DHP (but not calcicludine) binding and the coupling between DHP and calcicludine binding. These findings are described using an allosteric binding model. We propose that DHP and calcicludine binding to their respective binding sites blocks currents through L-type Ca2+ channels by promoting similar structural rearrangements in the outer pore that stabilize the channel in a nonconducting state.
MATERIALS AND METHODS
Construction and expression of mutant Ca2+ channels
cDNAs encoding wild-type (20) and mutant CaV1.2 Ca2+ channels were co-transfected with β2a (21) and α2δ (22) into HEK293 cells by calcium phosphate precipitation as described previously (23). All cDNAs were expressed in the expression plasmid, pCDNA3 (Invitogen, Carlsbad, CA). Mutant α1C subunits were constructed by site-directed mutagenesis using polymerase chain reaction. Briefly, a NheI site was introduced by silent mutagenesis into the pore of domain III at amino acid positions 1131 and 1132. Mutagenic reverse primers and a forward primer lying upstream of an existing EcoRI site in Domain II were used in single polymerase chain reactions. The resulting PCR products were amplified using the TOPO ZeroBlunt Cloning System (Invitrogen, Carlsbad, Ca, USA). Mutant inserts were excised by EcoRI/NheI digestion and ligated into EcoRI/NheI-digested CaV1.2 vector. The integrity of the resulting mutant cDNAs was confirmed DNA sequence analysis. The general gating properties of the mutant channels used in these studies are indistinguishable from those of wild-type.
Membrane preparation and radioligand binding
Membranes were harvested 2–3 days following transfection. Cells were washed twice, transferred to a glass-tephlon homogenizer and homogenized on ice in Binding Buffer (50 mM Tris, 100 μM phenylmethylsulfonyl fluoride, 100 μM benzamidine 1.0 μM pepstatin A, 1.0 μg/μl leupeptin, and 2.0 μg/ml aprotinin, pH 8.0). The homogenate was centrifuged at 1700 × g for 10 min at 4°C and the resulting pellet was re-homogenized and centrifuged. The final pellet was discarded and the supernatants were combined and centrifuged at 100,000-× g at 4°C for 30 min. The resulting membrane pellet was washed and homogenized in Binding Buffer. Membrane aliquots were stored at −70°C and remained stable for several months as determined by radioligand binding, but were typically used within one week of harvesting.
Saturation binding assays were performed in Binding Buffer, 1 mM CaCl2, 20–100 μg of membrane protein and 0.1–10 nM (+)-[3H]PN200-110 (PerkenElmer Life and Analytical Sciences, Boston, MA). Reactions were incubated 2 hours at 22–24°C and each data point was determined by three replicates. No specific binding was detected using membranes from untransfected cells. Assays designed to evaluate the cooperativity between DHP and calcicludine binding were performed in Binding Buffer, 1 mM CaCl2, 20–100 mg membrane protein and the indicated concentrations of calcicludine (Alomone Labs, Tel Aviv, Isreal) and each data point was determined by four replicates. After 30 minutes at 22–24°C, (+)-[3H]PN200-110 was added at a concentration equal to KDHP. This concentration of (+)-[3H]PN200-110 produces a fractional occupancy of 0.5 in the absence of calcicludine. Nonspecific binding was determined in all experiments by the addition of 1 μM (±)PN200-110, thus reducing kon for the radiolabeled ligand to an insignificant level. Bound radioligand was recovered by vacuum filtration through GF/C glass fiber filters.
DHP binding as a function of Calcicludine concentration was fit using Scheme 1 (Fig. 1) with the aid of the analysis and graphics programs EXCEL (Microsoft, Redmond, WA) and ORIGIN (OriginLab Corp., Northampton, MA). KDHP and KCaC can be determined from Scheme 1 using the following relationships:
where KDHP, KCaC and α correspond to the dissociation constants for [3H]PN200-110 (DHP) and calcicludine (CaC) and a unitless coupling factor (α) that is a measure of the strength of the cooperativity between the DHP and calcicludine binding sites. [CaV], [CaV·DHP], [CaV · CaC] and [CaV·DHP·CaC] correspond to binding states with no ligand, only DHP, only calcicludine and both DHP and calcicludine, respectively, bound to the channel. The population of CaV1.2 channels is distributed between each of the four binding states depicted in Scheme 1 and the fractional occupancies of all four states add to one:
| Equation 1 |
Scheme 1.
The above relationships can be used to describe each binding state depicted in Equation 1:
Of these four binding states, only [CaV·DHP] and [CaV·DHP·CaC] are represented in radioligand binding experiments—i.e., Total Bound = [CaV·DHP] + [CaV·DHP·CaC]. The occupancy of state [CaV·DHP] is solved by:
Divide by [CaV·DHP]:
| Equation 2 |
The occupancy of state [CaV·DHP·CaC] is solved by:
Divide by [CaV·DHP·CaC]:
| Equation 3 |
The sum of the inverses of equations 2 and 3 is a measure of the total bound [3H]PN200-110 at any given concentration of calcicludine and [3H]PN200-110:
| Equation 4 |
Equation 4 was used to fit the binding data depicted in Figs. 1B, 3A and 3B.
FIGURE 3. Three acidic amino acid residues, Glu-1122, Asp-1127 and Asp-1129, positioned proximal to the pore helix in domain III are important for calcicludine binding to CaV1.2 channels.

Wild-type, Mut-A, Mut-B and Mut-C (A) and Wild-type, E1122A, D1127A and D1129A (B) Ca2+ channels were incubated with [3H]PN200-110 and the indicated concentrations of calcicludine as described in the Materials and Methods. Data were fit using Scheme 1 (see Equation 4, Materials and Methods) and normalized such that the occupancy in zero calcicludine is equal to 1.0. The method for fitting binding data from the individual mutants, E1122A, D1127A and D1129A, is described in the text and Legend for Table 1. DHP binding to Mut-A and Mut-C membranes does not increase in the presence of calcicludine, but block of Mut-A and not Mut-C currents by calcicludine is similar to that of wild-type (Fig. 5; see text for details). Data are means ± SEM, and significant differences between binding parameters of wild-type and mutant channels were evaluated using a 2-tailed Students-t test P<0.05 (*). Error bars smaller than symbols do not appear in figures. See Table 1 for data summary and indications of statistical significance. (A) Wild-type (n = 6), Mut-A (n = 3), Mut-B (n = 3), Mut-C (n = 3); (B) Wild-type (n = 6), E1122A (n = 3), D1127A (n = 3), D1129A (n = 3).
Patch-clamp electrophysiology
Whole-cell currents were recorded at room temperature 2–3 days after transfection. Pipettes were pulled from borosilicate glass (1B150F-3; World Precision Instruments, Inc., Sarasota, FA) using a Sutter P-97 Flaming/Brown micropipette puller (Sutter Instruments Company, Novato, CA) and fire polished using a MF200 microforge (World Precision Instruments, Inc., Sarasota, FA). Pipette resistances were typically 2.5–3.0 MΩ. External solutions for whole-cell recordings contained (in mM): NMG-aspartate, 130; HEPES, 10; 4-aminopyridine, 10; glucose, 10; and BaCl2, 10. The internal solutions contained (in mM): NMG-MeSO3, 140; EGTA, 10; MgCl2, 1; MgATP, 4; and HEPES, 10. The osmolarity and pH of internal and external solutions were adjusted to 300 mmol/kg and 7.4, respectively. Data were acquired using a HEKA Epc9/2 amplifier and PULSE/PULSEFIT software (ALA Scientific Instruments, Inc., Westbury, NY). Currents were sampled at 10 kHz and filtered at 2 kHz. Series resistance was typically <6 MΩ and was compensated by ~70%. Leaks and capacitive transients were subtracted using a P/4 protocol. A homemade rapid-exchange, single-cell perfusion system was used to increase the external solution exchange rates, thus minimizing undesirable effects resulting from current rundown, and to minimize toxin consumption.
The statistical significance of the observed differences between the blocking parameters of wild-type and mutant channels was evaluated using a 2-tailed Students-t test. All data are means ± SEM and statistical significance was set at P<0.05 (*). Error bars smaller than symbols do not appear in figures.
RESULTS
Calcicludine is a positive allosteric modulator of DHP binding
Crude membranes from HEK 293 cells expressing wild-type CaV1.2 L-type Ca2+ channels were incubated with the DHP antagonist, [3H]PN200-110, and concentrations of calcicludine ranging from 3 nM to 3 μM (Fig. 1B). Calcicludine increases DHP binding by 51% with an EC50 of 372 nM, a value 4.5-fold higher than that reported for rat brain CaV1.2 Ca2+ channels expressed in HEK 293 cells (19). That calcicludine increases [3H]PN200-110 binding indicates that the two ligands bind to the Ca2+ channel simultaneously and do not compete for the same binding site. The binding data in Fig. 1B were fit using the allosteric binding model depicted in Fig. 1C (Scheme 1; Equation 4) where a single DHP receptor site is shown to transition between two affinity states (i.e., KDHP and αKDHP) and calcicludine binding to its receptor site shifts the equilibrium between these states to the right, thus stabilizing the high affinity state (i.e., αKDHP). DHP and calcicludine binding are linked in Scheme 1 by the coupling factor α, which is a unitless measure of the strength of the coupling between the two sites. Scheme 1 allows one to determine the dissociation constant for DHP binding at any calcicludine concentration and, conversely, the dissociation constant for calcicludine binding at any DHP concentration.
To better understand the positive cooperativity between the DHP and calcicludine binding sites and how each ligand interacts with specific structural determinants to alter the functional behavior of the Ca2+ channel, we used site-directed mutagenesis to localize individual amino acid determinants for calcicludine binding. We chose to limit this screen for calcicludine binding determinants to acidic residues in the segment connecting S5 to the selectivity filter in domain III (the IIIS5/P segment) for the following reasons: 1) our previous studies indicate that non-glutamate amino acid residues in the pore of domain III are associated with structural changes that occur upon DHP binding (12, 23); 2) Analysis of chimeric CaV1.2/CaV2.3 Ca2+ channels suggests that domain III possesses the most important determinants for calcicludine block (19). 3) Calcicludine is a highly basic peptide toxin that possesses a 12 positively charged lysine and arginine residues and only two acidic glutamate residues. Since basic residues of the BPTI/Kunitz dendrotoxins are known to interact with acidic residues in the S5/P segment of the outer vestibule of K+ channels (24, 25), we reasoned that similar interactions would be important for calcicludine binding to CaV1.2. In the following sections we use site directed mutagenesis, radioligand binding and whole-cell patch-clamp electrophysiology to localize individual molecular determinants for calcicludine binding.
Acidic residues proximal to the extracellular portion of IIIS5 are important binding determinants for [3H]PN200-110
The screen for amino acid residues critical for calcicludine binding was initiated by changing all nine acidic residues located in the IIIS5/P segment to alanine in three groups of three, resulting in the mutants Mut-A, Mut-B and Mut-C (Fig. 2A). Amino acids were changed to alanine in these studies because alanine substitutions eliminate the acidic properties of the substituted residues without causing global conformational changes in the channel protein (26). Initially, KDHP values were determined by performing saturation binding experiments on membranes derived from cells expressing wild-type and all three mutant Ca2+ channels using the DHP antagonist [3H]PN200-110. Fig. 2B demonstrates that [3H]PN200-110 binds to a single population of sites on membranes expressing wild-type CaV1.2 channels with a dissociation constant of 367 pM. In contrast, KDHP for [3H]PN200-110 binding to Mut-A membranes is increased to 1.7 nM (i.e., 4.5-fold larger than wild-type). KDHP values Mut-B and Mut-C are similar to that of wild-type (Fig. 2C).
FIGURE 2. Acidic residues proximal to the extracellular portion of IIIS5 are important binding determinants for [3H]PN200-110.

(A) Amino acid sequence of CaV1.2 beginning at the C-terminus of IIIS5 through the pore-loop. Peptide segments corresponding to regions spanned by Mut-A, Mut-B and Mut-C are indicated by lines above sequence. Glu-1122, Asp-1127 and Asp-1129 are indicated with arrows. The Pore Helix and P-Loop are indicated by lines below the sequence. (B) Saturation binding experiment using membranes derived form cells expressing wild-type CaV1.2 channels was performed as described in the Materials and Methods. The signal-to-noise can be assessed by comparing the relative levels of specific (open circles) and non-specific (solid circles) binding. (inset) Scatchard transformation of data from Panel B indicates that the cells express a single population of high affinity receptor sites. (C) Similar analyses were performed on independent membrane preparations derived from cells expressing Wild-type (n = 6), Mut-A (n = 3), Mut-B (n = 3) and Mut-C (n = 3) channels (see Table 1).
Three acidic amino acid residues, Glu-1122, Asp-1127 and Asp-1129, positioned proximal to the pore helix in domain III are important for calcicludine binding to CaV1.2 channels
The levels of [3H]PN200-110 binding to membranes prepared from cells expressing Mut-A, Mut-B and Mut-C channels were compared to wild-type in the presence of various concentrations of calcicludine using Scheme 1. In contrast to wild-type, DHP binding to Mut-A and Mut-C membranes is completely insensitive to calcicludine at concentrations up to 3 μM (Fig. 3A). Calcicludine enhances [3H]PN200-110 binding to Mut-B membranes with values for KCaC and α similar to that determined for wild-type (Fig. 3A; Table 1). Since block of Mut-A (but not Mut-C) currents by calcicludine is indistinguishable from that of wild-type (Fig 5A), efforts to localize individual determinants critical for calcicludine binding and block were limited to the segment spanning Mut-C.
TABLE 1.
Binding parameters for wild-type and mutant Ca2+ channels.
| KDHP (pM) | KCaC (%M) | % | |
|---|---|---|---|
| Wild-type | 367 ± 19 | 0.322 ± 0.055 | 0.48 ± 0.05 |
| Mut A | 1660 ± 120 * | 0.344 ± 0.065 | ~1 * |
| Mut B | 391 ± 52 | 0.216 ± 0.024 | 0.45 ± 0.05 |
| Mut C | 460 ± 50 | n.d. | n.d. |
| E1122A | 294 ± 42 | 1.14 ± 0.20 * | n.d. |
| D1127A | 511 ± 53 | 1.61 ± 0.94 * | 0.75 ± 0.07 * |
| D1129A | 289 ± 80 | 21.0 ± 6.0 * | n.d. |
KDHP values were determined by [3H]-PN200-110 binding in the absence of calcicludine, as described in Fig. 2. KCaC values were determined using whole-cell patch-clamp electrophysiology, as described in Figs 4–5. Values for the cooperativity factor alpha (α) for wild-type, Mut A and Mut B were determined by fitting the data depicted in Fig. 3. Mut C proved to be insensitive to calcicludine, so KCaC and α for this mutant were “not determined” (n.d.). Binding data from E1122A and D1129A were fit using the mutants’ respective KDHP and KCaC values and a value for α equal to that of wild-type (i.e., 0.48). In contrast to E1122A and D1129A, it was necessary to adjust α to 0.75 to achieve a reasonable fit for D1127A.
; P<0.05.
FIGURE 5. Glu-1122, Asp-1127 and Asp-1129 confer the channel’s sensitivity to block by calcicludine.

Currents through Mut-C, but not Mut-A or Mut-B, are insensitive to block by calcicludine. (A) The fraction of wild-type, Mut-A, Mut-B and Mut-C current remaining after the application of the indicated concentrations of calcicludine (I/Ino drug) is plotted. Lines are Logistic fits through wild-type (open circles) and Mut-C (solid squares). Only 63% of the maximal Ba2+ currents through wild-type channels is blocked by saturating concentrations of calcicludine. Data for Mut-A and Mut-B are fit nicely with the same logistic function used to fit wild-type data. Data are summarized in Panel C and Table 1. Wild-type (n = 19), Mut-A (n = 4), Mut-B (n = 3), Mut-C (n = 7). (B) Ba2+ currents through E1122A, D1127A and D1129A channels exhibit a reduced sensitivity to block by calcicludine. The fraction of Ba2+ current remaining (I/Ino drug) after the addition of 0, 100, 500 and 5000 nM calcicludine to wild-type and mutant Ca2+ channels is plotted. Best fits through the data were made assuming a Hill coefficient of 1.0 and, as was observed for wild-type (Panel A), that a maximum of 63% of the total current is blocked at saturating concentrations of calcicludine. Wild-type (n = 19), E1122A (n = 6), D1127A (n = 6), D1129A (n = 4). (C) Summary of data depicted in Panels A and B (also, see Table 1). An IC50 value for Mut-C could not be determined (n.d.). IC50 values from these experiments were used to generate fits through binding data shown in Fig. 3B (see text and Legend for table 1 for details). The statistical significance of the observed differences between the blocking parameters of wild-type and mutant channels was evaluated using a 2-tailed Students-t test. Patch-clamp data are means ± SEM and significance was set at P<0.05 (*). Error bars smaller than symbols do not appear in figures.
Three mutant channels containing single amino acid substitutions corresponding to those made in Mut-C, E1122A, D1127A and D1129A, were analyzed to determine which are important for modulating DHP binding and channel block (Fig. 3B, Table 1). As expected, KDHP values for all three mutant channels are similar to those of wild-type and Mut-C (Table 1). The ability of calcicludine to increase [3H]PN200-110 binding to the mutant channels was assessed as described above. Due to the relatively large increases in KCaC and the high cost of calcicludine, it was not possible to maximize the effect calcicludine has on DHP binding to E1122A, D1127A and D1129A membranes. Therefore, curve fitting data from the mutant channels proved unreliable. To circumvent this limitation, the fits through the mutant data points (solid lines) were constructed using KCaC values determined using the whole-cell patch-clamp experiments described below (Fig. 5 and Table 1). Initially, the value for the cooperativity factor, α, was set to equal 0.48 (i.e., the same value as wild-type). The binding data for wild-type, E1122A and D1129A were fit reasonably well using these constraints. This was not the case for D1127A (dashed line). It was necessary to increase α from 0.48 to 0.75 to obtain a reasonable fit through the data for D1127A (arrows). These results suggest that positively charged residues on calcicludine interact with Glu-1122, Asp-1127 and Asp-1129 in the outer vestibule of domain III of CaV1.2 and that, Asp-1127 may play an additional role in coupling DHP binding to calcicludine binding.
Glu-1122, Asp-1127 and Asp-1129 confer the channel’s sensitivity to block by calcicludine
In Fig. 4, whole-cell patch-clamp electrophysiology was used to characterize the basic functional and pharmacological properties of cells expressing wild-type and mutant channels. Initially, it was found that of Mut-A, Mut-B and Mut-C, only Mut-C is insensitive to block by calcicludine (Fig. 4). In Figs. 4A and 4D, cells were depolarized every 20 sec to −10 mV from a holding potential of −100 mV in the presence of 0, 100, 500 and 5000 nM calcicludine. Cells expressing wild-type channels are blocked by calcicludine with an IC50 of 322 nM and, in contrast to the findings of Stotz et al. (2000) and Schweitz et al. (1994), a small statistically insignificant amount of current (~8%) is typically recovered upon washout of the toxin. Consistent with the findings of Stotz et al. (2000), but not Schweitz et al. (1994), only 63% of the wild-type current is sensitive to calcicludine. Also in agreement with Stotz, et al. (2000), we did not observe a change in the half-activation potential of wild-type or mutant CaV1.2 currents upon application of 500 nM calcicludine (Figs. 4C). In contrast to wild-type, Mut-A and Mut-B, Mut-C is resistant to block by calcicludine at concentrations up to 5000 nM (Figs. 4D, 4E, and 4F). Thus, the IC50 for block of Mut-C by calcicludine must be at least 500 μM. These findings are summarized in Fig. 5A. Block of currents through the single mutants, E1122A, D1127A and D1129A, by calcicludine were increased 3.5-, 5.0- and ~65-fold, respectively, above wild-type, indicating that all three residues contribute to calcicludine binding (Figs. 5B and 5C).
FIGURE 4. Mut-C channels are insensitive to by calcicludine.

(A, D) Peak amplitudes of Ba2+ currents evoked by 300 msec step depolarizations from −100 to −10 mV every 20 seconds from cells expressing wild-type (A) and Mut-C (D) Ca2+ channels in the presence of 0, 100, 500 and 5000 nM calcicludine. Note that Mut-C currents are insensitive to block by calcicludine. (B, E) Sample traces resulting from voltage steps from −100 to −10 mV in the absence and presence of 500 nM calcicludine. (C, F) Current-voltage relations in the absence and presence of calcicludine were generated by depolarizing cells from a holding potential of −100 mV to 300 msec step depolarizations to potentials ranging from −60 to +80 mV. Peak currents were plotted against corresponding test voltages to give the current-voltage relationship (I-V). These data were fit using the equation, I=G(Vm−Vrev)/(1+exp[(Vh−Vm)/k]), where G is the maximal slope conductance, Vrev is the reversal potential, Vm is the membrane potential, Vh is the half activation potential and k is the slope factor. I-V measurements were made in each cell in the absence and presence of calcicludine, and were normalized by dividing the peak current at each step potential by the peak of the fit through I-V data acquired in the absence of calcicludine. Current voltage relations indicate that the gross gating properties of Mut-C (F) are similar to that of wild-type (D).
DISCUSSION
Here, we used calcicludine, a toxin from the venom of the green mamba snake, as a molecular probe to obtain a deeper understanding of the mechanism by which DHP antagonists block L-type Ca2+ channels. We found that the DHP, [3H]PN200-110, binds cooperatively with calcicludine to cardiac CaV1.2 Ca2+ channels expressed in HEK 293 cells (Fig. 1). The positive cooperativity observed between calcicludine and [3H]PN200-110 can only occur if both ligands bind simultaneously to the channel, so calcicludine appears to be an allosteric modulator of DHP binding. Scheme 1 was developed to model the interactions between DHP and calcicludine binding. Neutralization of three acidic amino acid residues, Glu-1122, Asp-1127 and Asp-1129, located adjacent to the pore helix of Repeat III results in mutant channels that have reduced sensitivity to block by calcicludine (Figs. 4 and 5; Table 1). Similar results were obtained using an allosteric binding assay (Fig. 3) where data were fit nicely using IC50 values for half maximal block of Ba2+ currents through the mutant channels. These independent experimental approaches (one a DHP-independent functional assay and the other a DHP-dependent binding assay) suggest that calcicludine interacts directly with acidic amino acid residues, Glu-1122, Asp-1127 and Asp-1129. Below, we use a domain-interface model to explain the molecular details that underlie the positive cooperativity between the DHP and calcicludine binding sites and propose a molecular mechanism to explain how calcicludine and DHPs modulate Ca2+ channel activity.
The molecular basis for positive cooperativity between DHP and calcicludine binding
The domain-interface theory predicts that allosteric ligands bind at structural interfaces, possibly because the receptor protein is most flexible in such regions (7, 27–31). The domain interface theory can be used to explain how the calcicludine and DHP binding sites are allosterically coupled, as well. Our findings indicate that calcicludine binds to a peptide segment including and proximal to the pore helix in repeat III. Structural models of L-type Ca2+ channels predict that this segment is nestled between IIIS5, IIIS6 and IVS6 (32–34). Thus, although DHPs and calcicludine possess very distinct chemical and physical properties, their respective binding sites share an important feature: both binding sites are located at the interface between repeats III and IV. Binding of either ligand would be expected to alter the interactions between repeats III and IV and, consequently, the functional properties of the channel. The positive cooperativity between DHP antagonists and calcicludine likely occurs because the two ligands bind at the interface between the pore motifs of repeats III and IV and stabilize a common conformational state in the outer pore. Consequently, DHP binding becomes more energetically favorable in the presence of calcicludine and, though not tested, vise versa. This model for DHP and calcicludine binding predicts that interdomain movements resulting from the binding of either ligand shift the channel from a permeant state to a blocked state. This scenario is consistent with our previous reports that DHP antagonists block Ca2+ channels by promoting structural rearrangements in the outer pore (12). In these studies, we postulated that the outer pore of an activated channel switches between conducting and nonconducting, blocked states and that the overall open probability (PO) of the channel is determined by the probability that the inner gate is open and whether the outer pore is in its conducting or non-conducting state. These findings further suggest that DHP antagonists (and perhaps calcicludine) block Ca2+ currents by promoting structural rearrangements in the outer pore that correspond to the nonconducting state.
The domain interface model for the allosteric interactions between the calcicludine and DHP receptor sites is further supported by the observation that the amino acid substitutions in Mut-A located in the extracellular loop proximal to IIIS5 are important determinants for DHP binding (Fig. 2D; Table 1). The DHP receptor site is thought to lie deep within the lipid bilayer, yet the segment containing the amino acid substitutions in Mut-A is predicted to be extracellular. Thus, the DHP receptor site and the amino acid residues altered in Mut-A are not spatially oriented in a way that would allow the two regions of the channel to form a single binding site. Interestingly, the dissociation constant for [3H]PN200-110 binding (i.e., KDHP) to Mut-A membranes is 4.5-fold larger than wild-type (Fig. 2). Since the sensitivity of Mut-A currents to calcicludine is similar to that of wild-type (Fig. 3A), it appears that the dissociation constant for calcicludine binding, KCaC to Mut-A is unaltered, but that KDHP increases 4.5-fold and the cooperativity between the DHP and calcicludine binding sites, α, increases from 0.5 to ~1.
It seems unlikely that the amino acid residues substituted in Mut-A contribute directly to the formation of the DHP receptor site, so these substitutions may affect KDHP and α via an allosteric mechanism. The most reasonable explanation for these findings is that one or more of the three acidic amino acid residues substituted in Mut-A plays is important for stabilizing the interaction between repeats III and IV in a way that is favorable for DHP binding. Amino acid substitutions at this position would then destabilize these interactions and DHP binding would become less favorable. Thus, drugs that target amino acid residues that lie within the Mut-A peptide segment may function as allosteric modulators of voltage-gated Ca2+ channels.
Calcicludine and δ-dendrotoxin share similar pharmacological properties
The selectivity filters of CaV1.2 differs from that of KcsA and other K+ channels, but the gross structural features of their respective pores are predicted to be conserved. Therefore, we compared the results of our localization studies with those of Imredy et al. who characterized the binding δ-dendrotoxin to the inward rectifying (Kir1.1) and voltage-gated (KV1.1) K+ channels (24, 25). Like calcicludine, δ-dendrotoxin is a member of the BPTI/Kunitz-like toxin family (15, 18). Neutralization of Glu-123 of Kir1.1 results in a mutant channel with an IC50 for half-maximal block by δ-dendrotoxin that is more than 250-fold larger than wild-type (24), and substitution of alanine for Asp-431 results in a 160-fold increase in the IC50 for half maximal block of KV1.1 channels by δ-dendrotoxin (25). As with Glu-1122, Asp-1127 and Asp-1129 of CaV1.2, Glu-123 of Kir1.1 and Asp-431 of KV1.1 are located within and proximal to the pore helix. Therefore, the interactions between calcicludine and δ-dendrotoxin and their respective channel targets appear to be conserved.
In their later study, Imredy and Mackinnon (2000) found that one face of δ-dendrotoxin interacts off-center in the pore with three residues on the turret and an aspartate residue (Asp-431 of KV1.1) positioned in the outer portion of the pore-helix between two K+ channel subunits. The similarities between our results and those of Imredy and MacKinnon (2000) and Stotz et al. (2000) indicate that calcicludine and δ-dendrotoxin bind to their respective receptor sites in a similar manner and suggests that the two toxins may alter channel activity via a similar modus operandi: i.e., by partially occluding the permeation pathway (19, 24, 25). As noted by Stotz et al. (2000), our findings can also be explained by an allosteric model. The selectivity filter appears to be a dynamic structure that undergoes structural rearrangements in response to changes in channel activity. For example, a pore containing no divalent cations is predicted to be 6 Å in diameter (35) and freely conducts monovalent cations. Occupancy of the pore by a single divalent cation introduces electrostatic forces that cause the selectivity filter to collapses to 2.8 Å (33), a structure that corresponds to the nonconducting, blocked state stabilized by DHP antagonists (12). Occupancy by a second divalent cation introduces repulsive forces in the selectivity filter and stabilizes a state that conducts divalent cations. Calcicludine binding to the outer vestibule of the pore may promote a pore structure that is structurally analogous to the non-conducting, blocked state stabilized by DHP antagonists.
Acknowledgments
We thank Cara Martinez-Williams for technical assistance.
Abbreviations
- DHP
dihydropyridine
- EEEE locus
selectivity filter
- PN200-110
4-(4-Benzofurazanyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid methyl 1-methylester
Footnotes
This work was supported by research grants from the American Heart Association (0230298N) and the National Institutes of Health (RO1 HL074143) to B.Z.P.
References
- 1.Hille B. Ion channels of excitable membranes. 3. Sinaur Associates, Inc.; Sunderland, MA: 2001. [Google Scholar]
- 2.Herbert LG. Membrane pathways for drug/ion channel interactions: molecular basis for pharmacokinetic properties. Drug Development and Research. 1994:214–224. [Google Scholar]
- 3.Bangalore R, Baindur N, Rutledge A, Triggle DJ, Kass RS. L-type calcium channels: asymmetrical intramembrane binding domain revealed by variable length, permanently charged 1,4-dihydropyridines. Mol Pharmacol. 1994:660–666. [PubMed] [Google Scholar]
- 4.He M, Bodi I, Mikala G, Schwartz A. Motif III S5 of L-type calcium channels is involved in the dihydropyridine binding site. A combined radioligand binding and electrophysiological study. J Biol Chem. 1997;272:2629–2633. doi: 10.1074/jbc.272.5.2629. [DOI] [PubMed] [Google Scholar]
- 5.Mitterdorfer J, Wang Z, Sinnegger MJ, Hering S, Striessnig J, Grabner M, Glossmann H. Two amino acid residues in the IIIS5 segment of L-type calcium channels differentially contribute to 1,4-dihydropyridine sensitivity. J Biol Chem. 1996:30330–30335. doi: 10.1074/jbc.271.48.30330. [DOI] [PubMed] [Google Scholar]
- 6.Peterson BZ, Johnson BD, Hockerman GH, Acheson M, Scheuer T, Catterall WA. Analysis of the dihydropyridine receptor site of L-type calcium channels by alanine-scanning mutagenesis. J Biol Chem. 1997:18752–18758. doi: 10.1074/jbc.272.30.18752. [DOI] [PubMed] [Google Scholar]
- 7.Peterson BZ, Tanada TN, Catterall WA. Molecular determinants of high affinity dihydropyridine binding in L- type calcium channels. J Biol Chem. 1996:5293–5296. doi: 10.1074/jbc.271.10.5293. [DOI] [PubMed] [Google Scholar]
- 8.Schuster A, Lacinova L, Klugbauer N, Ito H, Birnbaumer L, Hofmann F. The IVS6 segment of the L-type calcium channel is critical for the action of dihydropyridines and phenylalkylamines. Embo J. 1996;15:2365–2370. abs.html. [PMC free article] [PubMed] [Google Scholar]
- 9.Mitterdorfer J, Sinnegger MJ, Grabner M, Striessnig J, Glossmann H. Coordination of Ca2+ by the pore region glutamates is essential for high-affinity dihydropyridine binding to the cardiac Ca2+ channel alpha 1 subunit. Biochemistry. 1995:9350–9355. doi: 10.1021/bi00029a010. [DOI] [PubMed] [Google Scholar]
- 10.Peterson BZ, Catterall WA. Calcium binding in the pore of L-type calcium channels modulates high affinity dihydropyridine binding. J Biol Chem. 1995:18201–18204. doi: 10.1074/jbc.270.31.18201. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Ponoran TA, Rasmusson RL, Ragsdale DS, Peterson BZ. Amino Acid Substitutions in the Pore of the CaV1.2 Calcium Channel Reduce Barium Currents without Affecting Calcium Currents. Biophys J. 2005:1731–1743. doi: 10.1529/biophysj.104.058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson BZ, Catterall WA. Allosteric interactions required for high-affinity binding of dihydropyridine antagonists to Ca(V)1.1 Channels are modulated by calcium in the pore. Mol Pharmacol. 2006:667–675. doi: 10.1124/mol.105.020644. [DOI] [PubMed] [Google Scholar]
- 13.Menez A. Perspectives in Molecular Tocicology. John Wiley & Sons, Ltd; West Sussex: 2002. [Google Scholar]
- 14.Swaminathan P, Hariharan M, Murali R, Singh CU. Molecular structure, conformational analysis, and structure-activity studies of Dendrotoxin and its homologues using molecular mechanics and molecular dynamics techniques. J Med Chem. 1996:2141–2155. doi: 10.1021/jm950579p. [DOI] [PubMed] [Google Scholar]
- 15.Gilquin B, Lecoq A, Desne F, Guenneugues M, Zinn-Justin S, Menez A. Conformational and functional variability supported by the BPTI fold: solution structure of the Ca2+ channel blocker calcicludine. Proteins. 1999:520–532. [PubMed] [Google Scholar]
- 16.Schweitz H, Heurteaux C, Bois P, Moinier D, Romey G, Lazdunski M. Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for L-type channels in cerebellar granule neurons. Proc Natl Acad Sci U S A. 1994:878–882. doi: 10.1073/pnas.91.3.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zupunski V, Kordis D, Gubensek F. Adaptive evolution in the snake venom Kunitz/BPTI protein family. FEBS Lett. 2003:131–136. doi: 10.1016/s0014-5793(03)00693-8. [DOI] [PubMed] [Google Scholar]
- 18.Harvey AL. Twenty years of dendrotoxins. Toxicon. 2001:15–26. doi: 10.1016/s0041-0101(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 19.Stotz SC, Spaetgens RL, Zamponi GW. Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J Membr Biol. 2000:157–165. doi: 10.1007/s002320001040. [DOI] [PubMed] [Google Scholar]
- 20.Wei XY, Perez-Reyes E, Lacerda AE, Schuster G, Brown AM, Birnbaumer L. Heterologous regulation of the cardiac Ca2+ channel alpha 1 subunit by skeletal muscle beta and gamma subunits. Implications for the structure of cardiac L-type Ca2+ channels. J Biol Chem. 1991;266:21943–21947. [PubMed] [Google Scholar]
- 21.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L. Cloning and expression of a cardiac/brain beta subunit of the L-type calcium channel. J Biol Chem. 1992:1792–1797. [PubMed] [Google Scholar]
- 22.Tomlinson WJ, Stea A, Bourinet E, Charnet P, Nargeot J, Snutch TP. Functional properties of a neuronal class C L-type calcium channel. Neuropharmacology. 1993:1117–1126. doi: 10.1016/0028-3908(93)90006-o. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Ponoran TA, Rasmusson RL, Ragsdale DS, Peterson BZ. Amino acid substitutions in the pore of the Ca(V)1.2 calcium channel reduce barium currents without affecting calcium currents. Biophys J. 2005:1731–1743. doi: 10.1529/biophysj.104.058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imredy JP, Chen C, MacKinnon R. A snake toxin inhibitor of inward rectifier potassium channel ROMK1. Biochemistry. 1998:14867–14874. doi: 10.1021/bi980929k. [DOI] [PubMed] [Google Scholar]
- 25.Imredy JP, MacKinnon R. Energetic and structural interactions between delta-dendrotoxin and a voltage-gated potassium channel. J Mol Biol. 2000:1283–1294. doi: 10.1006/jmbi.2000.3522. [DOI] [PubMed] [Google Scholar]
- 26.Blaber M, Zhang XJ, Matthews BW. Structural basis of amino acid alpha helix propensity. Science. 1993:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- 27.Barford D, Johnson LN. The allosteric transition of glycogen phosphorylase. Nature. 1989;340:609–616. doi: 10.1038/340609a0. [DOI] [PubMed] [Google Scholar]
- 28.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamoylase: the molecular basis for a concerted allosteric transition. Trends Biochem Sci. 1990:53–59. doi: 10.1016/0968-0004(90)90176-c. [DOI] [PubMed] [Google Scholar]
- 29.Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Liang JY, Huang S, Lipscomb WN. Toward a mechanism for the allosteric transition of pig kidney fructose-1,6-bisphosphatase. J Mol Biol. 1994:609–624. doi: 10.1006/jmbi.1994.1755. [DOI] [PubMed] [Google Scholar]
- 31.Lipkind GM, Fozzard HA. Molecular modeling of interactions of dihydropyridines and phenylalkylamines with the inner pore of the L-type Ca2+ channel. Mol Pharmacol. 2003:499–511. doi: 10.1124/mol.63.3.499. [DOI] [PubMed] [Google Scholar]
- 32.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 33.Lipkind GM, Fozzard HA. Modeling of the outer vestibule and selectivity filter of the L-type Ca2+ channel. Biochemistry. 2001:6786–6794. doi: 10.1021/bi010269a. [DOI] [PubMed] [Google Scholar]
- 34.Zhorov BS, Folkman EV, Ananthanarayanan VS. Homology model of dihydropyridine receptor: implications for L-type Ca(2+) channel modulation by agonists and antagonists. Arch Biochem Biophys. 2001:22–41. doi: 10.1006/abbi.2001.2484. [DOI] [PubMed] [Google Scholar]
- 35.McCleskey EW, Almers W. The Ca channel in skeletal muscle is a large pore. Proc Natl Acad Sci U S A. 1985:7149–7153. doi: 10.1073/pnas.82.20.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng ZP, Hamid J, Doering C, Jarvis SE, Bosey GM, Bourinet E, Snutch TP, Zamponi GW. Amino acid residues outside of the pore region contribute to N-type calcium channel permeation. J Biol Chem. 2001:5726–5730. doi: 10.1074/jbc.C000791200. [DOI] [PubMed] [Google Scholar]
- 37.Williamson AV, Sather WA. Nonglutamate pore residues in ion selection and conduction in voltage- gated Ca(2+) channels. Biophys J. 1999:2575–2589. doi: 10.1016/s0006-3495(99)77092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]